
ABSTRACT
LncRNAs are novel noncoding RNAs involved in the epigenetic regulation of gene expression by recruiting ribonucleoprotein complexes to specific genomic loci to initiate histone methylation and/or other chromatin modifications. LncRNAs themselves function as tumor suppressors or oncogenes, depending on the gene regulatory networks they govern. We identified lnc00673 (ERRLR01) as a marker of overall survival (OS) in breast cancer patients. Specifically, ERRLR01 levels were elevated in triple-negative breast cancer (TNBC) as compared with Luminal-A, Luminal-B, and HER2 breast cancer subtypes. ERRLR01 levels were also inversely correlated with breast cancer survival across all breast cancer patients. Upon stratification, OS in ERα− tumors correlated with negative overall survival, while in ERα+ tumors, ERRLR01 correlated with positive outcomes. This suggests ERRLR01 is modulated by hormone signaling in breast cancer. Gene-network analysis revealed ERRLR01 correlated with distinct pathways including “epithelial development” and “cellular differentiation.” These data suggest ERRLR01 operates as an oncogene in TNBC, as well as a biomarker in breast cancer patients.
Introduction
Long-noncoding RNAs function as decoys, regulators of translation, and/or molecular scaffolds that recruit chromatin modifying enzymes to distinct genomic loci.Citation1-4 LncRNA transcripts are transcribed in sense and/or anti-sense orientation,Citation5,6,7 and lncRNA transcription depends upon a tightly controlled regulatory network that as of yet, is still not well understood. The ENCODE Consortium determined the abundance and genomic location of many lncRNAs across several species, which in various cases are conserved through positional synteny.Citation8,9 LncRNAs reside near protein coding regions and function in cis to maintain the surrounding chromatin in an open and epigenetically active state. In other cases, lncRNAs are transcribed in an anti-sense orientation to a protein-coding gene, which in turn recruits histone chromatin-modifying complexes that support gene silencing.Citation10,11 The nomenclature for a particular lncRNA is derived, in part, from the closest neighboring protein coding gene. For instance, lincRNA-p21 neighbors CDKN1A and regulates CDKN1A (p21) expression, as evidenced by knockdown studies where lincRNA-p21 loss phenocopies the effects imparted by the loss of CDKN1A.Citation12,13 Additionally, genes such as ANRIL (antisense noncoding RNA in the INK4 locus), which is an antisense transcript produced within the CDKN2B locus, recruits the PRC1/2 complex and inhibits CDKN2B gene expression.Citation14,15
There has been a recent effort to elucidate lncRNA function by identifying particular cellular states and/or pathophysiologies associated with dysregulated lncRNA expression.Citation16 In this context, modulating lncRNA abundance, either through gain- or loss-of-function approaches serves as an opportunity to understand which gene-regulatory networks a specific lncRNA is associated with. Furthermore, alteration of lncRNA expression that promotes changes in cellular phenotypes indicates a particular lncRNA could be a driver of certain disease or pathophysiological states. Several lncRNAs have been identified as important mediators in chronic diseases, such as PTCSC3 in papillary thyroid carcinoma, and ANRIL in type-2 diabetes.Citation17,18 Additionally, MALAT-1 is expressed in the heart, lung, and kidneys, but is elevated in lung tumor tissue and has elevated biologic activity in metastatic lung adenocarcinoma as compared with a benign or pre-malignant state.Citation19–22 HOTAIR, initially described in fibroblasts, regulates the cluster of HOX genes through cis- as well as trans-regulatory mechanisms,Citation16 and is associated with oncogenesis. HOTAIR expression levels are high in several tumors types including lung, breast, and prostate. Furthermore, ectopic expression of HOTAIR results in aberrant PRC2 function and improper recruitment of PRC2-associated complexes to the correct genomic loci. This process facilitates promiscuous gene regulation allowing for a pro-tumorigenic state.Citation23 Several other lncRNAs including PCAT-1 and GAS5 also have tumorigenic function; however, there is a growing need to elucidate the function of the nearly 118,000 recently annotated human lncRNAs. Given lncRNAs serve as decoys, function as scaffolds that mediate protein-protein interactions, and promote chromatin remodeling through recruitment of DNA-modifying enzymes to distinct genomic loci, the capability to tease out specific mechanisms associated with a particular lncRNA remains a challenge. However, determining putative lncRNA functions can be initiated through investigation of associative miRNA and mRNA gene regulatory networks, which could in turn be used to determine if a lncRNA harbors putative oncogenic potential when dysregulated.
In this study, we identified ERRLR01 (Estrogen-Receptor Related LincRNA 01), as a lncRNA whose abundance was differentially expressed across four breast cancer subtypes within Affymetrix and The Cancer Genome Atlas (TCGA) data sets. Furthermore, ERRLR01 expression correlated with overall survival (OS) and relapse-free survival (RFS) outcomes within an Affymetrix data set. Previous reports have described ERRLR01 as a prognostic marker in non-small cell lung cancer,Citation24 and as a pro-metastatic lncRNA in melanoma.Citation25 In our model, ERRLR01 was aberrantly expressed across breast cancer tumor subtypes and cell lines. Therefore, we performed gene network analysis and identified a series of pathways highly correlated with ERRLR01 expression, and further ascertained putative miRNA-lncRNA interactions that could be responsible for modulating ERRLR01 expression in specific cellular contexts. Overall, these studies indicate ERRLR01 may function as an oncogene in breast cancer, and that anti-sense strategies developed to target this lncRNA may be a new therapeutic approach to treat breast cancer patients.
Materials and methods
Gene chip database construction
To develop survival analysis software, a gene chip database was first established as described previously.Citation26 In brief, a GEO search was made to identify breast cancer gene expression datasets with available survival data, with each dataset containing at least 30 patient samples. The raw .CEL files were downloaded and MAS5 normalized in the R statistical environment (http://www.r-project.org) using the Affy Bioconductor library. Database quality control and removal of duplicate samples were performed as described.Citation27 This analysis methodology was then used on a curated Affymetrix data set described in Gyorffy, B et. al., 2013.Citation39 Patient samples from the Affymetrix data sets were primarily of Caucasian origin, and biopsies were obtained from the primary breast tumor. This curated data set contains over 2000 breast cancer patient samples, all containing survival endpoints, with a majority of samples being ERα+.Citation39
Specifically, Affymetrix data set analysis was performed whereby expression of ERRLR01 in each patient sample was determined using probe set 227452_at. The original signal was MAS5 normalized, and listed in rank-order via spearman rank analysis.Citation38,39 For ERRLR01, the probe set 227452_at was used. This probe set has specificity of one of the ERRLR01 transcripts and covers the full exonic region of the linc00673.1 variant.Citation28 ESR1 and HER2 status was determined for each sample using the probe sets 205225_at and 216836_s_at as described earlier.Citation29 To determine receptor status, the raw expression cutoff values of 500 and 1,150 were used for ESR1 and HER2, respectively.
RNA-Seq database construction
RNA-Seq measurement for breast cancer patients were published by The Cancer Genome Atlas (TCGA) of the National Cancer Institute (https://cancergenome.nih.gov/)Citation30 and we downloaded the pre-processed level 3 data generated using the Illumina HiSeq 2000 RNA Sequencing Version 2 platform. The primary tumor samples from this dataset were obtained from patients primarily of Caucasian origin. For these samples, expression levels were determined using a combination of MapSplice and RSEM. We have combined the individual patient files in R using the plyr package. TCGA data sets also underwent Kaplan-Meir survival analysis using the statistical methodologies highlighted below.
Quantitative real time PCR
For qPCR, cells were lysed in TRIzol (Life Technologies), and total RNA was used as the input for subsequent RT-PCR reactions. For lncRNA analysis, cDNA synthesis and qRT-PCR were performed according to the miScript II RT Kit (Qiagen) and miScript SYBR® Green PCR Kit (Qiagen) protocols respectively.Citation31 MiRNA levels were normalized to RNU6B levels. For mRNA analysis, cDNA synthesis and qRT-PCR were performed per the RTCitation2 First Strand Kit and RTCitation2 qPCR Primer assay (Qiagen) protocols respectively.
Genetic-network and Gene Ontology term analysis
For gene pathway analysis, Spearman-Rank analysis was performed on the entire Affymetrix data. We identified the top 200 genes most highly correlated with ERRLR01 across all patient samples. The top 200 genes are equivalent to the top 1% of protein coding genes in the Spearman-rank analysis. We used this gene set to perform Gene Ontology (GO) term analysis using the WebGestalt algorithm.Citation55 In these studies, we performed a series of data queries including GO term analysis, KEGG analysis, and miRNA target analysis. The algorithm compares the enrichment of genes uploaded in the program compared with reference gene names of the entire human genome. Utilizing hypergeometric analysis with Benjamini and Hochberg multiple test adjustment (i.e., FDR analysis), we identified significant gene pathways using a cutoff of p value < 0.05. Only pathways containing 6 genes or more from the genes loaded into the algorithm were analyzed. Spearman-Rank analysis was also used to identify genes that were the most anti-correlated with ERRLR01. Similar statistical analysis was performed, and the resulting data are highlighted in the supplemental data.
Statistical analysis
Molecular subtypes were designated per the StGallen guidelines utilizing expression of ESR1, HER2, and MKI67 (PMID: 23917950). This includes a TNBC cohort (ESR1- and HER2-negative patients), a HER2-enriched cohort (HER2-positive and ESR1-negative patients), a Luminal A cohort (ESR1-positive and HER2-negative patients with low MKI67), and a Luminal B cohort (ESR1-positive and HER2-negative patients with high MKI67 expression).
Survival analysis was performed in the R statistical environment (http://www.r-project.org) using the survival Bioconductor library. For the expression of ERRLR01, each percentile (of expression) between the lower and upper quartiles was computed and the best performing threshold was used as the final cutoff in the Cox regression analysis. Kaplan–Meier survival plot, and the hazard ratio with 95% confidence intervals and log-rank P Values were calculated and plotted in R. Kruskal-Wallis test was used to compare continuous expression among multiple cohorts and a Mann-Whitey test was used to compare each cohort tested. Statistical significance was set at p value < 0.05.
Results
ERRLR01 is highly expressed in estrogen receptor negative tumor subtypes
We performed an in-silico screen of lncRNAs that were differentially expressed across breast cancer patients, or between normal and breast cancer tissue samples. We used numerous databases including MiTranscriptome,Citation32 which is a meta-assembly of 6,503 RNA-Seq libraries (Figure S1). We identified ERRLR01 as a long noncoding RNA expressed at higher levels in breast cancer patient samples as compared with normal breast tissue. ERRLR01 is a recently described lncRNA that has sequence similarity to the steroid receptor RNA activator 1 (SRA-like-non-coding RNA), and hence termed ERRLR01. ERRLR01 is located on chromosome 17q24.3, a region associated with genomic breakpoints within a variety of cancer types including breast, lung, colorectal, and uterine leiomyomas.Citation33–37 Additionally, ERRLR01 was identified to play a role in melanoma invasion,Citation25 and was linked with worse overall survival in melanoma patients. ERRLR01 expression was also previously reported to be elevated in melanoma cell lines post invasion, suggesting ERRLR01 promotes invasion. Given the role of ERRLR01 in melanoma, and the finding that ERRLR01 was elevated in breast cancer specimens as compared with normal breast tissue, we decided to investigate whether ERRLR01 could serve as a biomarker of breast cancer progression and/or disease onset.
ERRLR01 is a predictor of overall survival in breast cancer
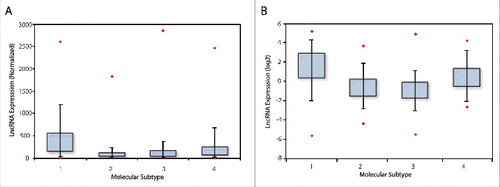
We analyzed Affymetrix U133 gene chip data, as well as the breast cancer TCGA data set and determined that across four distinct breast cancer subtypes, as determined by PAM50 classification, ERRLR01 was elevated in TNBC patient samples (). The analysis of the Affymetrix data set was performed by Spearman-rank analysis whereby expression of ERRLR01 in each patient sample was determined by probe set 227452_at and whereby the signal was MAS5 normalized, and listed in rank-order.Citation38,39 The mean rank-order across all patients within each sample subset was determined and is depicted as a box-whisker-plot (). ERRLR01 expression was elevated in both TNBC as well as HER2+ patient subtypes when compared with Luminal-A and Luminal-B subtypes. This indicated ERRLR01 was inversely correlated with ERα status in breast cancer patients. To validate this result, we assessed the expression of ERRLR01 in the breast cancer TCGA data set.Citation30 Here, ERRLR01 expression was determined using the MapSplice Algorithm,Citation40 and the resultant log(2) transformed normalized data was plotted as a box-whisker-plot (). In this datasets ERRLR01 expression was elevated in both TNBC and HER2+ patient samples. Together, results from both patient datasets indicated that ERRLR01 expression was elevated in ERα− tumor subtypes, as compared with ERα+ tumor subtypes (p value < 1 × 10−16, as determined by Kruskal-Wallis analysis).
Figure 1. LncRNA expression in breast cancer patient populations which was acquired using an Affymetrix U133A array dataset, and is depicted by box-whisker-plots. In this analysis, ERRLR01 expression was stratified into 4 subpopulations, and the mean rank expression was reported (A). 1 = TNBC, n = 577; 2 = Luminal A, n = 1432; 3 = Luminal B, n = 632; 4 = HER2+, n = 301. ERRLR01 expression in the TCGA data set (B), and data are presented in a log(2) transformed format. 1 = TNBC, n = 154; 2 = Luminal A, n = 91; 3 = Luminal B, n = 538; 4 = HER2+, n = 53. *denotes significance at P < 1 × 10−16 as determined by Kruskal-Wallis one-way variance analysis testing.
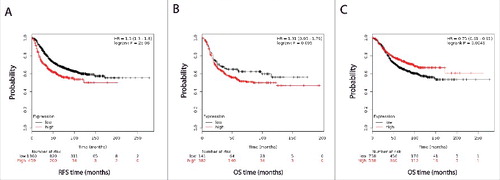
This analysis indicated that ERRLR01 may be inversely correlated with hormone signaling, as hormone-sensitive tumors expressed lower levels of ERRLR01 than those with hormone-independent tumors. To test this hypothesis Kaplan-Meir survival and Cox regression analyses were performed on the Affymetrix dataset described above. We determined that within all breast cancer patient samples, high ERRLR01 expression inversely correlated with RFS (); HR = 1.5 with a p value < 5 × 10−6. When segregating patients by ERα status, we found that ERα+ patients with high expression of ERRLR01 had better overall survival (OS) rates than those with low ERRLR01 expression (); HR = 0.75 with a p value < 0.0046. We did not perform RFS analysis after patient stratification due to smaller sample size and reduced statistical power. Interestingly, in patients with ERα− tumors, ERRLR01 expression was inversely correlated with OS (); HR = 1.31 with a p value < 0.095. This was a unique finding and suggests ERRLR01 is regulated by ERα or that ERα signaling controls ERRLR01 expression.
Figure 2. Kaplan-Meir and cox regression analysis of ERRLR01 levels and OS in breast cancer patients. Data was obtained from Affymetrix datasets, and analyzed on KM-Plotter software. (A) represents RFS in all breast cancer patients, (B) depicts OS in ER-negative breast cancer patients, and panel (C) depicts OS in ER-positive breast cancer patients. Hazard ratios and log- rank p values are reported for each analysis. ERRLR01 correlates with poor survival outcomes in “all patients” as well as “ER-negative patients,” while positively correlates with OS in “ER- positive patients.”
To test this notion further, we assessed ERRLR01–related OS in breast cancer patients after PAM50 sub-stratification. Within Basal-like (i.e., TNBC) and HER2-positive breast cancer subtypes high ERRLR01 expression inversely correlated with OS, while in Luminal-A and Luminal-B subtypes ERRLR01 expression correlated with OS (Figure S2). We attempted to validate this survival relationship using the TCGA dataset; however, we did not find any significant survival benefit based on differential ERRLR01 expression. It should be noted that overall survival analysis in TCGA is difficult to assess, given most samples are acquired from primary tumor samples with only 5–10 yr follow-up.
Characterization of the ERRLR01 locus and interacting transcription factors
Given the significant ERRLR01-related OS ratios in hormone-dependent and hormone-independent breast cancer patients, we wanted to elucidate a putative mechanism for such a relationship. To do this we first determined the genomic location of ERRLR01 and inquired as to which putative regulatory moieties could serve as signals to recruit potential RNA- and protein-binding components to the ERRLR01 transcript. ERRLR01 is positionally conserved via synteny within the human and mouse genome (). A moderate level of transcription was observed in both RNA-Seq data on nine cell lines from ENCODE, and from lncRNA RNA-Seq datasets where ERRLR01 was expressed highest in testes and brain tissue, but lower in normal breast and other tissues. Furthermore, evidence of CpG promoter methylation was observed (Methylation Score = 117) using the UCSC genome browser dataset. Overall these data support the notion that ERRLR01 is expressed at low to moderate levels in most normal tissue specimens or cell lines. These findings are also in line with the Affymetrix and TCGA datasets, where the raw FPKMs for ERRLR01 were ≤10 FPKM.
Figure 3. Genomic analysis of the ERRLR01 gene region and the regulatory factors that interact with ERRLR01 using the UCSC Genome browser. Transcription orientation is depicted from right (5’ end) to left (3’ end). (A) ERRLR01.1 is a 4-exon gene conserved between mouse and human, and is expressed at high levels in testis, placenta, and brain tissue. (B, Top Panel) Detectable transcription is present across a panel of cell lines with evidence of H3K4Me1 modification. (B, Bottom Panel) Potential estrogen-regulated transcription factor binding locations are denoted, such as the GATA family of transcription factors, CTCF, and ERα itself.
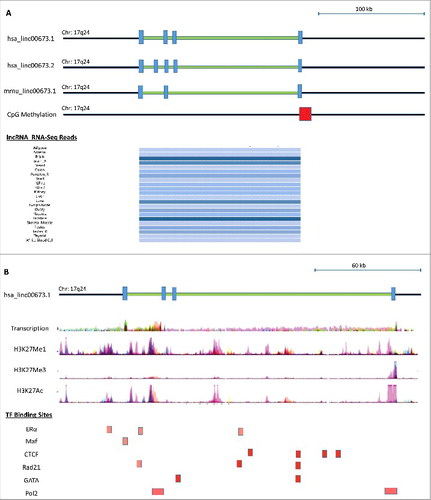
Upon further characterization of the ERRLR01 genomic loci we identified H3K4Me1 and H3K4Me3 marks between ERRLR01 exons 3 and 4 of the linc00673.1 transcript. In some cases this overlapped with CTCF binding sites, as well as other estrogen-related transcription factors (). The convergence of CTCF binding on the ERRLR01 genomic loci was of particular interest since CTCF is a well-documented 17β-estradiol regulated protein whose function is that of an insulator to prevent enhancer element binding to particular promoters.Citation41–47 As an example, CTCF is a factor known to be regulated by ERα in MCF-7 cells, and therefore could also affect ERRLR01 levels in these 17β-estradiol-sensitive cell lines. This notion is supported by GEO datasets, (see Figure S3). Therefore, ERRLR01 could operate as an oncogene as is suggested by some groups, yet in hormone-sensitive tumors a 17β-estradiol-CTCF regulatory axis could be reducing the activity of ERRLR01. In support of this, at least 3 ERα binding sites were identified on the 3’ end of the ERRLR01 transcript. Whether these sites are functional or serve as co-activator versus co- repressor binding sites is unclear. Further in silico analysis indicated that the family of GATA transcription factors (including GATA3) also bound to the exonic regions of ERRLR01 (). Further work is required to determine the functionality of these sites.
Potential RNA- and miRNA- binding sites within the ERRLR01 locus
To further understand the potential RNA-regulatory network that ERRLR01 is a part of, we used several databases to interrogate whether certain RNA-binding proteins interacted with ERRLR01, or whether specific miRNAs could bind the ERRLR01 transcript. The strongest RNA binding protein interaction we identified with was FUS (). FUS binding to ERRLR01 was confirmed through the StarBase 2.0 algorithmCitation45 and is denoted as a HHF3_128133 binding site (). Other RNA binding protein sites were identified through HITS-CLIP data; however, most these sites require confirmation through RNA-IP experiments to verify these interactions. It was interesting to note that DGCR8 bound to intronic regions of ERRLR01 indicating that a miRNA sequence may be derived from this host sequence.
Figure 4. (A) In silico assessment of RNA binding proteins that interact with ERRLR01 as determined by HITS-CLIP experiments. StarBase 2.0 was used to confirm FUS binding to the ERRLR01 locus. To focus on the 3’ UTR-miRNA binding sites the ERRLR01 transcript is depicted from right (3’ end) to left (5’ end), and only includes the 3’ UTR through Exon 2. Orientation is depicted in this manner since ERRLR01 is transcribed in the anti-sense orientation (derived from the minus strand of the DNA). The ERRLR01 transcript is depicted in the 3’ to 5’ orientation, as it is transcribed in the anti-sense orientation (minus strand of the DNA).
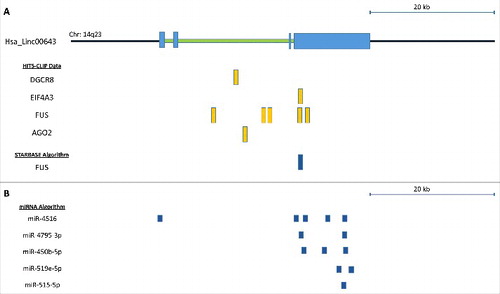
Next we asked whether there were any regulatory interactions between miRNAs and the ERRLR01 transcript. Using the miRdB algorithmCitation46 we observed several putative miRNA interactions across the ERRLR01 transcript, and the top 5 miRNA candidates are being depicted (). Many of these miRNAs have unknown functions, however miR-515-5p is a well-studied oncogene in breast cancer,Citation47–50 and is involved in mediating or regulating EMT.Citation51–53 miR-515 is also an estrogen regulated gene,Citation48 where ERα binds to the promoter of miR-515 and regulates gene expression in a 17β-estradiol-dependent manner.
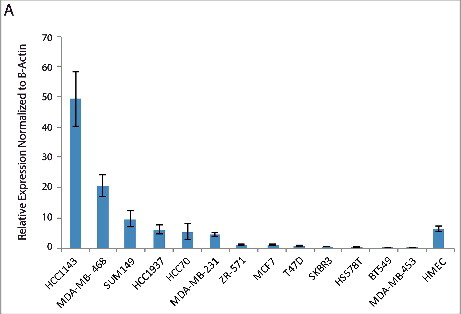
Overall these bioinformatic analyses indicated that ERRLR01 was a highly regulated lncRNA predictive of overall survival in breast cancer patients, and could be regulated by 17β-estradiol signaling in breast cancer. To test this notion, we screened for ERRLR01 expression across several breast cancer cell lines. We confirmed that several TNBC cell lines harbored high expression of ERRLR01 as compared with ERα+ cell lines, and normal HMECs (). The high expression of ERRLR01 in TNBC cell lines indicated that ERRLR01 may be repressed by 17β-estradiol signaling. However, this analysis was done under steady-state growth conditions whereby 17β-estradiol was present in the growth media. However, a recent study by Lin et al.,Citation54 indicated that in MCF-7 cells, ERRLR01 levels are low under hormone-depleted conditions yet upon 17β-estradiol addition ERRLR01 levels increase within 3 h (Figure S3). The consequences of this rise in ERRLR01 levels requires elucidation, as expression of numerous transcription factors and RNA binding proteins may also be modulated by this change in ERRLR01 levels. It would be interesting to determine in future studies whether direct modulation of ERRLR01 induces functional and/or phenotypic effects in breast cancer cells.
Figure 5. Quantitative PCR analysis of ERRLR01 expression across a panel of breast cancer cell lines. Analysis indicates a few TNBC cell lines express high levels of ERRLR01, as compared with normal HMEC lines, as well as ERα+ cell lines.
Gene regulatory network analysis associated with ERRLR01 expression
Given the evidence for ERRLR01 to be an estrogen-regulated gene in breast cancer, we wanted to identify signaling pathways that may be part of an ERRLR01-mediated RNA network. To do this we used GO term analysis using the WebGestalt algorithm.Citation55 We extracted the gene expression profiles from all patients within the Affymetrix dataset and performed Spearman-Rank analysis to determine which genes were most correlated with ERRLR01 expression (). We used as a cut-off the 200 most positively correlated ERRLR01 genes, which is approximately the top 1% genes derived from the analysis. We then performed a series of GO term and KEGG analysis using WebGestalt against the entire human reference genome, and found significantly enriched ERRLR01 pathways (). We identified pathways such as Wnt Receptor Signaling (p value < 7.70 × 10−2), Epithelial Differentiation (p value < 7.7 × 10−2), and Epithelial Development (p value < 7.70 × 10−2). Importantly, pathways such as Epithelial Development and Hormone Signaling were recurring pathways associated with ERRLR01-correlated genes. Therefore, we conclude that ERRLR01 is a lncRNA tightly regulated and serves to control proper developmental timing of mammary gland differentiation and development.
Figure 6. Go Term analysis of the top 200 positively correlated ERRLR01 genes via spearman rank analysis of the Affymetrix dataset (HGU133). (A) Analysis was performed in Basal-like breast tumors(n = 577), given ERRLR01 expression is high/detectable within those samples. Analysis indicated ERRLR01 levels correlated with “Central Nervous System Development,” “Stem Cell Differentiation,” “Protein Dimerization Activity,” and “Positive Regulation of Cell Proliferation.” (B) Go Term Analysis of the top 200 negatively correlated ERRLR01 genes via spearman rank analysis. Red Boxes highlight significant pathways and Black Boxes are highlighted as non-significant pathways.
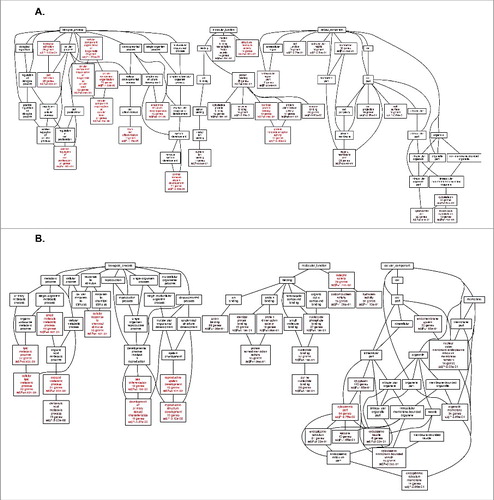
Similar analysis was performed on the 200 most inversely correlated ERRLR01-associated genes (). Pathways such as Small Molecule Metabolic Processes (p value < 8.80 × 10−3), Cellular Lipid Metabolic Processes (p value < 8.80 × 10−3), and Reproductive System Development (p value < 3.12 × 10−2) were identified. Similar analysis was performed on the TCGA dataset (see Figures S4-S5).
Discussion
ERRLR01 is located on chr17:70,396,217–70,590,488, and was first identified by Schmidt et al., where ERRLR01 transcriptionally upregulated MMP9 in melanoma cell lines.Citation25 In this study, ERRLR01 was compared across 150 clinical samples from TCGA and was found to be elevated in tumor samples as compared to normal. Furthermore, patient samples that expressed ERRLR01 ≥ 14.1 RPKM had a worse OS outcome than patients with ≤ 14.1 RPKM. This indicated that ERRLR01 operated as an oncogene in melanoma. ERRLR01 was described as an oncogene in other cancer systems as well, including lung and pancreatic cancer.Citation24,56 In lung cancer ERRLR01 expression associated with higher TMN stages as well as lymph node metastasis. ERRLR01 also functioned as a pro-metastatic factor in melanoma, since siRNA knockdown studies showed that loss of ERRLR01 resulted in reduced invasion as measured by Boyden-Chamber Matrigel assays.Citation25 In pancreatic cancer, the direct evidence for ERRLR01 as an oncogene is less well determined, as a particular pancreatic cancer risk variant was identified within the ERRLR01 transcript that generated a miR-1231 binding site, which presumably supports oncogenesis. More specifically, ERRLR01 modulated PTPN11 degradation by promoting E3 ligase induced ubiquitination of PTPN11 and diminished extracellular signal-related kinase (ERK) oncogenic signaling. Therefore, in pancreatic cancer the presumption is that ERRLR01 operates as a tumor suppressor by dampening the pro-proliferative mitogen-activated protein kinase (MAPK)/ERK signaling pathway.
ERRLR01 could also be a therapeutic target given knockdown of ERRLR01 in lung cancer cell lines and in mouse xenograft models resulted in reduced cell viability, and reduced cellular growth in vivo. ERRLR01 does this by binding directly to LSD1 (KDM1A) and functions as a chaperone protein to recruit this histone demethylase to NCALD.Citation24 LSD1 is known to regulate cellular differentiation and cell cycle progression, while NCALD is a visinin-like protein 1 subfamily of EF-hand calcium-binding proteins that is downregulated in patients with poor prognosis tumors and those harboring poorly differentiated ovarian tumors.Citation57 Therefore, the mechanism of action is such that ERRLR01 recruits LSD1 to epigenetically silence NCALD to promote tumorigenesis. The mechanism of action in melanoma is slightly different given ERRLR01 serves as a scaffold to bring 2 protein components in proximity to each other, Brn3a and AR. The interaction with ERRLR01 allows these protein heterodimers to bind to specific chromatin regions, within a site located in the promoter of MMP925. The transcriptional upregulation of MMP9 imparted by ERRLR01 promotes a metastatic phenotype in melanoma cell lines. Overall, these studies highlight the notion that ERRLR01 mediates oncogenic or tumor suppressor functions dependent upon the cellular context, which is true for many non-coding RNAs.
While ERRLR01 is described as a chaperone protein, our bioinformatics analysis identified ERRLR01 as part of a gene-regulatory network involving miRNA sponging and interactions with specific RNA-binding proteins. This was an important analysis given the biologic significance of both small and long noncoding RNAs are becoming increasingly appreciated. We further identified several SNPs across the ERRLR01 transcript, however it was unclear as to whether any of these polymorphisms were associated with disease risk and/or onset. We intend on pursuing this line of analysis given genome-wide association studies (GWAS) have identified cancer risk loci outside protein-coding regions, such as PTCSC3 in papillary thyroid carcinoma, and ANRIL in type-2 diabetes.Citation17,58,59
A major finding of this study was that 17β-estradiol regulated ERRLR01 expression. A few studies have identified specific lncRNAs to be regulated by 17β-estradiol. For instance, linc00160 expression can be modulated by 17β-estradiol, however the mechanism of action is unclear.Citation60 Similarly, well-studied lncRNAs have been identified to be regulated by 17β-estradiol, including H19, HOTAIR, and MALAT-1Citation61-63 In these studies, it is unclear how these lncRNAs relate to survival in breast cancer patients, and furthermore, if these lncRNAs predict overall survival based on ERα-stratification. In this study, we provide strong evidence that ERRLR01 is prognostic in breast cancer, that ERRLR01–related OS is dependent upon ERα status, and that ERRLR01 itself is regulated by ERα signaling and 17β-estradiol-associated coregulatory proteins in breast cancer. Overall, the continued efforts to understand this epigenetic regulation mediated by lncRNAs in the context of cancer development will aid in the generation of more effective therapeutics to treat the disease.Citation64
Disclosure of potential conflicts of interest
B.D.A holds patent interests with, and consults with AUM LifeTech. B.D.A is also the President of The Brain Institute of America, LLC. The other authors have no conflicts of interest to disclose.
Contributions
Manuscript Writing – Ubaidat Abdul-Rahman, Balázs Győrffy, and Brian D. Adams.
TGCA and Affymetrix Bioinformatic Analysis – Balázs Győrffy.
KEGG and Pathway Analysis – Brian D. Adams.
Concept and Design - Brian D. Adams.
Experimental Design - Ubaidat Abdul-Rahman and Brian D. Adams.
1329684_Supplemental_Material.zip
Download Zip (1.5 MB)Acknowledgments
We would like to thank our many colleagues for the review of this manuscript. We also thank Kim DeWeerd at the Molecular Core and Tissue Core for providing the equipment necessary to perform these experiments.
Funding
We thank the State of New York and The Research Foundation for startup funds to B.D.A for making this manuscript possible. B.G. was supported by the OTKA 108655 grant.
Related Research Data
References
- Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem 2012; 81:145-66; PMID:22663078; https://doi.org/10.1146/annurev-biochem-051410-092902
- Rinn JL. LncRNAs: Linking RNA to chromatin. Cold Spring Harb Perspect Biol 2014; 6:a018614; PMID:25085913; https://doi.org/10.1101/cshperspect.a018614
- Huarte M. The emerging role of lncRNAs in cancer. Nat Med 2015; 21:1253-61; PMID:26540387; https://doi.org/10.1038/nm.3981
- Goff LA, Rinn JL. Linking RNA biology to lncRNAs. Genome Res 2015; 25:1456-65; PMID:26430155; https://doi.org/10.1101/gr.191122.115
- Morris KV. Long antisense non-coding RNAs function to direct epigenetic complexes that regulate transcription in human cells. Epigenetics 2009; 4:296-301; https://doi.org/10.4161/epi.4.5.9282
- Villegas VE, Zaphiropoulos PG. Neighboring gene regulation by antisense long Non- Coding RNAs. Int J Mol Sci 2015; 16:3251-66; PMID:25654223; https://doi.org/10.3390/ijms16023251
- Morris KV, Vogt PK. Long antisense non-coding RNAs and their role in transcription and oncogenesis. Cell Cycle 2010; 9:2544-7; PMID:20581457; https://doi.org/10.4161/cc.9.13.12145
- Elnitski LL, Shah P, Moreland RT, Umayam L, Wolfsberg TG, Baxevanis AD. The ENCODEdb portal: Simplified access to ENCODE Consortium data. Genome Res 2007; 17:954-9; PMID:17568011; https://doi.org/10.1101/gr.5582207
- ENCODE Project Consortium, T. E. P. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004; 306:636-40; PMID:15499007; https://doi.org/10.1126/science.1105136
- Werner A. Biological functions of natural antisense transcripts. BMC Biol 2013; 11:31; PMID:23577602; https://doi.org/10.1186/1741-7007-11-31
- Werner A. Natural antisense transcripts. RNA Biol 2005; 2:53-62; PMID:17132938; https://doi.org/10.4161/rna.2.2.1852
- Tang SS, Zheng BY, Xiong XD. LincRNA-p21: Implications in human diseases. Int J Mol Sci 2015; 16:18732-40; PMID:26270659; https://doi.org/10.3390/ijms160818732
- Dimitrova N, Zamudio JR, Jong RM, Soukup D, Resnick R, Sarma K, Ward AJ, Raj A, Lee JT, Sharp PA, et al. LincRNA-p21 activates p21 in cis to promote polycomb target gene expression and to enforce the G1/S checkpoint. Mol Cell 2014; 54:777-90; PMID:24857549; https://doi.org/10.1016/j.molcel.2014.04.025
- Pasmant E, Sabbagh A, Vidaud M, Bièche I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J 2011; 25:444-8; PMID:20956613; https://doi.org/10.1096/fj.10-172452
- Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 2016; 17:47-62; PMID:26666209; https://doi.org/10.1038/nrg.2015.10
- Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discovery 2011; 1:391-407; PMID:22096659; https://doi.org/10.1158/2159-8290.CD-11-0209
- Cheetham SW, Gruhl F, Mattick JS, Dinger ME. Long noncoding RNAs and the genetics of cancer. Br J Cancer 2013; 108:2419-25; PMID:23660942; https://doi.org/10.1038/bjc.2013.233
- Jendrzejewski J, He H, Radomska HS, Li W, Tomsic J, Liyanarachchi S, Davuluri RV, Nagy R, de la Chapelle A. The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type. Proc Natl Acad Sci U S A 2012; 109:8646-51; PMID:22586128; https://doi.org/10.1073/pnas.1205654109
- Gutschner T, Hämmerle M, Eissmann M, Hsu J, Kim Y, Hung G, Revenko A, Arun G, Stentrup M, Gross M, et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res 2013; 73:1180-9; PMID:23243023; https://doi.org/10.1158/0008-5472.CAN-12-2850
- Zhu L, Liu J, MA S, Zhang S. Long Noncoding RNA MALAT-1 Can Predict Metastasis and a Poor Prognosis: a Meta-Analysis. Pathol Oncol Res 2015; 21:1259-64; PMID:26159858; https://doi.org/10.1007/s12253-015-9960-5
- Zhou Y, Xu X, Lv H, Wen Q, Li J, Tan L, Li J, Sheng X. The long noncoding RNA MALAT-1 is highly expressed in ovarian cancer and induces cell growth and migration. PLoS One 2016; 11:e0155250; PMID:27227769; https://doi.org/10.1371/journal.pone.0155250
- Arun G, Diermeier S, Akerman M, Chang KC, Wilkinson JE, Hearn S, Kim Y, MacLeod AR, Krainer AR, Norton L, et al. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev 2016; 30:34-51; PMID:26701265; https://doi.org/10.1101/gad.270959.115
- Hajjari M, Salavaty A. HOTAIR: an oncogenic long non-coding RNA in different cancers. Cancer Biol Med 2015; 12:1-9; PMID:25859406
- Shi X, Ma C, Zhu Q, Yuan D, Sun M, Gu X, Wu G, Lv T, Song Y. Upregulation of long intergenic noncoding RNA 00673 promotes tumor proliferation via LSD1 interaction and repression of NCALD in non-small-cell lung cancer. Oncotarget 2016; 7:25558-75; PMID:27027352
- Schmidt K, Joyce CE, Buquicchio F, Brown A, Ritz J, Distel RJ, Yoon CH, Novina CD. The lncRNA ERRLR011 Mediates Melanoma Invasion through a Conserved SRA1-like Region. Cell Rep 2016; 15:2025-37; PMID:27210747; https://doi.org/10.1016/j.celrep.2016.04.018
- Mihály Z, Kormos M, Lánczky A, Dank M, Budczies J, Szász MA, Győrffy B. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res Treatment 2013; 140:219-32; PMID:23836010; https://doi.org/10.1007/s10549-013-2622-y
- Győrffy B, Benke Z, Lánczky A, Balázs B, Szállási Z, Timár J, Schäfer R. RecurrenceOnline: an online analysis tool to determine breast cancer recurrence and hormone receptor status using microarray data. Breast Cancer Res Treat 2012; 132:1025-34; PMID:21773767; https://doi.org/10.1007/s10549-011-1676-y
- Li Q, Birkbak NJ, Gyorffy B, Szallasi Z, Eklund AC. Jetset: selecting the optimal microarray probe set to represent a gene. BMC Bioinformatics 2011; 12:474; PMID:22172014; https://doi.org/10.1186/1471-2105-12-474
- Gyorffy B, Benke Z, Lánczky A, Balázs B, Szállási Z, Timár J, Schäfer R. RecurrenceOnline: An online analysis tool to determine breast cancer recurrence and hormone receptor status using microarray data. Breast Cancer Res Treat 2012; 132:1025-34; PMID:21773767; https://doi.org/10.1007/s10549-011-1676-y
- Cancer T, Atlas G. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490:61-70; PMID:23000897; https://doi.org/10.1038/nature11412
- Adams BD, Wali VB, Cheng CJ, Inukai S, Booth CJ, Agarwal S, Rimm DL, Győrffy B, Santarpia L, Pusztai L, et al. MiR-34a silences c-SRC to attenuate tumor growth in triple-negative breast cancer. Cancer Res 2016; 76:927-39; PMID:26676753; https://doi.org/10.1158/0008-5472.CAN-15-2321
- Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet 2015; 47:199-208; PMID:25599403; https://doi.org/10.1038/ng.3192
- Cotterill S. Chromosome 14. Cancer Genet http://www.cancerindex.org/geneweb/clinkc14.htm- 2015
- Zhang X, Cowper-Sal-lari R, Bailey SD, Moore JH, Lupien M. Integrative functional genomics identifies an enhancer looping to the SOX9 gene disrupted by the 17q24.3 prostate cancer risk locus. Genome Res 2012; 22:1437-46; PMID:22665440; https://doi.org/10.1101/gr.135665.111
- Pezzolo A, Coco S, Raso A, Parodi F, Pistorio A, Valdora F, Capra V, Zollo M, Aschero S, Basso E, et al. Loss of 10q26.1-q26.3 in association with 7q34-q36.3 gain or 17q24.3- q25.3 gain predict poor outcome in pediatric medulloblastoma. Cancer Lett 2011; 308:215-24; PMID:21652146; https://doi.org/10.1016/j.canlet.2011.05.006
- Sun J, Purcell L, Gao Z, Isaacs SD, Wiley KE, Hsu FC, Liu W, Duggan D, Carpten JD, Grönberg H, et al. Association between sequence variants at 17q12 and 17q24.3 and prostate cancer risk in European and African Americans. Prostate 2008; 68:691-7; PMID:18361410; https://doi.org/10.1002/pros.20754
- Solomon E, Borrow J, Goddard AD. Chromosome aberrations and cancer. Science 1991; 254:1153-60; PMID:1957167; https://doi.org/10.1126/science.1957167
- Szász AM, Lánczky A, Nagy Á, Förster S, Hark K, Green JE, Boussioutas A, Busuttil R, Szabó A, Győrffy B. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget 2016; 7:49322-33; PMID:27384994
- Gyorffy B, Surowiak P, Budczies J, Lánczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 2013; 8:e82241; PMID:24367507; https://doi.org/10.1371/journal.pone.0082241
- Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al. MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res 2010; 38:e178; PMID:20802226; https://doi.org/10.1093/nar/gkq622
- Phillips JE, Corces VG. CTCF: Master weaver of the genome. Cell 2009; 137:1194-211; PMID:19563753; https://doi.org/10.1016/j.cell.2009.06.001
- de Wit E, Vos ES, Holwerda SJ, Valdes-Quezada C, Verstegen MJ, Teunissen H, Splinter E, Wijchers PJ, Krijger PH, de Laat W. CTCF binding polarity determines chromatin looping. Mol Cell 2015; 60:676-84; PMID:26527277; https://doi.org/10.1016/j.molcel.2015.09.023
- Ong C, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Publ Gr 2014; 15:234-46
- Ohlsson R, Bartkuhn M, Renkawitz R. CTCF shapes chromatin by multiple mechanisms: The impact of 20 years of CTCF research on understanding the workings of chromatin. Chromosoma 2010; 119:351-60; PMID:20174815; https://doi.org/10.1007/s00412-010-0262-0
- Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH. StarBase: A database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res 2011; 39:D202-9; PMID:21037263; https://doi.org/10.1093/nar/gkq1056
- Wong N, Wang X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res 2015; 43:D146-52; PMID:25378301; https://doi.org/10.1093/nar/gku1104
- Mattiske S, Suetani RJ, Neilsen PM, Callen DF. The oncogenic role of miR-155 in breast cancer. Cancer Epidemiol Biomarkers Prevention 2012; 21:1236-43; https://doi.org/10.1158/1055-9965.EPI-12-0173
- Pinho FG, Frampton AE, Nunes J, Krell J, Alshaker H, Jacob J, Pellegrino L, Roca-Alonso L, de Giorgio A, Harding V, et al. Downregulation of microRNA-515-5p by the estrogen receptor modulates Sphingosine kinase 1 and breast cancer cell proliferation. Cancer Res 2013; 73:5936-48; PMID:23928990; https://doi.org/10.1158/0008-5472.CAN-13-0158
- Pardo OE, Castellano L, Munro CE, Hu Y, Mauri F, Krell J, Lara R, Pinho FG, Choudhury T, Frampton AE, et al. miR-515-5p controls cancer cell migration through MARK4 regulation. EMBO Rep 2016; 17:570-84; PMID:26882547; https://doi.org/10.15252/embr.201540970
- Gilam A, Edry L, Mamluk-Morag E, Bar-Ilan D, Avivi C, Golan D, Laitman Y, Barshack I, Friedman E, Shomron N. Involvement of IGF-1R regulation by miR-515-5p modifies breast cancer risk among BRCA1 carriers. Breast Cancer Res Treat 2013; 138:753-60; PMID:23549953; https://doi.org/10.1007/s10549-013-2502-5
- De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 2013; 13:97-110; PMID:23344542; https://doi.org/10.1038/nrc3447
- Abdelmohsen K, Srikantan S, Kuwano Y, Gorospe M. miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proc Natl Acad Sci U S A 2008; 105:20297-302; PMID:19088191; https://doi.org/10.1073/pnas.0809376106
- Abdelmohsen K, Kim MM, Srikantan S, Mercken EM, Brennan SE, Wilson GM, Cabo Rd, Gorospe M. miR-519 suppresses tumor growth by reducing HuR levels. Cell Cycle 2010; 9:1354-9; PMID:20305372; https://doi.org/10.4161/cc.9.7.11164
- Lin Z, Reierstad S, Huang CC, Bulun SE. Novel estrogen receptor-?? binding sites and estradiol target genes identified by chromatin immunoprecipitation cloning in breast cancer. Cancer Res 2007; 67:5017-24; PMID:17510434; https://doi.org/10.1158/0008-5472.CAN-06-3696
- Zhang B, Kirov S, Snoddy J. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 2005; 33:W741-8; PMID:15980575; https://doi.org/10.1093/nar/gki475
- Zheng J, Huang X, Tan W, Yu D, Du Z, Chang J, Wei L, Han Y, Wang C, Che X, et al. Pancreatic cancer risk variant in LINC00673 creates a miR-1231 binding site and interferes with PTPN11 degradation. Nat Genet 2016; 48:747-57; PMID:27213290; https://doi.org/10.1038/ng.3568
- Nymark P, Guled M, Borze I, Faisal A, Lahti L, Salmenkivi K, Kettunen E, Anttila S, Knuutila S. Integrative analysis of microRNA, mRNA and aCGH data reveals asbestos- and histology-related changes in lung cancer. Genes Chromosom Cancer 2011; 50:585-97; PMID:21563230; https://doi.org/10.1002/gcc.20880
- Pasmant E, Sabbagh A, Vidaud M, Bièche I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J 2011; 25:444-8; PMID:20956613; https://doi.org/10.1096/fj.10-172452
- Jendrzejewski J, He H, Radomska HS, Li W, Tomsic J, Liyanarachchi S, Davuluri RV, Nagy R, de la Chapelle A. The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type. Proc Natl Acad Sci 2012; 109:8646-51; PMID:22586128; https://doi.org/10.1073/pnas.1205654109
- Jonsson P, Coarfa C, Mesmar F, Raz T, Rajapakshe K, Thompson JF, Gunaratne PH, Williams C. Single-molecule sequencing reveals estrogen-regulated clinically relevant lncRNAs in breast cancer. Mol Endocrinol 2015; 29:1634-45; PMID:26426411; https://doi.org/10.1210/me.2015-1153
- Sun H, Wang G, Peng Y, Zeng Y, Zhu QN, Li TL, Cai JQ, Zhou HH, Zhu YS. H19 lncRNA mediates 17β-estradiol-induced cell proliferation in MCF-7 breast cancer cells. Oncol Rep 2015; 33:3045-52; PMID:25846769
- Zhao Z, Chen C, Liu Y, Wu C. 17β-Estradiol treatment inhibits breast cell proliferation, migration and invasion by decreasing MALAT-1 RNA level. Biochem Biophys Res Commun 2014; 445:388-93; PMID:24525122; https://doi.org/10.1016/j.bbrc.2014.02.006
- Bhan A, Mandal SS. Estradiol-Induced Transcriptional Regulation of Long Non-Coding RNA, HOTAIR. Methods Mol Biol 2016; 1366:395-412; PMID:26585152
- McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A, et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015; 521:232-6; PMID:25915022; https://doi.org/10.1038/nature14443