
ABSTRACT
The fact that many cancer types display transcriptional addiction driven by dysregulation of oncogenic enhancers and transcription factors has led to increased interest in a group of protein kinases, known as transcriptional cyclin dependent kinases (tCDKs), as potential therapeutic targets. Despite early reservations about targeting a process that is essential to healthy cell types, there is now evidence that targeting tCDKs could provide enough therapeutic window to be effective in the clinic. Here, we discuss recent developments in this field, with an emphasis on highly-selective inhibitors and the challenges to be addressed before these inhibitors could be used for therapeutic purposes.
Abbreviations: CAK: CDK-activating kinase;CDK: cyclin-dependent kinase;CMGC group: CDK-, MAPK-, GSK3-, and CLK-like;CTD: C-terminal repeat domain of the RPB1 subunit of RNA polymerase II;DRB: 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole;mCRPC: metastatic castration-resistant prostate cancer;NSCLC: non-small cell lung cancer;P-TEFb: positive elongation factor b;RNAPII: RNA polymerase II;S2: serine-2 of CTD repeats;S5: serine-5 of CTD repeats;S7: serine-7 of CTD repeats;SEC: super elongation complex;tCDK: transcriptional cyclin-dependent kinase;TNBC: triple-negative breast cancer
Introduction
The cyclin-dependent kinases (CDKs) form an evolutionarily distinct branch of the CMGC (CDK, MAPK, GSK3 and CLK) group and possess several common regulatory features including the requirement of cyclin binding for activity, phosphorylation of the activating T-loop by a CDK-activating kinase (CAK), and control by subcellular localization and/or allosteric binding partners [Citation1,Citation2]. Although originally discovered in yeast based on their critical roles in regulating the cell cycle, mammalian CDKs are comprised of subfamilies with specialized functions related to the cell cycle (CDK1, −2, −4, −6) or transcription regulation (CDK7, −8, −9, −12, −13, −19) [Citation2–Citation4]. These important roles in cell proliferation and gene expression, and the deregulation of many CDKs in cancer, have motivated considerable efforts aimed at therapeutic targeting of the CDKs. With the exception of CDK7, which also serves as a CAK, the roles of the transcriptional CDKs (tCDKs) are largely distinct from those of the cell cycle-related CDKs, and their functions and substrates are much less well-understood. The recent recognition of transcriptional addiction as a potential vulnerability in many cancer types has led to increased interest in this group of CDKs, despite earlier reservations about targeting a process essential to all cells. Here, we discuss recent developments in this field, with an emphasis on highly-selective inhibitors and the challenges to be addressed before these inhibitors could be used for therapeutic purposes.
Figure 1. Summary of the major known roles of the transcriptional CDKs during the RNAPII transcription cycle. Black circles denote positive roles, grey circles denote indirect roles of CDK7 (via phosphorylation of CDK9), and the open circle denotes the kinase-independent inhibitory effect of CDK8 on Mediator-RNAPII interaction and preinitiation complex formation. Not depicted are CDK19 or the phosphorylation of transcription-related targets such as transcription factors.
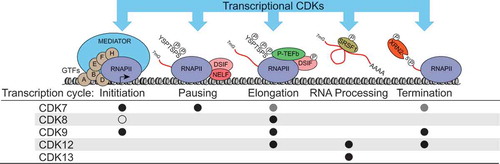
The RNAPII transcription cycle and tCDKs
Transcription of protein-coding genes by RNA polymerase II (RNAPII) proceeds via a “transcription cycle” with distinct phases – initiation, pausing, elongation, and termination – each of which is associated with changes in the composition and activity of the transcription machinery and associated regulatory factors. Genomic studies have helped to reveal that promoter-proximal transcriptional pausing is prevalent at metazoan genes and represents a key regulatory event [Citation5]. Importantly, dysregulation of transcription is a hallmark of many cancer types [Citation6]. The tCDKs are central to orchestration of the transcription cycle and its coordination with cotranscriptional processes such as RNA capping, splicing, 3ʹ end formation, export, and regulation of the chromatin landscape [Citation7] (). One of the best-characterized tCDK substrates is the C-terminal domain (CTD) of RPB1, the largest RNAPII subunit, which is subject to sequential and dynamic phosphorylation of multiple residues within its 52 heptad repeats (consensus YSPTSPS) as the transcription cycle proceeds (reviewed in [Citation8–Citation11]).
In contrast to cell cycle CDKs, tCDKs associate with their cyclin partners constitutively, and are often recruited to chromatin as components of much larger complexes, with defined patterns of recruitment and T-loop phosphorylation [Citation3]. Historically, only three CDKs were thought to be involved in regulation of the transcription cycle in humans. CDK7 (as part of the TFIIH general transcription factor complex) was thought to primarily control initiation and serve as the major kinase for Serine-5 (S5) in the CTD repeats [Citation12]. CDK9 (as part of P-TEFb, the Positive Transcription Elongation Factor b) was thought to primarily control elongation and serve as the major CTD Serine-2 (S2) kinase [Citation13]; and CDK8 was thought to repress transcription by preventing association of the Mediator coactivator complex with RNAPII and by phosphorylating TFIIH [Citation14,Citation15]. This early view has evolved considerably due to recent research, and it is now clear that there is significant crosstalk among tCDKs, and that each kinase impacts multiple phases of the transcription cycle in a context-dependent fashion as discussed below [Citation7,Citation16,Citation17]. The identification of three additional tCDKs adds further complexity to this evolving picture: CDK19 is a paralog of CDK8 with non-redundant functions that also associates with the Mediator complex [Citation14,Citation18]; and CDK12 and CDK13 can phosphorylate the CTD at S2, S5, and Serine-7 (S7), with differing effects on the transcriptome [Citation19–Citation21]. While our understanding of the tCDKs continues to evolve, the field has been hampered by various limitations of the experimental approaches employed. For example, knockdown or knockout studies are subject to the effects of losing scaffold functions and/or compensation by related proteins, and small molecule CDK inhibitors which were once thought to be specific have later been revealed to affect multiple CDKs [Citation21–Citation23]. These issues have led to contradictory reports about the contributions of the tCDKs to RNAPII CTD phosphorylation, and whether each tCDK regulates RNAPII activity globally [Citation21,Citation24]. Although the focus of this review is on recent developments in therapeutic targeting of the tCDKs, the development and adoption of well-characterized chemical probes and drug-like compounds with improved selectivity will provide essential tools for resolving the specialized functions of the tCDKs.
Early therapeutic efforts
Unrestrained cell proliferation is a hallmark of cancer and, due to their central role in cell cycle control, many cancers deregulate the activity of one or more CDKs, commonly through cyclin overexpression or silencing of CDK inhibitors [Citation25]. In the last ~ 20 years numerous small molecule CDK inhibitors have been developed as potential therapeutics and tested in multiple cancer types and clinical trials [Citation26]. Early efforts to develop CDK inhibitors were focused on targeting cell cycle CDKs, which we will briefly discuss, but we refer readers to [Citation26] for in-depth discussion. First-generation pan-CDK inhibitors such as flavopiridol (alvocidib, active against CDK1, −2, −4, −6, −7, −9) and R-roscovitine (seliciclib, active against CDK1, −2, −5, −7, −9), and even second-generation inhibitors such as dinaciclib (active against CDK1, −2, −5, −9) showed promising results in in vitro models but have displayed largely disappointing results in clinical trials, with the exception of certain hematological malignancies [Citation26–Citation28]. Several inter-related factors have likely contributed to the lack of effectiveness of pan-CDK inhibitors [Citation26,Citation27]: 1) a poor understanding of mechanisms of action -including which CDK(s) are inhibited in vivo and a consequent inability to accurately measure target engagement; 2) a lack of biomarkers of sensitivity and thus no selection of patients by cancer type or genetic features; 3) limited therapeutic windows -due to pan-CDK inhibition and/or low potency. These factors make a strong case for development and thorough characterization of CDK inhibitors with greater potency and selectivity. Indeed, results with inhibitors of CDK4/6 have been more encouraging and three such compounds (palbociclib, ribociclib, abemaciclib) are now FDA-approved for treatment of certain types of breast cancer, with further clinical trials ongoing in other cancer types [Citation26,Citation29]. In the following sections, we will focus on rationales for targeting the tCDKs and on recent efforts to develop selective tCDK inhibitors as cancer therapeutics.
CDK7
Among the tCDKs, CDK7 has perhaps the most diverse functions. The minimal complex comprised of CDK7, CCNH, MAT1 complex serves as the CAK for essentially all cell cycle CDKs, as well as CDK9 and CDK11, and as a critical component of the 10-subunit TFIIH complex that plays an essential role in transcription by RNAPI/II and nucleotide excision repair (reviewed in [Citation30]).
During transcription by RNAPII, CDK7/TFIIH is the last of the general transcription factors to be recruited to the preinitiation complex where it phosphorylates the CTD, at S5 and S7, and TFIIE [Citation22,Citation30,Citation31]. For some time, it has been generally accepted that CDK7 activity is required for initiation, wherein CTD phosphorylation is thought to destabilize the interaction between RNAPII and Mediator, thus facilitating promoter escape [Citation32–Citation34]. However, recent evidence suggests that this is not the only function of CDK7, as inhibition with the covalent inhibitor THZ1 (see below) has little effect on initiation in vitro but rather results in decreased DSIF and NELF recruitment [Citation35], consistent with other reports [Citation22,Citation36]. CDK7 inhibition also affects capping, pausing, elongation, and termination, largely through phosphorylation of both the CTD and the CDK9 activation T-loop [Citation22,Citation24,Citation35,Citation37]. Finally, CDK7 activity affects chromatin modification during transcription by facilitating recruitment of histone methyltransferases SETD1A/B and SETD2 through CTD phosphorylation and/or by activation of CDK9/P-TEFb [Citation24]. In addition to SPT5, TFIIE, and the CTD, CDK7 phosphorylates several transcription factors including p53 and multiple nuclear hormone receptors (RAR, RAR, AR, ER) to promote their full activation and subsequent degradation [Citation38]. Despite this relative paucity of known substrates, it is likely that additional phosphorylation targets contribute to the functions of CDK7 in transcription and other processes.
Validity of CDK7 as a therapeutic target
Regulation of the cell cycle, transcription, and DNA repair are all critical for rapidly proliferating cells. Interestingly, deletion of Cdk7 in mouse embryonic fibroblasts causes cell cycle arrest without an accompanying reduction in bulk CTD S5 phosphorylation, suggesting the existence of redundant/compensatory kinase activity [Citation39]. In vivo, Cdk7 loss causes early embryonic lethality and in adult mice leads to depletion of stem cell pools in proliferative tissues but has little effect on non-proliferating tissues [Citation39]. In keeping with a role in maintaining high rates of proliferation, elevated CDK7 expression has been observed in gastric, breast, and hepatocellular cancers [Citation40–Citation43]. Many cancer types rely on high-levels of transcription, commonly driven by oncogenic transcription factors and/or so-called super-enhancers that are especially sensitive to perturbation [Citation6,Citation44,Citation45], and promising results have now been reported for CDK7 inhibitors in various cancer types. Finally, several pathogenic viruses produce proteins that modulate TFIIH activity [Citation30]. For example, the HIV Tat protein can recruit and activate CDK7 to help drive transcription of the viral genome [Citation46,Citation47]. Accordingly, inhibition of CDK7 appears to have broad-spectrum antiviral activity [Citation48]. Thus, despite the important role of CDK7 in processes essential to all cells, a therapeutic window may exist for targeting CDK7 in cancer, and potentially other pathologies.
Available CDK7 inhibitors
In addition to the early-generation pan-CDK inhibitors with activity against CDK7, several classes of small molecule compounds with improved selectivity towards CDK7 have been described (). Here we give a brief overview of the development and selectivity of some of these compounds, and a summary of their therapeutic potential as determined by preclinical and/or clinical testing.
Table 1. CDK inhibitors discussed in this review. * = where reported; note that different standards to define off-target binding and/or effects may have been used by each study; 1 = In vitro kinase activity assay; 2 = Binding assay; 3 = In vivo kinase activity assay.
BS-181 and ICEC0942
BS-181 is a pyrazolopyrimidine derivative with side chains based on the pan-CDK inhibitor roscovitine [Citation49]. In vitro testing against various kinases indicates excellent selectivity for CDK7 over other CDKs and additional kinases and growth inhibitory activity was demonstrated against multiple cancer cell lines (breast, lung, prostate, and colorectal) [Citation49]. BS-181 treatment inhibited phosphorylation of CTD S5, as well as CTD S2 and RB (retinoblastoma protein), cell cycle analysis indicated G1 accumulation, and apoptosis was apparent at higher concentrations (≥ 25µM). In a mouse MCF7 xenograft model, intraperitoneal injection resulted in dose dependent growth inhibition of 25–50% over 14 days with no apparent toxicity. However, BS-181 was rapidly cleared and bioavailability was low, indicating the need for further refinement of pharmacological properties [Citation49].
Recently, in silico modeling of BS-181 and related compounds with the crystal structure of CDK7 led to the development of ICEC0942, an analog of BS-181 with improved cell permeability and oral bioavailability [Citation50,Citation51]. ICEC0942 inhibited growth of all NCI-60 cancer cell lines, with a median GI50 of 0.25 µM [Citation51]. ICEC0942 treatment produced dose-dependent inhibition of CTD (S2/5/7) and RB phosphorylation in MCF7 cells and in the colorectal carcinoma line HCT116, which was especially sensitive to growth inhibition, and reduced xenograft tumors of these cell lines by ~ 60% over 14 days. Treatment led to cell cycle delay at G2M in both cell lines, with apoptotic markers apparent by 24 hrs. The drug appeared to be well tolerated in mice, however reduced lymphocyte counts were observed. In December 2017, a phase I/II clinical trial of ICEC0942 as a single agent was initiated by Carrick Therapeutics (NCT03363893), with an estimated completion date of December 2018.
LDC3140 and LDC4297
Kelso et al. [Citation36] used the CDK7/Cyclin H/Mat1 complex to screen a compound library based on BS-181 and other ATP-competitive kinase inhibitor scaffolds to identify candidates that were then subjected to medicinal chemistry optimization. This led to the identification of the lead compounds LDC3140 and LDC4297 with potent activity against CDK7 and selectivity against other CDKs and kinases [Citation36]. LDC3140 was shown to inhibit CTD S5/S7 phosphorylation and transcription in vitro, and steady-state mRNA expression in A549 cells. Further analysis using ChIP- and qRT-PCR confirmed effects on RNAPII distribution across candidate genes and inhibition of nascent transcript production in vivo [Citation36]. Treatment of A549, HeLa, and HCT116 cells with either compound elicited an apoptotic response, and was associated with cell type-specific G1/G2M delay [Citation36]. In addition to activity against cancer cell lines, LDC4297 has also been demonstrated to have activity against a range of pathogenic viruses [Citation48]. In this context, although LDC4297 displayed varying levels of antiproliferative and cytotoxic activity in primary human fibroblasts, these effects were at higher concentrations than required for antiviral activity [Citation48].
THZ1/THZ2
THZ1 is a phenylaminopyrimidine that acts as a covalent inhibitor of CDK7 by modification of cysteine-312 [Citation52], and has been extensively validated both in vitro and in cell-based models. Although none of the other CDKs possess a cysteine residue at the equivalent position, CDK12 and CDK13 do have cysteines within four amino acids, and THZ1 was confirmed to inhibit CDK12 at slightly higher concentrations [Citation52]. Treatment of T-ALL Jurkat cells with 250 nM THZ1, but not a non-reactive analog THZ1-R, was shown to inhibit phosphorylation of CTD S5, S7, and S2, as well as reducing T-loop phosphorylation of CDK1, −2, and −9 [Citation52]. Expression of a CDK7 C312S mutant partially rescued these effects, confirming C312 as a critical target of THZ1. THZ1 was subsequently screened against 1,000 cancer cell lines, with over half showing IC50 values < 200 nM, and sensitivity was associated with deregulation of oncogenic transcription factors. Consistent with a core gene expression network driven by misregulation of transcription factors, THZ1 treatment of Jurkat cells (250 nM) led to a global reduction in steady-state mRNA levels and RNAPII occupancy at promoters and gene bodies, with specific genes such as RUNX1 sensitive at lower concentrations (50 nM). RUNX1 expression is part of an auto regulatory loop that drives its own expression and, in Jurkat cells, is associated with a super-enhancer. Importantly, super-enhancers are commonly acquired at oncogenic transcription factor genes in cancer cells to drive high-level expression [Citation44] and are more sensitive to perturbation of the transcriptional machinery than typical enhancers or promoters [Citation45].
In further support of the therapeutic potential of targeting cancers driven by aberrant transcription via CDK7, THZ1 has also proven to be effective in selectively targeting several other aggressive cancer types in preclinical cell and mouse tumor models, including MYCN-amplified neuroblastoma [Citation53], small-cell lung cancer [Citation54] and triple-negative breast cancer (TNBC) [Citation55,Citation56]. Importantly, these studies demonstrated differential sensitivity to THZ1 over non-MYCN-amplified, non-small cell lung cancer (NSCLC), and hormone receptor-positive breast cancer cell lines, respectively, as well as non-transformed human and mouse cell lines [Citation45,Citation53,Citation54,Citation56].
Several additional preclinical studies further highlight the therapeutic potential of CDK7 inhibitors, alone or in combination with other targeted agents. High CDK7 mRNA and protein levels were found to be correlated with poor prognosis in TNBC, and a comparison of THZ1 and BS-181 demonstrated sensitivity to CDK7 inhibition, with THZ1 having a more potent effect on cell proliferation, apoptosis, and CTD phosphorylation compared to BS-181 [Citation43]. Interestingly, BH3 profiling indicated that CDK7 inhibition induced an increased dependency on the anti-apoptotic proteins BCL-2 and BCL-XL. Combination treatment with THZ1 and the BH3 mimetic ABT-263 gave synergistic inhibition of proliferation and induction of apoptosis in multiple TNBC cell lines [Citation43]. Combinatorial treatment with THZ1 and BH3 mimetics has also been reported to produce synergistic growth inhibition in peripheral T-cell lymphoma cell lines and mouse xenograft models [Citation57]. Fisher and colleagues recently demonstrated that inhibition of CDK7 in HCT116 cells synergizes with non-genotoxic activation of p53 using MDM2 inhibitors to promote apoptosis [Citation58]. Importantly, combination treatment did not trigger apoptosis in a non-transformed colon epithelial cell line [Citation58].
In addition to THZ1, several additional covalent CDK7 inhibitors have been developed with improved pharmacodynamic properties (THZ2 [Citation56]) and/or selectivity (YKL-1–116 [Citation58] and SY-1365). Both THZ1 and THZ2 are licensed to Syros Pharmaceuticals which is currently running a phase I clinical trial of SY-1365 (NCT03134638), although few details are available.
CDK9
CDK9, first described as PITALRE based on its conserved cyclin-binding peptide motif, is the human ortholog of the budding yeast Bur1 and associates with cyclins T1, T2a, T2b and K to form P-TEFb [Citation38]. Two CDK9 isoforms, CDK942 and CDK955, are expressed from the CDK9 locus, thus giving rise to as many as eight distinct P-TEFb heterodimers, the functional significance of which have yet to be elucidated [Citation59]. The Price lab was the first to demonstrate the role of P-TEFb in transcription and CTD phosphorylation, using studies in Drosophila, and early on P-TEFb was known to interact with the HIV Tat protein to activate viral transcription [Citation60–Citation62]. In addition to binding with its partner cyclins, CDK9 requires phosphorylation at threonine-186 within its T-loop for activity, carried out by CDK7/CAK in the context of transcription complexes, and is subject to additional regulatory phosphorylations and modifications [Citation38]. CDK9 activity is further regulated by association with various factors and multi-subunit complexes. The 7SK snRNP complex sequesters and inhibits a large fraction of P-TEFb [Citation63], that can then be released upon gene activation by the action of factors such as KAP1 or BRD4 [Citation64–Citation66]. The most transcriptionally active P-TEFb-containing complexes are the super elongation complex (SEC) and the BRD4-P-TEFb complex [Citation5]. Furthermore, P-TEFb can also be recruited by transcription factors, including MYC, MYOD, PPAR, CIITA, and HIV Tat protein, to stimulate transcription of their respective target genes [Citation38]. These multiple modes of recruitment underscore the importance of P-TEFb in transcription and the prevailing model, established by Price et al., is that CDK9 phosphorylates the NELF-E subunit of NELF and the SPT5 subunit of DSIF, allowing the release of promoter-proximal paused RNAPII into the productive elongation phase [Citation5]. Although this model has largely held true, many details remain to be elucidated, including the degree to which CDK9/P-TEFb has global versus gene-specific effects on transcription, and its preference for phosphorylating S2 versus S5 in vivo [Citation5,Citation38].
Recently, a chemical genetics approach was used to identify numerous additional putative substrates of CDK9, which are enriched for factors involved in transcription and RNA processing, including the exoribonuclease XRN2 which was validated as a bona fide target of CDK9 [Citation67]. XRN2 is involved in RNAPII transcription termination and phosphorylation by CDK9 enhances its enzymatic activity. Inhibition or depletion of CDK9 leads to decreased chromatin localization of XRN2 and increased read-through transcription, clearly implicating CDK9 in efficient transcription termination.
Validity of CDK9 as a therapeutic target
Initial interest in CDK9 as a therapeutic target in cancer was based on: 1) data suggesting that inhibition of CDK9, rather than cell-cycle CDKs, was required for cell death caused by pan-CDK inhibitors such as flavopiridol and roscovitine [Citation68,Citation69]; 2) its role in maintaining expression of short-lived anti-apoptotic transcripts such as MCL1 and XIAP in leukemias and other cancer types [Citation68,Citation69]; and 3) the dysregulation of CDK9 during other diseases and pathologies, including viral infection and cardiac hypertrophy [Citation68,Citation70]. More recently, the concept of transcriptional addiction in cancer has led to renewed interest in CDK9/P-TEFb as a therapeutic target that may offer selectivity over normal/untransformed cells.
Dysregulation of CDK9/P-TEFb function is especially common in several hematological malignancies driven by oncogenic fusions of the MLL gene on chromosome 11. Many of the most frequent translocation partners of MLL are components of the SEC, resulting in aberrant recruitment of active CDK9 to MLL target genes [Citation71,Citation72]. The transcription factor c-MYC plays a key role in cellular proliferation and MYC is one of the most frequently amplified oncogenes in human tumors [Citation73]. Importantly, c-MYC can recruit CDK9/P-TEFb to facilitate transcriptional pause-release, and overexpression of c-MYC drives transcriptional amplification in tumor cells [Citation74,Citation75]. In addition to copy number amplification, high MYC expression is often driven by cancer specific super-enhancers, requiring both BRD4 and CDK9/P-TEFb [Citation76–Citation78]. CDK9/P-TEFb can also be directly recruited by additional transcription factors, including androgen receptor in prostate cancer, and NFKB or STAT3 during inflammation [Citation68].
Available CDK9 inhibitors
Although both flavopiridol and the adenosine analog 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) have historically been used to inhibit CDK9 activity for the study of transcriptional mechanisms, these compounds suffer from a lack of specificity and potency, which renders them unsuitable for therapeutic purposes and of limited use as chemical probes. A large number of scaffolds/core structures have been explored for the development of CDK9 inhibitors [Citation79] and numerous compounds with activity against CDK9 have been described (). Unfortunately, for many of these compounds there is a lack of data concerning their selectivity over other tCDKs and their pharmacological properties. We highlight here several compound classes for which the available data appears promising and/or that are currently in clinical trials.
LDC067
One of the first compounds with improved CDK9 selectivity to be described, LDC067, is based on a 2,4-aminopyrimidine scaffold and has 55–230-fold selectivity over other CDKs [Citation80]. LDC067 was shown to inhibit transcription in vitro and treatment of mouse ESCs and HeLa cells reduced CTD S2 phosphorylation levels. Treatment with LDC067 induced apoptosis in several cancer cell lines and primary AML blasts, which was assumed to be due to loss of short-lived apoptotic transcripts. Further analysis confirmed dose-dependent depletion of primary transcripts and steady-state mRNA levels for select genes, as well as increased RNAPII levels at the 5ʹ end of MYC, consistent with increased transcriptional pausing. Unfortunately, LDC067 is ~ 20-fold less potent against CDK9 than flavopiridol, indicating that further optimization is required.
i-CDK9
i-CDK9 is a more potent inhibitor of CDK9 and was shown to be selective for CDK9 over several other CDKs, by either in vitro kinase assay (AlphaScreen, Perkin Elmer) (≥ 600-fold vs. CDK1, −2, −4, −7, −8) or Kinomescan competition assay (CDK3, −5, −11, −13) [Citation77]. Although some activity was observed against several non-CDK kinases, the next-best targets, DYRK1A and 1B, had in vitro IC50 values at least 100-fold higher. i-CDK9 inhibits CDK9 CTD S2 phosphorylation activity in vitro, although S5 and S7 phosphorylation levels were not tested, and dose-dependent inhibition of CTD S2 and SPT5 phosphorylation was demonstrated in HeLa cells, along with downregulation of expression of MCL1 and induction of apoptosis. ChIP-seq with an antibody against RNAPII showed increased pausing at > 50% of genes in HeLa cells. Interestingly, a small group of genes, including MYC, displayed increased expression upon i-CDK9 treatment, albeit at lower concentrations and earlier times than required for full inhibition of S2 phosphorylation. Experimental evidence indicated that this could be attributed to release of CDK9 from the inhibitory 7SK complex and recruitment to MYC by BRD4. Importantly, the increase in MYC expression was blocked by the BET/BRD4 inhibitor JQ1 and combination treatment of HeLa and H1792 non-small cell lung cancer cells synergistically inhibited growth and induced apoptosis. Unfortunately, there are not yet any reports of the effect of i-CDK9 as a single agent, or in combination with JQ1, on untransformed cells, nor have its pharmacological properties been reported.
BAY1143752/atuveciclib
Starting with a triazine lead compound, BAY-958, which is potent and selective against CDK9 but has poor pharmacological properties, a group from Bayer AG recently developed the benzyl sulfoximine BAY1143752 as a clinical candidate [Citation81]. BAY1143752 displays impressive potency against CDK9 and selectivity against the tested CDKs, with sub-micromolar activity against only two non-CDK kinases, and with improved pharmacological properties. Antiproliferative activity was demonstrated against HeLa and MOLM-12 AML cells in vitro, BAY1143752 significantly reduced the growth of MOLM-13 and MV4-11 tumor xenografts in nude mice and rats, respectively, and was well-tolerated in both models. Based on these results, two Phase I clinical trials in patients with advanced cancer (NCT01938638) and leukemia (NCT02345382) were recently completed, with results yet to be reported. Two additional Phase I trials (NCT02745743 and NCT02635672) of a related compound, BAY1251152, are currently underway, although details of this compound have not yet been published.
THAL-SNS-032 and NVP-2
NVP-2 is an aminopyrimidine-based inhibitor developed by Novartis and was recently shown to have sub-nanomolar potency towards CDK9/CycT and good selectivity against a panel of 468 kinases (Kinomescan), with ≥ 700-fold lower activity against DYRK1B and CDK7 [Citation82]. NVP-2 displays anti-proliferative activity against multiple leukemia cell lines and induces MCL-1 loss and apoptosis within four hours in MOLT4 ALL cells. As an alternative to traditional kinase inhibitors, this group also explored the potential of NVP-2 and the multi-CDK inhibitor SNS-032 to serve as targeting moieties for thalidomide-induced recruitment of the E3 ligase CRBN, a strategy that has been used for selective degradation of other targets, including BRD4 and CDK8 [Citation83,Citation84]. Interestingly, while thalidomide-NVP-2 conjugates had no effect on CDK9 levels, THAL-SNS-032 selectively induced CDK9 degradation with little effect on the protein levels of other CDKs, despite retaining its ability to block their kinase activity. THAL-SNS-032 displayed more potent anti-proliferative and apoptotic activity than the parent compound SNS-032 against multiple leukemia cell lines. Both NVP-2 and THAL-SNS-032 were shown to significantly downregulate steady-state mRNA levels in MOLT4 cells, to increase levels of promoter-proximal RNAPII, and to decrease RNAPII and SPT5 levels across gene bodies, consistent with inhibition of pause-release and elongation.
CDK8 and CDK19
The conserved Mediator complex is a global regulator of RNA polymerase II (RNAPII) activity and is required for transcription of most protein-coding genes [Citation18]. Mediator interacts directly with transcription factors, the core transcriptional machinery, and chromatin architectural proteins, thus integrating multiple regulatory inputs to ensure appropriate control of RNAPII activity. In humans, the 26-subunit core Mediator is reversibly associated with a four-subunit “CDK module” that plays an important role in conveying positive and negative regulatory signals to core Mediator and the transcription machinery [Citation14]. The CDK module contains the only known catalytic activity within Mediator, in the form of CDK8 or its close paralog CDK19 [Citation85,Citation86]. Genetic and biochemical evidence indicates that these CDKs are functionally specialized and assemble with Mediator in a mutually-exclusive fashion [Citation87]. However, the precise mechanism(s) by which CDK8 and CDK19 regulate transcription remain unclear, as do their relative contributions to cellular signaling, proliferation, and survival. While originally reported as a negative regulator of transcription, multiple studies have demonstrated that the Mediator CDK module also positively regulates transcription [Citation14,Citation88]. As a negative regulator of transcription, CDK8 is thought to block transcription initiation by phosphorylation of the Cyclin H subunit of TFIIH [Citation15] or by preventing association of Mediator with RNAPII in a kinase activity-independent manner [Citation89], and has been implicated in limiting high levels of transcription driven by super-enhancer-controlled genes [Citation90]. As a positive regulator of transcription, CDK8 promotes the recruitment of the SEC to facilitate release of transcriptionally-paused RNAPII, particularly at signal-responsive genes such as those regulated by growth factor stimulation and HIF1A [Citation91,Citation92], but the role of kinase activity in this process remains undefined. Little is known about the functions of CDK19 and it remains unclear to what extent the two Mediator kinases have shared versus specialized roles in transcription regulation. Given their association with Mediator and the transcription machinery, it is likely that CDK8 and CDK19 also influence multiple phases of the transcription cycle via phosphorylation of proteins within the transcription and RNA processing machineries.
Validity of mediator-associated CDKs as therapeutic targets
The context-dependent effects of the CDK module on transcription are highlighted by the implication of various CDK module subunits as promotors or suppressors of tumor development [Citation93]. CDK8 in particular has been shown to be capable of activating expression of tumor-promoting gene networks [Citation91,Citation92,Citation94,Citation95], but appears to have a tumor-suppressive role in endometrial and bladder cancers, and esophageal squamous cell carcinoma [Citation93,Citation96]. CDK8 has also been implicated in regulation of the immune response, where it has been shown to phosphorylate both STAT1 and STAT3, potentiate NFKB transcriptional activity, influence natural killer cell activity, and suppress production of IL-10 [Citation97,Citation98]. In contrast, few functions of CDK19 have been described: CDK19 is likely to have both redundant and unique functions, and we recently discovered a kinase-independent role for CDK19 in the response to activation of the tumor suppressor p53 [Citation99]. Furthermore, although numerous dual CDK8/19 kinase inhibitors have shown promise, there is evidence that simultaneous inhibition of the Mediator kinases may cause toxicity (see below). These observations underscore the importance of developing a detailed understanding of the roles of CDK8 and CDK19 kinase activity in transcription regulation and cancer biology for developing effective CDK8/19-based therapeutic strategies.
Available CDK8/19 inhibitors
The interest in CDK8 as a therapeutic target, especially in melanoma, colon, and breast cancer, has led to the development of numerous small molecule Type I and Type II kinase inhibitors (that bind to active and inactive conformations, respectively) as potential therapeutics for CDK8-driven malignancies () [Citation100,Citation101].
Cortistatin A
The first inhibitor with selectivity for CDK8/19 to be described was the marine natural product Cortistatin A, isolated on the basis of anti-angiogenic activity [Citation102]. Cortistatin A was subsequently shown to bind with high affinity to CDK8 and CDK19 [Citation103]. More extensive studies by the Shair and Taatjes groups confirmed the potency and selectivity of Cortistatin A for CDK8/19, demonstrated inhibition of phosphorylation of known CDK8 targets, such as the CTD, STAT1, and SMAD2/3, in vivo, and have used Cortistatin A to identify additional putative targets of CDK8/19 [Citation90,Citation104,Citation105]. Cortistatin A has been shown to have anti-proliferative activity against multiple leukemia cell lines of different origin in vitro and in two mouse models of acute myeloid leukemia. Interestingly, this anti-leukemic activity appears to be due to de-repression of super-enhancer-regulated genes involved in differentiation, indicating that in this context CDK8/19 activity promotes oncogenesis via negative regulation of transcription [Citation90].
Senexin A and B
SNX2-class compounds are 4-aminoquinazolines that were identified in a high-throughput screen for inhibitors of p21-activated transcription and subjected to chemical optimization. The screen led to the generation of CDK8/19-selective compounds, including Senexin A, which inhibits CDK8 with an IC50 of 0.28 µM, although activity against other CDKs was not reported [Citation106]. Senexin A was demonstrated to inhibit β-catenin transcriptional activity and induction of EGR1 by serum stimulation, consistent with known roles of CDK8, and subsequently to abrogate CDK8-dependent tumor promoting paracrine activity in an A549 xenograft model of NSCLC. Senexin A and an optimized derivative, Senexin B, have been shown to suppress estrogen-dependent transcription in ER-positive breast cancer cell lines in vitro and suppress growth of ER-positive breast cancer xenografts in mice [Citation107]. However, few details have been published regarding Senexin B and it is therefore difficult to predict its therapeutic potential.
Azabenzothiophene- and sorafenib-based compounds
A Genentech-led group has reported the development of two structurally different CDK8/19 inhibitor series: Type I inhibitors based on a 6-azabenzothiophene scaffold and Type II inhibitors based on the multi-kinase inhibitor sorafenib [Citation108,Citation109]. Both classes demonstrated potency and selectivity and were confirmed to reduce phosphorylation of STAT1. However, these compounds had weak anti-proliferative activity when tested on HCT116 colorectal cancer cells, highlighting the complexity of establishing the therapeutic potential of the Mediator-associated kinases [Citation108,Citation109].
Pyridine-based compounds
Several additional classes of CDK8/19 inhibitors have been developed by investigators at Cancer Research UK and Merck KGaA in Germany and have undergone thorough validation [Citation110–Citation115]. Two of these classes, the CCT series based on a 3,4,5-trisubstituted pyridine scaffold and the MSC series based on a 3-methyl-1H-pyrazolo[3,4-b]pyridine scaffold, are among the most extensively characterized of the dual CDK8/19 inhibitors. Both classes have potent and selective activity against CDK8 and CDK19, inhibit phosphorylation of known CDK8 targets, and exhibit effects on gene expression consistent with inhibition of the Mediator kinases. However, only modest effects against human cancer cell lines and patient-derived xenograft and mouse models were observed. Of greater concern, despite apparently being well-tolerated in mice, both compound classes exhibited multiple toxicities in rats and dogs at doses required for therapeutic effects [Citation110]. Because these compounds are structurally distinct, this was interpreted as on-target toxicity, suggesting that the therapeutic window for dual CDK8/19 inhibition could be limited. However, given the relative chemical simplicity of these compounds, off-target effects beyond CDK8/19 cannot be discounted. Development of small molecule inhibitors selective for either of the Mediator kinases is likely to be challenging due to their high homology and it remains to be established if targeting of either kinase has improved results. Furthermore, the fact that these compounds all inhibit CDK8 and CDK19 with similar potency prevents the assignment of putative substrates, or biological effects, such as efficacy against cancer cells, to either kinase.
With these problems in mind, we recently engineered HCT116 cells to express ATP analog-sensitive CDK8 (AS-CDK8) [Citation116]. Selective inhibition of CDK8 led to significant changes in steady-state mRNA levels in HCT116 cells; downregulated genes were enriched for the glycolysis pathway and CDK8 inhibition, alone or in combination with CDK19, sensitized these cells to the glycolysis inhibitor 2-deoxyglucose [Citation116]. Thus, combination treatment strategies may allow for use of CDK8/19 inhibitors at lower concentrations that do not cause toxicity [Citation117]. Interestingly, a phenotypic screen for small molecules that enhance IL-10 secretion led to the identification of BRD6989 which displays some selectivity for CDK8 over CDK19 [Citation98] and may therefore be useful in further establishing the therapeutic potential of targeting CDK8 alone.
A number of other inhibitors of CDK8/19 have recently been described or reported () including JH-VII-49 and JH-XI-10–02 [Citation84], SEL120-34A from Selvita, and patented compounds from Hoffman La Roche and Nimbus Therapeutics [Citation101].
CDK12 and CDK13
CDK12 (also known as CRKRS) and CDK13 are the human orthologs of the budding yeast CTD S2 kinase Ctk1 and associate with Cyclin K to form active kinase complexes [Citation118,Citation119]. CDK12/13 share nearly identical kinase domains but largely differ outside this region, especially their unstructured C-terminal tails. While both kinases are known to phosphorylate the CTD and are likely to share some functions, CDK12 is the best-studied, while the function(s) of CDK13 are less clear. In vitro, CDK12 can phosphorylate the CTD on both S2 and S5, with the highest activity towards S5 on S7 pre-phosphorylated CTD [Citation20]. In contrast, there is evidence that CDK12, rather than CDK9, is the major CTD S5 kinase during transcription elongation in vivo [Citation118,Citation120], and its recruitment to transcription units is mediated by the PAF1 complex [Citation121]. CDK12 has also been implicated in recruitment of the transcription termination factors CSTF64 and CSTF77 [Citation122]. Interestingly, depletion of CDK12 (or CDK13) appears to have effects on the transcriptome without alterations in bulk CTD phosphorylation levels [Citation21,Citation118,Citation120], and it remains unclear the extent to which CDK12 (or CDK13) serve as global versus gene-specific regulators of transcription elongation. Moreover, both CDK12 and CDK13 possess N-terminal arginine-serine (RS)-rich domains characteristic of RNA binding and splicing factors and have been shown to interact with splicing factors and regulate splicing and/or 3ʹ-end processing [Citation21,Citation122].
Validity of CDK12 and CDK13 as therapeutic targets
CDK12 is subject to both gain- and loss-of-function alterations in human cancers, suggesting that it has oncogenic or tumor-suppressive functions, depending on context [Citation122–Citation124]. Of particular relevance in cancer, depletion of CDK12 appears to preferentially affect the expression of genes involved in DNA damage response and homologous recombination repair [Citation21,Citation120], and CDK12 is required for maintenance of genomic integrity during embryonic development [Citation125]. Accordingly, CDK12 loss-of-function mutations found in high-grade serous ovarian cancer are associated with genomic instability [Citation126,Citation127]. Potential loss-of-function mutations in CDK12 also occur at ~ 1.5% in invasive breast cancer where they may contribute to characteristic genomic instability of this cancer type [Citation122,Citation128]. Interestingly, loss or depletion of CDK12 is synthetic lethal with inhibitors of PARP1, an enzyme involved in the DDR [Citation129,Citation130]. Along similar lines, inactivating CDK12 mutations found in a subset of metastatic castration-resistant prostate cancer (mCRPC) samples were associated with genomic instability, higher levels of neoantigens, and increased T-cell infiltration; two of four mCRPC patients with CDK12 mutations treated with anti-PD-1 immunotherapy displayed strong decreases in prostate-specific antigen levels [Citation131,Citation132]. These results indicate that CDK12 loss of function can expose cancer vulnerabilities and suggest that targeting CDK12 activity in CDK12 wild-type cells might also confer sensitivity to these targeted therapies.
In contrast, CDK12 is frequently co-amplified with ERBB2 in HER2-positive breast cancer where it has been shown to regulate alternative last exon splicing and CDK12 expression is correlated with increased migration and invasiveness of breast cancer cell lines [Citation133,Citation134]. Finally, CDK13 amplification and overexpression has been observed in primary hepatocellular carcinoma and colon tumor samples and confers oncogenic properties when expressed in NIH-3T3 cells [Citation135].
Available CDK12 and CDK13 inhibitors
There is a distinct lack of selective small molecule chemical probes and drug-like compounds targeting CDK12/13, which hampers mechanistic studies and investigation of their potential as therapeutic targets. HeLa cells expressing ATP analog-sensitive CDK12 have been generated to facilitate dissection of CDK12-specific functions [Citation136], however this approach is not useful therapeutically. The covalent CDK7 inhibitor THZ1 also has activity against CDK12/13 at higher doses and was used as a starting point to develop a covalent inhibitor selective for CDK12/13, THZ531 [Citation137]. Treatment of Jurkat T-ALL cells with THZ531 led to a reduction in CTD S2 phosphorylation, both globally and specifically at THZ531-affected genes. In keeping with previous studies, low doses of THZ531 downregulated expression of DDR-related genes, while higher doses of THZ531 suppressed super-enhancer-regulated transcription factors and induced apoptosis [Citation137]. More recently, two new classes of noncovalent CDK12/13 inhibitors with good potency and selectivity for other CDKs and additional kinases have been described [Citation138,Citation139]. These compounds were reported to inhibit CTD S2 phosphorylation and inhibit the growth of ovarian or breast cancer cells and could serve as useful chemical probes and/or lead compounds for further validation of CDK12 and −13 as therapeutic targets.
Similar to CDK7, inhibitors of CDK12/13 may offer an advantage by targeting two processes important to many cancer cells, aberrant transcription and genomic instability. This may be an effective single-agent therapeutic strategy for select cancer types, while synergizing with other targeted therapies in other cancers. As described above, CDK12/13 inhibition may be effective in combination with drugs targeting the DNA damage response or immune checkpoint blockade. Indeed, THZ531 synergizes with PARP inhibitors in both in vitro and in vivo models of Ewing sarcoma [Citation140].
Emerging themes
The transcriptional CDKs play important roles in regulating every stage of the RNAPII transcription cycle, from initiation and pause-release, to elongation and termination, as well as coordinating co-transcriptional events including chromatin modification and RNA processing. Aberrant transcription is a hallmark of many tumor types and is often driven by oncogenic alterations of transcription factors or cofactors (e.g. MYC, AR, ER, CDK8) [Citation6,Citation74] and the establishment of cancer-specific super-enhancers [Citation44]. In cancer types, such as TNBC, for which no clear drivers have been identified, consistent patterns of gene expression are nonetheless established and maintained [Citation55]. Recent studies have demonstrated that this transcriptional addiction creates vulnerabilities that might be targeted therapeutically and tCDKs thus represent logical targets [Citation55]. These studies have established that, despite transcription being an essential process common to all cells, highly proliferative cells may be especially susceptible to transcriptional inhibitors [Citation55], similar to DNA damaging agents. Interestingly, hemizygous deletion of POLR2A (by virtue of its proximity to TP53) occurs relatively frequently in human cancers and sensitizes to transcription inhibition [Citation141], suggesting that many tumors with p53 genomic loss may also be targeted in this way. Moreover, targeting the general transcription machinery may help prevent adaptive responses and emergence of resistance to existing targeted agents [Citation142].
In surveying the recent and historical literature on inhibitors of the tCDKs, several themes emerge:
Caution in interpretation of results should be exercised when employing compounds that inhibit multiple kinases as either potential therapeutics or as chemical probes, as there may be a narrow therapeutic window between target engagement and toxicity, and there is likely to be difficulty in confirming mechanism of action and thus predicting response. Even where the affected kinases are close paralogs, such as dual inhibitors of CDK8/19 or MED12/13, the use of these compounds without validation of effects due to individual paralogous kinases can obscure their true functions.
Covalent kinase inhibitors show great potential for selective targeting of tCDKs. Although these small molecules may still bind to kinase active sites and retain their ability to inhibit kinase activity, they possess electrophilic moieties capable of forming covalent bonds with nucleophiles such as unique cysteines lying outside the kinase active site. These compounds are likely to be effective at lower doses than traditional inhibitors.
Another strategy that shows promise is the use of proteolysis-targeting chimeras, or PROTACs), such as thalidomide-conjugated compounds that recruit E3 ligases to tag the target protein for proteasomal degradation [Citation143]. Potential advantages of these compounds include the ability to simultaneously inhibit and degrade kinases; targeting of non-kinase/scaffolding functions and lowering of effective doses. These compounds may also offer greater selectivity over their parent compounds, as in the case of THAL-SNS-032 [Citation82].
Many of the latest generation of selective kinase inhibitors only display potent single-agent efficacy against specific cancer types such as hematopoietic lineages. Rational combination therapies are likely to greatly extend the therapeutic applications of selective inhibitors of tCDKs. Mechanistic studies and synthetic lethality screens have already numerous revealed pathways, other targeted agents, or conventional chemotherapeutics that can synergize with tCDK inhibitors [Citation58,Citation116,Citation140,Citation144–Citation147], and additional examples are likely to exist.
A number of challenges continue to limit progress in developing therapeutic inhibitors of tCDKs, not least of which is the difficulty imposed by the high degree of homology within the ATP binding pockets of CDKs and other kinases. Thorough biochemical, structural and biological validation of new compounds is essential to identify their true in vivo targets, to forestall emergence of resistance, and to identify biomarkers of sensitivity and response. Nonetheless, progress continues to be made and the recent development of novel targeting strategies shows great promise.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Manning G, Whyte DB, Martinez R, et al. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. Epub 2002/12/10. PubMed PMID: 12471243.
- Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15(6):122. Epub 2014/09/03. PubMed PMID: 25180339; PubMed Central PMCID: PMC4097832.
- Fisher RP. The CDK network: linking cycles of cell division and gene expression. Genes Cancer. 2012;3(11–12):731–738. Epub 2013/ 05/02. PubMed PMID: 23634260; PubMed Central PMCID: PMC3636752.
- Guo Z, Stiller JW. Comparative genomics of cyclin-dependent kinases suggest co-evolution of the RNAP II C-terminal domain and CTD-directed CDKs. BMC Genomics. 2004;5:69. Epub 2004/09/24. PubMed PMID: 15380029; PubMed Central PMCID: PMC521075.
- Chen FX, Smith ER, Shilatifard A. Born to run: control of transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol. 2018;19(7):464–478. Epub 2018/ 05/10. PubMed PMID: 29740129.
- Bradner JE, Hnisz D, Young RA. Transcriptional addiction in cancer. Cell. 2017;168(4):629–643. Epub 2017/02/12. PubMed PMID: 28187285; PubMed Central PMCID: PMC5308559.
- Fisher RP. CDK regulation of transcription by RNAP II: not over ‘til it’s over? Transcription. 2017;8(2):81–90. Epub 2016/ 12/23. PubMed PMID: 28005463; PubMed Central PMCID: PMC5423476.
- Zaborowska J, Egloff S, Murphy S. The pol II CTD: new twists in the tail. Nat Struct Mol Biol. 2016;23(9):771–777. Epub 2016/ 09/09. PubMed PMID: 27605205.
- Custodio N, Carmo-Fonseca M. Co-transcriptional splicing and the CTD code. Crit Rev Biochem Mol Biol. 2016;51(5):395–411. Epub 2016/10/25. PubMed PMID: 27622638.
- Harlen KM, Churchman LS. The code and beyond: transcription regulation by the RNA polymerase II carboxy-terminal domain. Nat Rev Mol Cell Biol. 2017;18(4):263–273. Epub 2017/ 03/02. PubMed PMID: 28248323.
- Jeronimo C, Bataille AR, Robert F. The writers, readers, and functions of the RNA polymerase II C-terminal domain code. Chem Rev. 2013;113(11):8491–8522. Epub 2013/07/11. PubMed PMID: 23837720.
- Orphanides G, Lagrange T, Reinberg D. The general transcription factors of RNA polymerase II. Genes Dev. 1996;10(21):2657–2683. Epub 1996/11/01. PubMed PMID: 8946909.
- Bartkowiak B, Greenleaf AL. Phosphorylation of RNAPII: to P-TEFb or not to P-TEFb? Transcription. 2011;2(3):115–119. Epub 2011/ 08/10. PubMed PMID: 21826281; PubMed Central PMCID: PMC3149687.
- Galbraith MD, Donner AJ, Espinosa JM. CDK8: A positive regulator of transcription. Transcription. 2010;1(1):4–12. PubMed PMID: 21327159; PubMed Central PMCID: PMC3035184.
- Akoulitchev S, Chuikov S, Reinberg D. TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature. 2000;407(6800):102–106. Epub 2000/ 09/19. PubMed PMID: 10993082.
- Sanso M, Fisher RP. Pause, play, repeat: cDKs push RNAP II’s buttons. Transcription. 2013;4(4):146–152. Epub 2013/ 06/13. PubMed PMID: 23756342; PubMed Central PMCID: PMC3977912.
- Devaiah BN, Singer DS. Cross-talk among RNA polymerase II kinases modulates C-terminal domain phosphorylation. J Biol Chem. 2012;287(46):38755–38766. Epub 2012/10/03. . PubMed PMID: 23027873; PubMed Central PMCID: PMC3493918.
- Poss ZC, Ebmeier CC, Taatjes DJ. The mediator complex and transcription regulation. Crit Rev Biochem Mol Biol. 2013;48(6):575–608. PubMed PMID: 24088064; PubMed Central PMCID: PMC3852498.
- Greifenberg AK, Honig D, Pilarova K, et al. Structural and functional analysis of the Cdk13/Cyclin K complex. Cell Rep. 2016;14(2):320–331. Epub 2016/01/11. PubMed PMID: 26748711.
- Bosken CA, Farnung L, Hintermair C, et al. The structure and substrate specificity of human Cdk12/Cyclin K. Nat Commun. 2014;5:3505. Epub 2014/03/26. PubMed PMID: 24662513; PubMed Central PMCID: PMC3973122.
- Liang K, Gao X, Gilmore JM, et al. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol Cell Biol. 2015;35(6):928–938. Epub 2015/01/07. PubMed PMID: 25561469; PubMed Central PMCID: PMC4333096.
- Larochelle S, Amat R, Glover-Cutter K, et al. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol. 2012;19(11):1108–1115. Epub 2012/10/16. PubMed PMID: 23064645; PubMed Central PMCID: PMC3746743.
- Laitem C, Zaborowska J, Isa NF, et al. CDK9 inhibitors define elongation checkpoints at both ends of RNA polymerase II-transcribed genes. Nat Struct Mol Biol. 2015;22(5):396–403. Epub 2015/04/08. PubMed PMID: 25849141; PubMed Central PMCID: PMC4424039.
- Ebmeier CC, Erickson B, Allen BL, et al. Human TFIIH Kinase CDK7 regulates transcription-associated chromatin modifications. Cell Rep. 2017;20(5):1173–1186. Epub 2017/08/03. PubMed PMID: 28768201; PubMed Central PMCID: PMC5564226.
- Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. Epub 2014/ 01/07. PubMed PMID: 24390350; PubMed Central PMCID: PMC4048962.
- Asghar U, Witkiewicz AK, Turner NC, et al. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14(2):130–146. Epub 2015/01/31. PubMed PMID: 25633797; PubMed Central PMCID: PMC4480421.
- Guha M. Cyclin-dependent kinase inhibitors move into Phase III. Nat Rev Drug Discov. 2012;11(12):892–894. Epub 2012/12/01. PubMed PMID: 23197022.
- Khalil HS, Mitev V, Vlaykova T, et al. Discovery and development of seliciclib. how systems biology approaches can lead to better drug performance. J Biotechnol. 2015;202:40–49. Epub 2015/03/10. PubMed PMID: 25747275.
- Vijayaraghavan S, Moulder S, Keyomarsi K, et al. Inhibiting CDK in cancer therapy: current evidence and future directions. Target Oncol. 2018;13(1):21–38. Epub 2017/ 12/09. PubMed PMID: 29218622.
- Rimel JK, Taatjes DJ. The essential and multifunctional TFIIH complex. Protein Sci. 2018;27(6):1018–1037. Epub 2018/04/18. PubMed PMID: 29664212; PubMed Central PMCID: PMC5980561.
- Akhtar MS, Heidemann M, Tietjen JR, et al. TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol Cell. 2009;34(3):387–393. Epub 2009/05/20. PubMed PMID: 19450536; PubMed Central PMCID: PMC2757088.
- Jeronimo C, Robert F. Kin28 regulates the transient association of mediator with core promoters. Nat Struct Mol Biol. 2014;21(5):449–455. Epub 2014/04/08. PubMed PMID: 24704787; PubMed Central PMCID: PMC3997488.
- Sogaard TM, Svejstrup JQ. Hyperphosphorylation of the C-terminal repeat domain of RNA polymerase II facilitates dissociation of its complex with mediator. J Biol Chem. 2007;282(19):14113–14120. Epub 2007/03/23. PubMed PMID: 17376774.
- Wong KH, Jin Y, Struhl K. TFIIH phosphorylation of the Pol II CTD stimulates mediator dissociation from the preinitiation complex and promoter escape. Mol Cell. 2014;54(4):601–612. Epub 2014/04/22. PubMed PMID: 24746699; PubMed Central PMCID: PMC4035452.
- Nilson KA, Guo J, Turek ME, et al. THZ1 reveals roles for Cdk7 in co-transcriptional capping and pausing. Mol Cell. 2015;59(4):576–587. Epub 2015/08/11. PubMed PMID: 26257281; PubMed Central PMCID: PMC4546572.
- Kelso TW, Baumgart K, Eickhoff J, et al. Cyclin-dependent kinase 7 controls mRNA synthesis by affecting stability of preinitiation complexes, leading to altered gene expression, cell cycle progression, and survival of tumor cells. Mol Cell Biol. 2014;34(19):3675–3688. Epub 2014/07/23. PubMed PMID: 25047832; PubMed Central PMCID: PMC4187722.
- Glover-Cutter K, Larochelle S, Erickson B, et al. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol Cell Biol. 2009;29(20):5455–5464. Epub 2009/08/12. PubMed PMID: 19667075; PubMed Central PMCID: PMC2756882.
- Paparidis NF, Durvale MC, Canduri F. The emerging picture of CDK9/P-TEFb: more than 20 years of advances since PITALRE. Mol Biosyst. 2017;13(2):246–276. Epub 2016/ 11/12. PubMed PMID: 27833949.
- Ganuza M, Saiz-Ladera C, Canamero M, et al. Genetic inactivation of Cdk7 leads to cell cycle arrest and induces premature aging due to adult stem cell exhaustion. Embo J. 2012;31(11):2498–2510. Epub 2012/04/17. PubMed PMID: 22505032; PubMed Central PMCID: PMC3365431.
- Wang Q, Li M, Zhang X, et al. Upregulation of CDK7 in gastric cancer cell promotes tumor cell proliferation and predicts poor prognosis. Exp Mol Pathol. 2016;100(3):514–521. Epub 2016/05/08. PubMed PMID: 27155449.
- Patel H, Abduljabbar R, Lai CF, et al. Expression of CDK7, Cyclin H, and MAT1 Is elevated in breast cancer and is prognostic in estrogen receptor-positive breast cancer. Clin Cancer Res. 2016;22(23):5929–5938. Epub 2016/11/04. PubMed PMID: 27301701; PubMed Central PMCID: PMC5293170.
- Wang C, Jin H, Gao D, et al. A CRISPR screen identifies CDK7 as a therapeutic target in hepatocellular carcinoma. Cell Res. 2018;28(6):690–692. Epub 2018/03/07. PubMed PMID: 29507396; PubMed Central PMCID: PMC5993748.
- Li B, Ni Chonghaile T, Fan Y, et al. Therapeutic rationale to target highly expressed CDK7 conferring poor outcomes in triple-negative breast cancer. Cancer Res. 2017;77(14):3834–3845. Epub 2017/04/30. . PubMed PMID: 28455421.
- Hnisz D, Abraham BJ, Lee TI, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–947. Epub 2013/10/15. PubMed PMID: 24119843; PubMed Central PMCID: PMC3841062.
- Loven J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. Epub 2013/04/16. PubMed PMID: 23582323; PubMed Central PMCID: PMC3760967.
- Cujec TP, Okamoto H, Fujinaga K, et al. The HIV transactivator TAT binds to the CDK-activating kinase and activates the phosphorylation of the carboxy-terminal domain of RNA polymerase II. Genes Dev. 1997;11(20):2645–2657. Epub 1997/10/23. PubMed PMID: 9334327; PubMed Central PMCID: PMC316603.
- Kim YK, Bourgeois CF, Pearson R, et al. Recruitment of TFIIH to the HIV LTR is a rate-limiting step in the emergence of HIV from latency. Embo J. 2006;25(15):3596–3604. Epub 2006/07/29. PubMed PMID: 16874302; PubMed Central PMCID: PMC1538560.
- Hutterer C, Eickhoff J, Milbradt J, et al. A novel CDK7 inhibitor of the Pyrazolotriazine class exerts broad-spectrum antiviral activity at nanomolar concentrations. Antimicrob Agents Chemother. 2015;59(4):2062–2071. Epub 2015/01/28. PubMed PMID: 25624324; PubMed Central PMCID: PMC4356785.
- Ali S, Heathcote DA, Kroll SH, et al. The development of a selective cyclin-dependent kinase inhibitor that shows antitumor activity. Cancer Res. 2009;69(15):6208–6215. Epub 2009/07/30. PubMed PMID: 19638587; PubMed Central PMCID: PMC2875168.
- Hazel P, Kroll SH, Bondke A, et al. Inhibitor selectivity for cyclin-dependent kinase 7: A structural, thermodynamic, and modelling study. ChemMedChem. 2017;12(5):372–380. Epub 2017/ 01/27. PubMed PMID: 28125165.
- Patel H, Periyasamy M, Sava GP, et al. ICEC0942, an orally bioavailable selective inhibitor of CDK7 for cancer treatment. Mol Cancer Ther. 2018;17(6):1156–1166. Epub 2018/03/17. PubMed PMID: 29545334; PubMed Central PMCID: PMC5985928.
- Kwiatkowski N, Zhang T, Rahl PB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511(7511):616–620. Epub 2014/ 07/22. PubMed PMID: 25043025; PubMed Central PMCID: PMC4244910.
- Chipumuro E, Marco E, Christensen CL, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159(5):1126–1139. Epub 2014/11/25. PubMed PMID: 25416950; PubMed Central PMCID: PMC4243043.
- Christensen CL, Kwiatkowski N, Abraham BJ, et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell. 2014;26(6):909–922. Epub 2014/12/10. PubMed PMID: 25490451; PubMed Central PMCID: PMC4261156.
- Franco HL, Kraus WL. No driver behind the wheel? Targeting transcription in cancer. Cell. 2015;163(1):28–30. Epub 2015/ 09/26. PubMed PMID: 26406367.
- Wang Y, Zhang T, Kwiatkowski N, et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell. 2015;163(1):174–186. Epub 2015/09/26. PubMed PMID: 26406377; PubMed Central PMCID: PMC4583659.
- Cayrol F, Praditsuktavorn P, Fernando TM, et al. THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nat Commun. 2017;8:14290. Epub 2017/01/31. PubMed PMID: 28134252; PubMed Central PMCID: PMC5290269.
- Kalan S, Amat R, Schachter MM, et al. Activation of the p53 transcriptional program sensitizes cancer cells to Cdk7 inhibitors. Cell Rep. 2017;21(2):467–481. Epub 2017/10/12. PubMed PMID: 29020632; PubMed Central PMCID: PMC5687273.
- Shore SM, Byers SA, Maury W, et al. Identification of a novel isoform of Cdk9. Gene. 2003;307:175–182. Epub 2003/04/23. PubMed PMID: 12706900.
- Marshall NF, Price DH. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem. 1995;270(21):12335–12338. Epub 1995/05/26. PubMed PMID: 7759473.
- Peng J, Marshall NF, Price DH. Identification of a cyclin subunit required for the function of Drosophila P-TEFb. J Biol Chem. 1998;273(22):13855–13860. Epub 1998/06/05. PubMed PMID: 9593731.
- Zhu Y, Pe’ery T, Peng J, et al. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 1997;11(20):2622–2632. Epub 1997/10/23. PubMed PMID: 9334325; PubMed Central PMCID: PMC316609.
- Li Q, Price JP, Byers SA, et al. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J Biol Chem. 2005;280(31):28819–28826. Epub 2005/06/21. PubMed PMID: 15965233.
- Jang MK, Mochizuki K, Zhou M, et al. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19(4):523–534. Epub 2005/08/20. PubMed PMID: 16109376.
- McNamara RP, Reeder JE, McMillan EA, et al. KAP1 recruitment of the 7SK snRNP complex to promoters enables transcription elongation by RNA polymerase II. Mol Cell. 2016;61(1):39–53. Epub 2016/01/05. PubMed PMID: 26725010; PubMed Central PMCID: PMC4714561.
- Zhou Q, Li T, Price DH. RNA polymerase II elongation control. Annu Rev Biochem. 2012;81:119–143. Epub 2012/03/13. PubMed PMID: 22404626; PubMed Central PMCID: PMC4273853.
- Sanso M, Levin RS, Lipp JJ, et al. P-TEFb regulation of transcription termination factor Xrn2 revealed by a chemical genetic screen for Cdk9 substrates. Genes Dev. 2016;30(1):117–131. PubMed PMID: 26728557; PubMed Central PMCID: PMC4701974.
- Krystof V, Baumli S, Furst R. Perspective of cyclin-dependent kinase 9 (CDK9) as a drug target. Curr Pharm Des. 2012;18(20):2883–2890. Epub 2012/ 05/11. PubMed PMID: 22571657; PubMed Central PMCID: PMC3382371.
- Lam LT, Pickeral OK, Peng AC, et al. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2001;2(10):RESEARCH0041. Epub 2001/10/13. PubMed PMID: 11597333; PubMed Central PMCID: PMC57796.
- Franco LC, Morales F, Boffo S, et al. CDK9: A key player in cancer and other diseases. J Cell Biochem. 2018;119(2):1273–1284. Epub 2017/ 07/20. PubMed PMID: 28722178.
- Lin C, Smith ER, Takahashi H, et al. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010;37(3):429–437. Epub 2010/02/18. PubMed PMID: 20159561; PubMed Central PMCID: PMC2872029.
- Luo Z, Lin C, Shilatifard A. The super elongation complex (SEC) family in transcriptional control. Nat Rev Mol Cell Biol. 2012;13(9):543–547. Epub 2012/ 08/17. PubMed PMID: 22895430.
- Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. Epub 2010/ 02/19. PubMed PMID: 20164920; PubMed Central PMCID: PMC2826709.
- Lin CY, Loven J, Rahl PB, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151(1):56–67. Epub 2012/10/02. PubMed PMID: 23021215; PubMed Central PMCID: PMC3462372.
- Rahl PB, Lin CY, Seila AC, et al. c-Myc regulates transcriptional pause release. Cell. 2010;141(3):432–445. Epub 2010/05/04. PubMed PMID: 20434984; PubMed Central PMCID: PMC2864022.
- Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. Epub 2011/09/06. PubMed PMID: 21889194; PubMed Central PMCID: PMC3187920.
- Lu H, Xue Y, Yu GK, et al. Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4-dependent mechanism. Elife. 2015;4:e06535. . PubMed Central PMCID: PMC4490784.
- Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54(5):728–736. Epub 2014/06/07. PubMed PMID: 24905006; PubMed Central PMCID: PMC4236231.
- Sonawane YA, Taylor MA, Napoleon JV, et al. Cyclin dependent kinase 9 inhibitors for cancer therapy. J Med Chem. 2016;59(19):8667–8684. Epub 2016/05/14. PubMed PMID: 27171036; PubMed Central PMCID: PMC5636177.
- Albert TK, Rigault C, Eickhoff J, et al. Characterization of molecular and cellular functions of the cyclin-dependent kinase CDK9 using a novel specific inhibitor. Br J Pharmacol. 2014;171(1):55–68.
- Lücking U, Scholz A, Lienau P, et al. Identification of Atuveciclib (BAY 1143572), the first highly selective, clinical PTEFb/CDK9 Inhibitor for the treatment of cancer. ChemMedChem. 2017;12(21):1776–1793. PubMed Central PMCID: PMC5698704
- Olson CM, Jiang B, Erb MA, et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol. 2017;14(2):163–170. PubMed PMID: 29251720; PubMed Central PMCID: PMC5912898.
- Lu J, Qian Y, Altieri M, et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem Biol. 2015;22(6):755–763. Epub 2015/06/09. PubMed PMID: 26051217; PubMed Central PMCID: PMC4475452.
- Hatcher JM, Wang ES, Johannessen L, et al. Development of highly potent and selective steroidal inhibitors and degraders of CDK8. ACS Med Chem Lett. 2018;9(6):540–545. Epub 2018/06/26. PubMed PMID: 29937979; PubMed Central PMCID: PMC6004574.
- Sato S, Tomomori-Sato C, Parmely TJ, et al. A set of consensus mammalian mediator subunits identified by multidimensional protein identification technology. Mol Cell. 2004;14(5):685–691. Epub 2004/06/04. PubMed PMID: 15175163.
- Bourbon HM. Comparative genomics supports a deep evolutionary origin for the large, four-module transcriptional mediator complex. Nucleic Acids Res. 2008;36(12):3993–4008. Epub 2008/06/03. PubMed PMID: 18515835; PubMed Central PMCID: PMC2475620.
- Daniels DL, Ford M, Schwinn MK, et al. Mutual exclusivity of MED12/MED12L, MED13/13L, and CDK8/19 paralogs revealed within the CDK-mediator kinase module. J Proteomics Bioinform. 2013;S2(004). DOI:10.4172/jpb.S2-004
- Nemet J, Jelicic B, Rubelj I, et al. The two faces of Cdk8, a positive/negative regulator of transcription. Biochimie. 2014;97:22–27. Epub 2013/ 10/22. PubMed PMID: 24139904.
- Knuesel MT, Meyer KD, Bernecky C, et al. The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev. 2009;23(4):439–451. Epub 2009/02/26. PubMed PMID: 19240132; PubMed Central PMCID: PMC2648653.
- Pelish HE, Liau BB, Nitulescu II, et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature. 2015;526(7572):273–276. PubMed PMID: 26416749; PubMed Central PMCID: PMC4641525.
- Donner AJ, Ebmeier CC, Taatjes DJ, et al. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat Struct Mol Biol. 2010;17(2):194–201. PubMed PMID: 20098423; PubMed Central PMCID: PMC2920286.
- Galbraith MD, Allen MA, Bensard CL, et al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell. 2013;153(6):1327–1339. PubMed PMID: 23746844; PubMed Central PMCID: PMC3681429.
- Clark AD, Oldenbroek M, Boyer TG. Mediator kinase module and human tumorigenesis. Crit Rev Biochem Mol Biol. 2015;50(5):393–426. Epub 2015/07/17. PubMed PMID: 26182352; PubMed Central PMCID: PMC4928375.
- Firestein R, Bass AJ, Kim SY, et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature. 2008;455(7212):547–551. Epub 2008/ 09/17. PubMed PMID: 18794900; PubMed Central PMCID: PMC2587138.
- Kapoor A, Goldberg MS, Cumberland LK, et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature. 2010;468(7327):1105–1109. Epub 2010/ 12/24. PubMed PMID: 21179167; PubMed Central PMCID: PMC3057940.
- Xu W, Ji JY. Dysregulation of CDK8 and Cyclin C in tumorigenesis. J Genet Genomics. 2011;38(10):439–452. PubMed PMID: 22035865.
- Chen M, Liang J, Ji H, et al. CDK8/19 Mediator kinases potentiate induction of transcription by NFkappaB. Proc Natl Acad Sci U S A. 2017;114(38):10208–10213. Epub 2017/09/01. PubMed PMID: 28855340; PubMed Central PMCID: PMC5617299.
- Johannessen L, Sundberg TB, O’Connell DJ, et al. Small-molecule studies identify CDK8 as a regulator of IL-10 in myeloid cells. Nat Chem Biol. 2017;13(10):1102–1108. Epub 2017/08/15. PubMed PMID: 28805801; PubMed Central PMCID: PMC5693369.
- Audetat KA, Galbraith MD, Odell AT, et al. A kinase-independent role for cyclin-dependent kinase 19 in p53 response. Mol Cell Biol. 2017;37(13). Epub 2017/04/19. DOI:10.1128/MCB.00626-16. PubMed PMID: 28416637; PubMed Central PMCID: PMC5472832.
- Philip S, Kumarasiri M, Teo T, et al. Cyclin-dependent kinase 8: a new hope in targeted cancer therapy? J Med Chem. 2018;61(12):5073–5092. Epub 2017/ 12/22. PubMed PMID: 29266937.
- Rzymski T, Mikula M, Wiklik K, et al. CDK8 kinase–an emerging target in targeted cancer therapy. Biochim Biophys Acta. 2015;1854(10 Pt B):1617–1629. Epub 2015/ 05/27. PubMed PMID: 26006748.
- Aoki S, Watanabe Y, Sanagawa M, et al. Cortistatins A, B, C, and D, anti-angiogenic steroidal alkaloids, from the marine sponge Corticium simplex. J Am Chem Soc. 2006;128(10):3148–3149. Epub 2006/03/09. PubMed PMID: 16522087.
- Cee VJ, Chen DY, Lee MR, et al. Cortistatin A is a high-affinity ligand of protein kinases ROCK, CDK8, and CDK11. Angew Chem Int Ed Engl. 2009;48(47):8952–8957. Epub 2009/10/22. PubMed PMID: 19844931.
- Poss ZC, Ebmeier CC, Odell AT, et al. Identification of mediator kinase substrates in human cells using cortistatin a and quantitative phosphoproteomics. Cell Rep. 2016;15(2):436–450. PubMed PMID: 27050516; PubMed Central PMCID: PMC4833653
- Nitulescu II, Meyer SC, Wen QJ, et al. Mediator kinase phosphorylation of STAT1 S727 promotes growth of neoplasms with JAK-STAT activation. EBioMedicine. 2017;26:112–125. Epub 2017/12/15. PubMed PMID: 29239838; PubMed Central PMCID: PMC5832629.
- Porter DC, Farmaki E, Altilia S, et al. Cyclin-dependent kinase 8 mediates chemotherapy-induced tumor-promoting paracrine activities. Proc Natl Acad Sci U S A. 2012;109(34):13799–13804. Epub 2012/08/08. PubMed PMID: 22869755; PubMed Central PMCID: PMC3427077.
- McDermott MS, Chumanevich AA, Lim CU, et al. Inhibition of CDK8 mediator kinase suppresses estrogen dependent transcription and the growth of estrogen receptor positive breast cancer. Oncotarget. 2017;8(8):12558–12575. Epub 2017/02/02. PubMed PMID: 28147342; PubMed Central PMCID: PMC5355036.
- Bergeron P, Koehler MF, Blackwood EM, et al. Design and development of a series of potent and selective type II inhibitors of CDK8. ACS Med Chem Lett. 2016;7(6):595–600. Epub 2016/06/22. PubMed PMID: 27326333; PubMed Central PMCID: PMC4904263.
- Koehler MF, Bergeron P, Blackwood EM, et al. Development of a Potent, specific CDK8 kinase inhibitor which phenocopies CDK8/19 knockout cells. ACS Med Chem Lett. 2016;7(3):223–228. Epub 2016/03/18. PubMed PMID: 26985305; PubMed Central PMCID: PMC4789660.
- Clarke PA, Ortiz-Ruiz MJ, TePoele R, et al. Assessing the mechanism and therapeutic potential of modulators of the human mediator complex-associated protein kinases. Elife. 2016;5. Epub 2016/12/10. DOI:10.7554/eLife.20722. PubMed PMID: 27935476; PubMed Central PMCID: PMC5224920.
- Czodrowski P, Mallinger A, Wienke D, et al. Structure-based optimization of potent, selective, and orally bioavailable CDK8 inhibitors discovered by high-throughput screening. J Med Chem. 2016;59(20):9337–9349. Epub 2016/08/05. PubMed PMID: 27490956.
- Mallinger A, Crumpler S, Pichowicz M, et al. Discovery of potent, orally bioavailable, small-molecule inhibitors of WNT signaling from a cell-based pathway screen. J Med Chem. 2015;58(4):1717–1735. Epub 2015/02/14. PubMed PMID: 25680029; PubMed Central PMCID: PMC4767141.
- Mallinger A, Schiemann K, Rink C, et al. 2,8-disubstituted-1,6-naphthyridines and 4,6-disubstituted-isoquinolines with potent, selective affinity for CDK8/19. ACS Med Chem Lett. 2016;7(6):573–578. Epub 2016/06/22. PubMed PMID: 27326329; PubMed Central PMCID: PMC4904262.
- Mallinger A, Schiemann K, Rink C, et al. Discovery of potent, selective, and orally bioavailable small-molecule modulators of the mediator complex-associated kinases CDK8 and CDK19. J Med Chem. 2016;59(3):1078–1101. Epub 2016/01/23. PubMed PMID: 26796641; PubMed Central PMCID: PMC5362750.
- Schiemann K, Mallinger A, Wienke D, et al. Discovery of potent and selective CDK8 inhibitors from an HSP90 pharmacophore. Bioorg Med Chem Lett. 2016;26(5):1443–1451. Epub 2016/02/08. PubMed PMID: 26852363.
- Galbraith MD, Andrysik Z, Pandey A, et al. CDK8 kinase activity promotes glycolysis. Cell Rep. 2017;21(6):1495–1506. Epub 2017/11/09. PubMed PMID: 29117556.
- Fisher RP. Taking aim at glycolysis with CDK8 inhibitors. Trends Endocrinol Metab. 2018;29(5):281–282. Epub 2018/02/25. PubMed PMID: 29475579; PubMed Central PMCID: PMC5911231.
- Bartkowiak B, Liu P, Phatnani HP, et al. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010;24(20):2303–2316. Epub 2010/10/19. PubMed PMID: 20952539; PubMed Central PMCID: PMC2956209.
- Cheng SW, Kuzyk MA, Moradian A, et al. Interaction of cyclin-dependent kinase 12/CrkRS with cyclin K1 is required for the phosphorylation of the C-terminal domain of RNA polymerase II. Mol Cell Biol. 2012;32(22):4691–4704. Epub 2012/09/19. PubMed PMID: 22988298; PubMed Central PMCID: PMC3486194.
- Blazek D, Kohoutek J, Bartholomeeusen K, et al. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011;25(20):2158–2172. Epub 2011/10/21. PubMed PMID: 22012619; PubMed Central PMCID: PMC3205586.
- Yu M, Yang W, Ni T, et al. RNA polymerase II-associated factor 1 regulates the release and phosphorylation of paused RNA polymerase II. Science. 2015;350(6266):1383–1386. Epub 2015/ 12/15. PubMed PMID: 26659056.
- Paculova H, Kohoutek J. The emerging roles of CDK12 in tumorigenesis. Cell Div. 2017;12:7. Epub 2017/11/02. PubMed PMID: 29090014; PubMed Central PMCID: PMC5658942.
- Chila R, Guffanti F, Damia G. Role and therapeutic potential of CDK12 in human cancers. Cancer Treat Rev. 2016;50:83–88. Epub 2016/09/24. PubMed PMID: 27662623.
- Zhang Y, Yang L, Kucherlapati M, et al. A pan-cancer compendium of genes deregulated by somatic genomic rearrangement across more than 1,400 cases. Cell Rep. 2018;24(2):515–527. Epub 2018/07/12. . PubMed PMID: 29996110.
- Juan HC, Lin Y, Chen HR, et al. Cdk12 is essential for embryonic development and the maintenance of genomic stability. Cell Death Differ. 2016;23(6):1038–1048. Epub 2015/ 12/15. PubMed PMID: 26658019; PubMed Central PMCID: PMC4987723.
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. Epub 2011/ 07/02. PubMed PMID: 21720365; PubMed Central PMCID: PMC3163504.
- Ekumi KM, Paculova H, Lenasi T, et al. Ovarian carcinoma CDK12 mutations misregulate expression of DNA repair genes via deficient formation and function of the Cdk12/CycK complex. Nucleic Acids Res. 2015;43(5):2575–2589. Epub 2015/02/26. . PubMed PMID: 25712099; PubMed Central PMCID: PMC4357706.
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. Epub 2012/ 09/25. PubMed PMID: 23000897; PubMed Central PMCID: PMC3465532.
- Bajrami I, Frankum JR, Konde A, et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014;74(1):287–297. Epub 2013/ 11/19. PubMed PMID: 24240700; PubMed Central PMCID: PMC4886090.
- Naidoo K, Wai PT, Maguire SL, et al. Evaluation of CDK12 protein expression as a potential novel biomarker for DNA damage response-targeted therapies in breast cancer. Mol Cancer Ther. 2018;17(1):306–315. Epub 2017/ 11/15. PubMed PMID: 29133620.
- Caruso C. CDK12 alterations telling for some with prostate cancer. Cancer Discov. 2018. Epub 2018/07/15. DOI:10.1158/2159-8290.CD-NB2018-093. PubMed PMID: 30006379.
- Wu YM, Cieslik M, Lonigro RJ, et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell. 2018;173(7):1770–82 e14. Epub 2018/06/16. PubMed PMID: 29906450; PubMed Central PMCID: PMC6084431.
- Mertins P, Mani DR, Ruggles KV, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534(7605):55–62. Epub 2016/06/03. PubMed PMID: 27251275; PubMed Central PMCID: PMC5102256.
- Tien JF, Mazloomian A, Cheng SG, et al. CDK12 regulates alternative last exon mRNA splicing and promotes breast cancer cell invasion. Nucleic Acids Res. 2017;45(11):6698–6716. Epub 2017/03/24. PubMed PMID: 28334900; PubMed Central PMCID: PMC5499812.
- Kim HE, Kim DG, Lee KJ, et al. Frequent amplification of CENPF, GMNN and CDK13 genes in hepatocellular carcinomas. PLoS One. 2012;7(8):e43223. Epub 2012/08/23. PubMed PMID: 22912832; PubMed Central PMCID: PMC3418236.
- Bartkowiak B, Yan C, Greenleaf AL. Engineering an analog-sensitive CDK12 cell line using CRISPR/Cas. Biochim Biophys Acta. 2015;1849(9):1179–1187. Epub 2015/ 07/21. . PubMed PMID: 26189575; PubMed Central PMCID: PMC4556607.
- Zhang T, Kwiatkowski N, Olson CM, et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat Chem Biol. 2016;12(10):876–884. Epub 2016/08/30. PubMed PMID: 27571479; PubMed Central PMCID: PMC5033074.
- Ito M, Tanaka T, Toita A, et al. Discovery of 3-benzyl-1-(trans-4-((5-cyanopyridin-2-yl)amino)cyclohexyl)-1-arylurea derivatives as novel and selective cyclin-dependent kinase 12 (CDK12) inhibitors. J Med Chem. 2018;61:7710–7728. Epub 2018/ 08/02. PubMed PMID: 30067358.
- Johannes JW, Denz CR, Su N, et al. Structure-based design of selective noncovalent CDK12 inhibitors. ChemMedChem. 2018;13(3):231–235. Epub 2017/ 12/22. PubMed PMID: 29266803.
- Iniguez AB, Stolte B, Wang EJ, et al. EWS/FLI confers tumor cell synthetic lethality to CDK12 inhibition in ewing sarcoma. Cancer Cell. 2018;33(2):202–16 e6. Epub 2018/01/24. PubMed PMID: 29358035; PubMed Central PMCID: PMC5846483.
- Liu Y, Zhang X, Han C, et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature. 2015;520(7549):697–701. Epub 2015/ 04/23. PubMed PMID: 25901683; PubMed Central PMCID: PMC4417759.
- Wong RWJ, Ishida T, Sanda T. Targeting general transcriptional machinery as a therapeutic strategy for adult T-cell leukemia. Molecules. 2018;23(5):1057. Epub 2018/05/05. PubMed PMID: 29724031.
- Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16(2):101–114. Epub 2016/ 11/26. PubMed PMID: 27885283; PubMed Central PMCID: PMC5684876.
- Albert TK, Antrecht C, Kremmer E, et al. The establishment of a hyperactive structure allows the tumour suppressor protein p53 to function through P-TEFb during limited CDK9 kinase inhibition. PLoS One. 2016;11(1):e0146648. Epub 2016/01/09. PubMed PMID: 26745862; PubMed Central PMCID: PMC4706356.
- Baker EK, Taylor S, Gupte A, et al. BET inhibitors induce apoptosis through a MYC independent mechanism and synergise with CDK inhibitors to kill osteosarcoma cells. Sci Rep. 2015;5:10120. Epub 2015/05/07. PubMed PMID: 25944566; PubMed Central PMCID: PMC4421868.
- Gerlach D, Tontsch-Grunt U, Baum A, et al. The novel BET bromodomain inhibitor BI 894999 represses super-enhancer-associated transcription and synergizes with CDK9 inhibition in AML. Oncogene. 2018;37(20):2687–2701. Epub 2018/03/02. PubMed PMID: 29491412; PubMed Central PMCID: PMC5955861.
- Moreno N, Holsten T, Mertins J, et al. Combined BRD4 and CDK9 inhibition as a new therapeutic approach in malignant rhabdoid tumors. Oncotarget. 2017;8(49):84986–84995. Epub 2017/11/22. PubMed PMID: 29156698; PubMed Central PMCID: PMC5689588.