Abstract
Understanding the function of chromatin has become an exciting field that interests researchers from a wide variety of disciplines. Chromatin is composed of fundamental nucleosome building blocks, and the ability to assemble defined, positioned mononucleosomes is central to experimental investigation of chromatin structure, stability and dynamics. However, the molecular principles, historical background and biochemistry of experimental techniques underlying positioned nucleosomes are sometimes unclear. This review provides an introduction from the perspective of the core histone octamer as a surface for histone–DNA interactions that directs a unique conformation for wrapped DNA. The origins and properties of the most widely used nucleosome positioning DNA sequences including the Widom 601, 5S rRNA and MMTV LTR as well as other less well-known examples are described. An overview of the main experimental methods for preparing nucleosomes and mapping their positions is also provided. These should be suitable for researchers entering the field of chromatin and enable an appreciation of the principles and practical aspects of experimental investigations using positioned nucleosomes in vitro.
Introduction
Chromatin is the universal packaging system used by eukaryotic genomes, and is based on highly conserved structures known as nucleosomes. The genetic strategies and molecular mechanisms that operate on chromatin as their substrate require a predictable packaging of DNA by these nucleosome complexes. Understanding the behaviour of the fundamental nucleosome units is therefore crucial to explaining the eukaryotic genome and its transactions.
This challenge has interested researchers from many experimental and theoretical fields, and led to fruitful research over several decades. Huge strides have been made in understanding the molecular properties of chromatin and its organisation, which depend substantially on our knowledge of the biochemical properties of nucleosomes. This review provides a general explanation of the background and concepts underlying the study of nucleosomes in vitro, with references to the original advances and links to recent reviews.
Chromatin research has always been strongly influenced by availability of techniques, with early investigations of genome packaging concentrating on polymer and chemical perspectives to explain DNA compaction (Van Holde Citation1989). Advances in biochemical purification and analysis in the 1970s subsequently revealed nucleosomes as beads on a string (Kornberg Citation1974; Olins and Olins Citation1974). This view of an array of subunit nucleosomes ( still anchors the prevailing model of chromatin as a contorted street map-like network of genome addresses that can be targeted and manipulated (Kornberg and Lorch Citation2007).
Figure 1. The nucleosome is the fundamental repeating unit of chromatin structure. (a) Schematic illustrating nucleosome core particle with core histone octamer, chromatosome with linker histone and nucleosome repeat including linker DNA. (b) Histone octamer provides a spiralling superhelical path of DNA interactions, shown looking along dyad axis towards central four helix bundle (upper) or down superhelical axis (lower). Only histone fold motif structural elements are shown for simplicity.
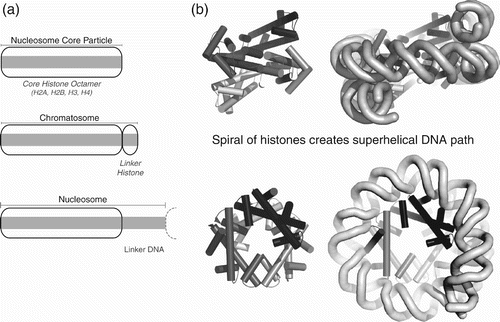
Structural information at progressively higher resolutions in turn revealed the ‘beads’ to have a tuna can-like appearance with DNA spooled around a semi-cylindrical histone octamer core (Klug et al. Citation1980), and eventually at atomic resolution to be a spiral of histone fold dimer units providing a ramp of specialised atomic interactions for DNA binding (Luger, Mäder, et al. 1997; Luger and Richmond Citation1998a).
Concepts of nucleosome positioning
Subsequent interpretation using a wide variety of biochemical and biophysical techniques has led to three interrelated principles of nucleosome positioning and dynamics: firstly, DNA sequence accessibility depends on its position within the nucleosome structure. Secondly, DNA sequences can influence their location in the nucleosome. And thirdly, DNA and the histone octamer core together control the potential for structural changes in the nucleosome.
These concepts are reflected in terminology. The location of DNA relative to the histone octamer in a stable nucleosome is known as nucleosome positioning, and so sequences that are able to direct the histone octamer location are known as nucleosome positioning sequences. The propensity for nucleosomes to resist or undergo structural changes is referred to as nucleosome stability or, conversely, as nucleosome dynamics. The well-known but ill-defined process of chromatin remodelling usually refers to enzyme-catalysed processes that interchange or disrupt stable nucleosomes.
Definition of a nucleosome position
The nucleosome is defined as the repeating unit of chromatin (Oudet et al. Citation1975). However, the term nucleosome is also frequently used as a shorthand for the nucleosome core particle (NCP), comprising a structured octamer of the core histones H2A, H2B, H3 and H4 wrapped with ∼147 bp DNA (.
The strict definition of the nucleosome as a repeating unit encompasses both the NCP and additional linker DNA (. This linker varies characteristically from 10 to 70 bp in ∼10 bp steps, depending on species and tissue (Widom Citation1992), and influences the repeating higher order chromatin structure (Grigoryev and Woodcock Citation2012). The linker DNA may be associated with a fifth linker histone, such as H1 in compacted chromatin, thus forming a stable complex of the core histone octamer, H1 and ∼165 bp DNA known as the chromatosome (Simpson Citation1978) (. The exact arrangement and number of linker histones per nucleosome has been controversial.
The fundamental specification of nucleosome positioning is based on the location of the NCP dyad axis (Weintraub et al. Citation1976) because the histone octamer has dyad symmetry due to mapping of the structure onto itself via a 180° rotation about a dyad axis as a consequence of the symmetric H3–H3 four helix bundle interaction at the centre of the histone octamer (. The location where the NCP dyad axis passes through the DNA duplex therefore provides a convenient coordinate from which to define the nucleosome position. DNA coordinates themselves are usually defined relative to a nearby genomic feature, such as a transcription start site.
Figure 2. Nomenclature for nucleosome position and DNA wrapping. (a) Nucleosome position is specified by where its pseudodyad axis passes through DNA, illustrated as a schematic (upper) or with structure (lower). (b) Superhelical locations (SHL) are defined from the dyad as SHL 0 and describe where minor groove faces away (integral) or towards (half integral) histone octamer (Klug et al. Citation1980).
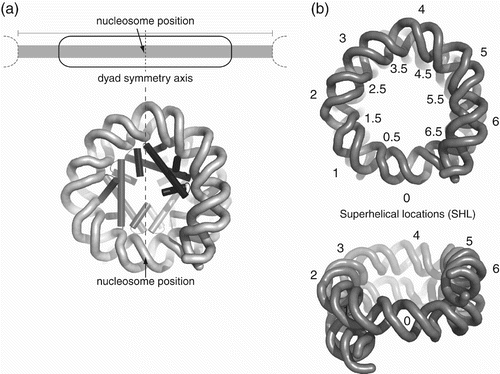
DNA wraps in 1.65 turns of left-handed superhelix around the histone octamer to create two superhelical gyres. Atomic resolution nucleosome structures and solution experiments show that the dyad axis normally passes through the plane of a single base pair at the dyad. Having a single central base implies that the symmetric nucleosome must wrap an odd number of base pairs (Flaus et al. Citation1996; Luger, Mäder, et al. 1997), nominally defined as 147 bp, and in principle this value supersedes an earlier widely used estimate of 146 bp derived from the limit of nuclease of linker DNA (Lutter Citation1978). However, the exact instantaneous number of wrapped base pairs may fluctuate due to stochastic thermal motion or during remodelling. It has also been pointed out that, since nucleosomes appear readily able to accommodate 1–2 bp of DNA overtwist, the most common instantaneous value in bulk genomic DNA could actually be 145–146 bp (Negri et al. Citation2001; Richmond and Davey Citation2003; Edayathumangalam et al. Citation2005).
The DNA turn encompassing the dyad axis base pair has its minor groove facing outwards and is defined as superhelical location 0 (SHL 0; ; Klug et al. Citation1980). Subsequent outward-facing minor grooves are numbered SHL ±1–6, and the inward-facing minor grooves where histone–DNA contacts occur are numbered SHL ±0.5–6.5 (.
Directionality arrives only from the DNA sequence because the histone octamer is in principle dyad symmetric, and so an SHL value can take either sign and is often prefixed ‘±’ to indicate this. Likewise, DNA projections from either side of the NCP cannot be distinguished and are referred to as entry/exit DNA. However, even if directionality is undefined, the DNA sequence is almost never truly palindromic and strict symmetry is broken. For this reason the NCP is often described as pseudodyad symmetric (Lutter Citation1978).
Rotational and translational positioning
At the molecular level, the regular pattern of beads on a string implies nucleosomes take up regular positions along DNA, and this was originally referred to as phasing (Lohr et al. Citation1977). Phasing can be explained as rotational positioning ( to define which helical face of DNA is exposed outwards from the nucleosome surface, combined with translational positioning ( to define where the nucleosome is located along the DNA (Drew and Travers Citation1985). Rotational phases are related by multiples of 10–11 bp because of the typical ∼10.5 bp/turn free DNA pitch, and will affect accessibility for binding factors at a general level. Rotational phasing is therefore a less stringent structural requirement, whereas precise translational positioning must distinguish between multiple locations with overlapping biophysical properties differing by 1 bp.
Figure 3. Nucleosome positioning concepts. (a) Rotational phasing, where nucleosomes at multiple locations maintain same exposed DNA face. (b) Translational positioning, where nucleosome maintains a specific histone–DNA relationship and location. (c) Statistical positioning, where a barrier arranges a series of nucleosomes by exclusion, typically with increasing variability along the array.
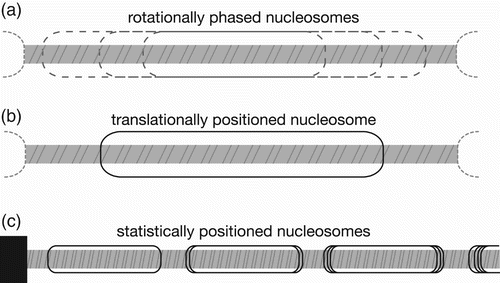
The fundamental observation of phased arrays led to the suggestion that a single initial barrier with a defined translational position could drive statistical positioning ( by biasing the locations of adjacent nucleosomes through exclusion (Kornberg and Stryer Citation1988). Barriers could be provided by a positioned nucleosome, by another DNA bound factor, or by a sequence that excludes nucleosomes. Spacing could be influenced by the deposition machinery, including ATP-dependent chromatin remodelling factors and polymerases, or inter-nucleosomal interactions such as higher order chromatin structure contacts. Such nucleosomal phasing has been observed empirically to persist for at least 4–6 nucleosomes (Yodh et al. Citation2002; Mavrich et al. Citation2008; Schones et al. Citation2008; Blacketer et al. Citation2010).
In vivo positioning as combination of effects
Nucleosome positioning in vivo is a multifactorial property that is influenced but not dictated by DNA properties alone (Iyer Citation2012). In Saccharomyces cerevisiae, genome-wide mapping of nucleosomes suggests that only a few DNA sequence-based properties such as absence of A/T rich regions, or conversely a high G/C content, have a strong effect on general positioning (discussed in Hughes and Rando Citation2009; Tillo and Hughes Citation2009; Radman-Livaja and Rando Citation2010; Zhang et al. Citation2010), so the dominant mechanism for defining the location of nucleosomes may be statistical positioning (Gkikopoulos et al. Citation2011; Zhang et al. Citation2011). Earlier individual nucleosome mapping approaches in S. cerevisiae at single sites had already shown that barrier effects provided a major positioning effect, with DNA properties and histone–DNA interactions acting to fine tune local accessibility and behaviour (Thoma Citation1992).
This complex behaviour in vivo means that recapitulating native nucleosome positioning is a challenge. However, the ability to reliably assemble nucleosomes at specific positions in vitro does allow specific contributions of DNA sequence to positioning to be studied in isolation and enables homogenous defined substrates to be prepared for assaying nucleosome dynamics and chromatin active enzymes.
The nucleosome as a surface of histone–DNA interactions
Sequence-dependent nucleosome positioning is generated by the choice of path for wrapping DNA around the core histone octamer. Although it is often represented as a smooth tuna can, the histone octamer in fact provides a feature-rich spiral of histone fold dimers (), with a specific path for combinatorial histone–DNA interactions that is crucial for nucleosome positioning.
Figure 4. Nomenclature of histone octamer structure. (a) Three alpha helices (α1, α2 and α3) define the common histone fold motif for all four core histones (upper), with histone fold dimers packing through specific hydrophobic interactions between helices (lower). (b) Specific interactions stabilise four helix bundle (4HB) interactions for H3:H3, which leads to the dyad symmetry axis of the nucleosome (central filled circle). H4:H2B forms an equivalent 4HB (not shown). (c) The two possible 4HBs and their geometry generates the spiral of histone fold dimers. (d) Outer faces of spiralled histone fold dimers presents α1α1 and L1L2 motifs for DNA interaction in specific order within nucleosome (see for details.)
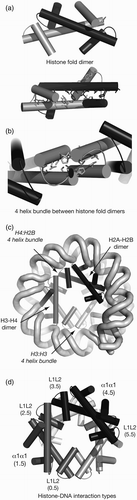
Histone octamer is a spiral of histone fold dimers
The histone fold motif is the common secondary structure pattern in all four core histones H2A, H2B, H3 and H4, and comprises a long central α2 helix flanked by shorter α1 and α3 helices ( and ; Dechassa and Luger Citation2012). The histones associate by specific mainly hydrophobic interactions in antiparallel pairs H2A–H2B and H3–H4 along the α2 helix, with the α1 and α3 helices folded back across the long α2 helix (.
Table 1. Histone amino acid residue numbering by structure elements.
Table 2. Direct histone–DNA hydrogen bonding interactions by superhelical location (SHL).
This modular arrangement presents protein–protein interactions, which define the histone octamer (Mariño-Ramírez et al. Citation2005): the α2–α3 surfaces of two H3 molecules self-associate tightly to create a symmetric four helix bundle (4HB; and generate the dyad axis. The α2–α3 surfaces of H2B and H4 then associate in another 4HB to complete the octamer (. Because no other 4HBs are stable (D'Anna and Isenberg Citation1974), the H2A–H2B dimer acts as a cap to limit the histone complex to four histone fold dimers, two each of H3–H4 and H2A–H2B (; Eickbush and Moudrianakis Citation1978).
The stronger H3:H3 4HB interaction is stable under physiologically relevant conditions in vitro, so the combined (H3–H4)2 pair is isolated biochemically as a tetramer, whereas the H2B:H4 4HB is more readily dissociated to release separate H2A–H2B dimers. When all eight histones are associated into an octamer, the angles of the 4HBs generate a left-handed spiral of histone fold dimers to form a ramp on which the DNA superhelix can wind (, 4c and 4d). A number of other internal interactions such as a contact between H2As of the two H2A–H2B dimer also stabilise the octamer structure (White et al. Citation2001; Mariño-Ramírez et al. Citation2005).
Histone octamer is a scaffold for DNA contacts
The folding back of the α1 and α3 helices also generates the two types of DNA interaction surfaces (. The first is formed by the combination of pairs of loops connecting alpha helices known as L1L2 motifs because the pairs involve the loops between α1 and α2 (L1), and α2 and α3 (L2). The second type of interaction surface is formed by paired N-terminal ends of α1 helices, and is known as the α1α1 motif.
The nomenclature for these interaction surfaces was first defined for the histone-like TAF42/62 complex whose structure was determined before the nucleosome (Xie et al. Citation1996). A number of other protein complexes have since been shown to share the same histone fold complex structure, although not all of these necessarily bind DNA (Kamada et al. Citation2001; Hartlepp et al. Citation2005; Nishino et al. Citation2012).
Because of the repeating nature of the histone fold dimer spiral, eight L1L2 and four α1α1 motifs provide a scaffold that presents a regular pattern of DNA contact interactions (, . These are almost exclusively made across the DNA minor groove as it faces inwards towards the histone octamer, and are responsible for wrapping SHL ±0.5–5.5 or ∼120 bp of the 147 bp of DNA in the NCP (; reviewed in Luger and Richmond Citation1998a).
Despite the underlying pattern of the histone fold dimer spiral, the DNA superhelix does not follow a perfectly smooth path due to variations in the histone sequences, which affect each SHL contact point. An example is the longer H3 and H2B L1 loops at SHL ±2.5 and SHL ±5.5, respectively, which affect the conformation of paired H4 and H2A L2 loops, and in turn the bend of their α2 helices (Luger, Mäder, et al. 1997).
Detailed structural analysis is therefore required to rationalise patterns of histone–DNA contacts. Some general principles have arisen from these considerations of DNA binding in the nucleosome.
Firstly, there is a general trend of increasing numbers of histone–DNA bonds nearer the dyad axis (; Davey et al. Citation2002). This is often interpreted as implying DNA is more tightly bound, although it is impossible to calculate bond strengths directly. Nevertheless, the trend is consistent with the higher ionic strength required to dissociate DNA from the inner (H3–H4)2 tetramer compared to the outer H2A–H2B dimers (Germond et al. Citation1976; Jorcano and Ruiz-Carrillo Citation1979) and with other biophysical observations (Hall et al. Citation2009). The consequence of more interactions being made between DNA and H3–H4 is that the region of DNA inside SHL +3.5 dominates DNA sequence effects on positioning and stability. It has been proposed that the energetic significance of contact points can be ranked in descending order: SHL (Chua et al. Citation2012).
Secondly, histone–DNA bonds do not simply involve charge–charge interactions and there is a major contribution of hydrogen bonding between the phosphates and the histone protein backbone (). Because these interactions are short range and provided by the polypeptide backbone, which is in principle more rigid than sidechains, the bonds enforce a stable scaffold on DNA binding that is likely to drive the wide variety of different DNA sequences to be wrapped into a limited number of conformations (Luger and Richmond Citation1998a). This in turn leads to major variations in the energetics of binding for different DNA sequences.
Thirdly, the function of basic histone sidechains is not simply to stabilise DNA binding by ionic interactions, but also to induce DNA bending through charge neutralisation on the inner DNA face, reducing the spine of phosphate repulsions that normally makes DNA linear (Strauss and Maher Citation1994). Arginine sidechains located at each of the 14 minor grooves make interactions with phosphates, although 8 of these are modulated by interacting threonine sidechains (Luger and Richmond Citation1998a).
Unique histone features interact with DNA
In addition to the regular DNA interaction scaffold provided by the histone fold-based structure, several non-histone fold parts of the histone polypeptides also contribute to DNA binding. The outermost DNA contact points at SHL ±6.5 nearest to the entry/exit DNA are made by the additional H3 αN helix, which runs parallel with this turn of DNA, providing multiple charge–charge interactions with the phosphate backbone (. This helix in turn makes interactions along other faces with the H2A C-terminal extension and H4 α1 helix (Ferreira et al. Citation2007). The H3 N-terminal tail from residues 38–44 immediately upstream of H3 αN threads between the central and entry/exit gyres and makes direct contacts at SHL ±0.5 and ±6.5 (. H3 αN therefore controls the entry/exit DNA while simultaneously interacting with structures around the dyad, and is potentially a key region for dynamic properties such as DNA site exposure and nucleosome disassembly (Ferreira et al. Citation2007).
Figure 5. Non-histone fold helices of H3 and H2B also bind DNA. (a) H3 αN organises entry/exit DNA at SHL±6.5 as well as contacting dyad DNA at SHL±1, H2A C-extension and H4 α1. H2B αC helix sits above DNA superhelix at SHL±3.7. (b) Histone tails exit locations for H2A N-tail (SHL±4.2), H2A C-tail (SHL ±0.7), H2B N-tail (between gyres at SHL±2.7/4.7), H3 N-tail (entry/exit DNA) and H4 N-tail (SHL±2.2).
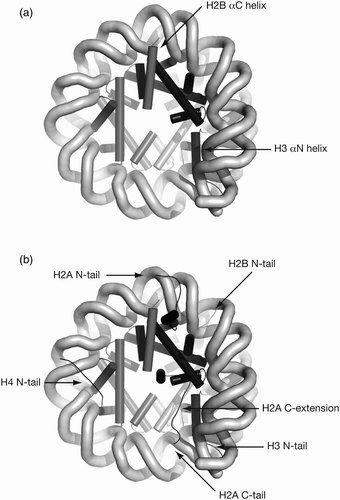
Additional irregular features include the H2B αC helix, which flops across the minor groove at SHL ±3.7 (, the N-terminal tail of H2B, which threads through the DNA superhelix making contact simultaneously at SHL ±2.7 and ±4.7 (, and the H2A and H4 tails, which pass over the DNA superhelix at SHL ±4.2 and SHL ±2.2, respectively (; reviewed in Luger and Richmond Citation1998a).
DNA conformation in the nucleosome
The scaffold of interaction surfaces provided by histones make up the set of 14 SHL contact points ( and ) or ‘binding platforms’ (Chua et al. Citation2012). Each induces a set of responses in the wrapped DNA conformation, which have been analysed in detail (Richmond and Davey Citation2003; Olson and Zhurkin Citation2011; Chua et al. Citation2012).
To fit the stereochemistry of histone interactions offered at the contact points, DNA generally undergoes large roll and also slide, twist and shift deformations, some of which are unusual in other DNA binding proteins (Richmond and Davey Citation2003). The energetics of these deformations gives rise to sequence specific effects on DNA binding, and many authors have addressed the question of how they are linked with choice of nucleosome position (see Segal and Widom Citation2009a; Travers et al. Citation2009; Radman-Livaja and Rando Citation2010; Olson and Zhurkin Citation2011; Trifonov Citation2011; Zhurkin Citation2011; Chua et al. Citation2012).
It has turned out to be unexpectedly difficult to understand the detailed principles of binding because wrapped DNA behaves as an irregular and cooperative superhelix that eludes simple causal explanations. The cooperative deformations across multiple base pairs at each contact point adapt in a variety of ways that do not fall into simple classes for the small number of wrapped DNAs so far observed in nucleosome crystal structures.
Adding to the complexity of interpretation is that DNA base sequence itself acts only via an indirect readout through the backbone conformation since DNA binding does not involve base-specific contacts. Furthermore, small differences at multiple histone–DNA contact points can be amplified when effects are in phase across the 14 SHL locations since each contacts approximately four phosphate groups. This means that the energetics of all the individual DNA deformation sites needs to be understood in detail in order to understand the wrapping. Unfortunately, the effect of any base pair is small and difficult to measure experimentally, and DNA takes up relatively extreme and poorly understood conformations that complicate computational approaches (Olson and Zhurkin Citation2011).
Overall, the deformations in localised bending and twisting suggest that sequence-dependent DNA bendability and twistability are central parameters in the energetics of binding and positioning. The most obvious and well-understood examples of these effects are localised ‘stretch’ sites and biased locations of A/T composition, respectively.
DNA twisting and localised stretch sites
One of the most distinctive properties of nucleosomal DNA is the remarkable one base pair stretch discontinuities observed at either SHL ±2 or SHL ±5 in most crystal structures. These involve base pair steps across a region of ∼10 bp stabilising increased twist and rise (distance between bases) to yield an overall stretching that simulates the presence of an extra base pair in the DNA helix geometry. This is referred to equivalently either as overtwisting because the angle between successive base pairs is increased or as underwinding because less bases are used to accomplish a turn of DNA helix. Both SHL ±2 and SHL ±5 are flanked by α1α1 and L1L2 motifs on their internal and external sides (; Tan and Davey Citation2010).
The stretch phenomenon was originally thought to be induced by crystal packing of exposed 146 bp DNA termini (Luger, Mäder, et al. 1997), but a recent structure without end packing also shows the same effect (Makde et al. Citation2010), suggesting that the phenomenon may be inherent to nucleosome wrapping.
Paradoxically, high-resolution nuclease footprinting in vitro on a defined positioning sequence shows that the innermost three turns of DNA inside SHL ±2 disturb the regular periodicity of outer regions and shift the phase of DNA helical parameters between the outer arms by 2 bp (Hayes et al. Citation1990). This is not influenced by the presence of histone tails (Hayes et al. Citation1991; Vitolo et al. Citation2004). Similar effects have also been observed in patterns from bulk populations of nucleosomes (Satchwell et al. Citation1986) and it has been hypothesised to represent a different arrangement of DNA in the dyad region (Travers and Klug Citation1987).
However, the discontinuity is not apparent in any of the DNA of crystal structures solved so far and may stem either from variation of stretching of actual genome sequences or from a dynamic equilibrium in solution that is not captured in crystals (Negri et al. Citation2001). Understanding the stabilisation and dynamics of stretching and its behaviour around the dyad region is likely to be crucial to nucleosome positioning, sliding and remodelling.
DNA bending and the role of A/T sequences
DNA is very tightly bent in the nucleosome, implicating the energetics of bending as an important property for positioning. Bending can be facilitated either with a static bend, where the naked DNA already has a bend, or with anisotropic bendability, where the DNA bends more readily in a particular direction when wrapped. A contribution from bending is implicit in the dominant DNA sequence-dependent effects on nucleosome positioning in vivo and appears to have significant links with the distribution of A/T nucleotides (Hughes and Rando Citation2009; Tillo and Hughes Citation2009).
Firstly, a pronounced preference for A/T containing sequences in the minor groove facing towards the histone octamer was observed in the original statistical sequencing experiments of Travers and colleagues (Drew and Travers Citation1985; Satchwell et al. Citation1986), and has been substantiated as a more general role for pyrimidine–purine steps such as TA and TG using synthetic constructs, modelling and observed patterns of genome-wide nucleosome positioning. The reason for this bias is that pyrimidine–purine steps are more readily compressible at the narrow inward facing minor groove, whereas G/C-containing sequences preferentially occupy the widened outward facing groove (Travers and Klug Citation1987).
It is important to note that although the least energetic cost of deformations is seen in TA base step stacking energy and A/T base pairs, and can be used to enable the required DNA conformation, sequences containing G/C base pairs can nevertheless be integrated in a histone–DNA contact point (Chua et al. Citation2012).
Secondly, G/C content may be required to counteract the pronounced effect of A/T tracts longer than 4–6 bp on nucleosome wrapping. These tracts can stack in a distorted conformation allowing special bifurcated hydrogen bonding between bases and stable minor groove hydration (Nelson et al. Citation1987; Woods et al. Citation2004; Segal and Widom Citation2009b). This additional stability induces a rigid DNA helix, which is less desirable for bending around the nucleosome, and such A/T tracts sequences have been shown to drive nucleosome positioning in vitro (Rhodes Citation1979; Simpson and Shindo Citation1979) and in vivo (Yuan et al. Citation2005). However, the stabilisation of rigid A/T tracts is moderate and environment dependent and therefore at elevated temperatures the tract structure is destabilised and such A/T tracts sequences give rise to flexible DNA, which is more readily wrapped around the nucleosome (Puhl et al. Citation1991; Puhl and Behe Citation1995). Sliding of nucleosomes over these tracts by remodelling machinery can also overcome the rigidity and enable nucleosome wrapping in vivo (Whitehouse et al. Citation2007).
Effect on DNA flexibility of nicks, gaps and small molecules
Both in vitro experimental designs and the biological process of DNA damage recognition are affected by DNA nicks and gaps. Although their detailed effect on the already taut nucleosomal DNA wrapping is unclear, these DNA backbone modifications have surprisingly small effects on free DNA. Introduction of nicks in the sugar–phosphate backbone does not cause significant effects on twistability or bendability because base pair stacking energies are very high and stabilise the duplex.
The DNA duplex can even tolerate single base pair gaps in one strand by restacking the flanking base pair planes (Mills et al. Citation1994; Yakovchuk et al. Citation2006; Peters and Maher Citation2010). Conversely, binding of small molecule reagents to the minor groove can have a strong influence on nucleosome positioning. For example, the naturally occurring A/T-specific antifungal agent distamycin has been shown to reverse the rotational positioning (Portugal and Waring Citation1986, Citation1987). Using this mode of binding and the ‘supergroove’ alignment of minor grooves in the stacked superhelical gyres, polyamide reagents that cooperatively bound to adjacent sites were shown to block nucleosome sliding (Edayathumangalam et al. Citation2004, Citation2005). This illustrates the requirement for cooperative DNA flexibility in nucleosome dynamics and the potential for DNA binding proteins to affect nucleosome structure.
In vitro nucleosome positioning sequences
A number of DNA sequences have been commonly used to direct histone octamer assembly to strongly biased locations during in vitro studies of nucleosome behaviour (). These are very useful tools for testing DNA binding preferences and can provide homogenous defined substrates for assaying nucleosome dynamics and chromatin active enzymes.
Table 3. DNA sequences commonly used for nucleosome positioning.
Significance of nucleosome positioning sequences
The equilibration determining nucleosome positioning during typical in vitro salt gradient assembly is dominated by the (H3–H4)2 tetramer (Dong and Van Holde Citation1991) and occurs at non-physiological salt concentrations (Drew Citation1991). Some subsequent repositioning must also occur since most reconstitutions also obey the full 147 bp interaction potential.
Furthermore, most DNA fragments without pronounced positioning signals will direct nucleosome assembly to terminal positions to minimise repulsion of the entry/exit DNA duplexes (Sakaue et al. Citation2001), although only a tiny proportion of nucleosomes occur at telomeres in vivo.
In vitro nucleosome sliding under physiologically relevant conditions often ends with redistribution to terminal positions for the same reasons, so nucleosomes assembled at internal positions by salt gradient dialysis have been described as disequilibrium nucleosomes (Anderson et al. Citation2002). The reason for this disequilibrium is that the nucleosome position is selected at an elevated salt concentration and dominated by the (H3–H4)2 tetramer (Drew Citation1991; Thåström et al. Citation2004). The nucleosomes are later able to re-explore their thermodynamic stability at physiologically relevant ionic strength by sliding along DNA in a non-dissociative mechanism if supplied with sufficient activation energy.
Even the largest eukaryotic genomes with 109–1010 base pairs and large fractions of repetitive DNA do not sample the potential complexity of 4147=1088 different possible sequences that could be wrapped around the nucleosome, and so an ultimate positioning sequence is unlikely to ever be determined.
Nucleosomes will almost always assemble in vitro to bind 147 bp DNA, meaning that a 200 bp DNA fragment has ∼50 possible nucleosome positions separated in 1 bp steps. For positioning, one of these must dominate to achieve strong positioning, and this makes unique nucleosome translational positioning an unlikely and valuable property.
Three strategies have led to the identification of the in vitro nucleosome positioning sequences that are in use. Firstly, screening of libraries of DNA fragments has given rise to positioning sequences like the popular 601. Secondly, biochemical interest in genome locations has led to the observation of natural positioning on 5S, mouse mammary tumour virus (MMTV) and alpha satellite sequences. And finally, de novo design and mutagenesis has led to the development of artificial sequences such as the TG repeated unit.
Widom 601 sequence
Probably the most common DNAs used for in vitro nucleosome positioning are the ‘601’ sequences popularised by Widom and colleagues (; Lowary and Widom Citation1998). The original 601 sequence was isolated by SELEX (Systematic Evolution of Ligands by Exponential Enrichment) from a seed pool of 1012 chemically randomised artificial 220 bp fragments, using 15 rounds of selection by competitive salt dialysis assembly with chicken histone octamer. This selection yielded ∼50 sequences of which 9 were ranked by competitive nucleosome reconstitution, revealing isolate 601 as highest (Lowary and Widom Citation1998). Analysis of the selected population revealed the dominance of TA steps at DNA helical periods in the family of sequences.
Subsequently, Widom and colleagues made 10 changes in the 147 bp nucleosomal region of the original 601 sequence to insert restriction sites, creating the 174 bp 601.2 fragment (Anderson and Widom Citation2000). 601.1 was an intermediate in the mutagenesis that has not been used experimentally. 601.2 was shown to retain high affinity in competitive reconstitution assays, albeit with reduced relative stability over the original 601. A 152 bp 601.3 fragment was later constructed with different flanking sequences but lacked 4 bp of the 147 bp core region (Anderson and Widom Citation2001).
Two recent crystal structures include the original, higher affinity 601 sequence (Makde et al. Citation2010; Vasudevan et al. Citation2010) and a detailed understanding of the conformation of the 601 sequence has been developed (Vasudevan et al. Citation2010; Chua et al. Citation2012).
This reveals that the crucial SHL ±1.5 region has two changes in 601.2 compared to 601. This location is known to be a significant feature of nucleosomal wrapping in solution (Fitzgerald and Anderson Citation1999; Fernandez and Anderson Citation2007) and structural analysis has identified this as the site of a unique ‘sugar clamp’ conformation (Wu et al. Citation2010). In fact, one of the differences between 601 and 601.2 is within the sugar clamp. Other sequence differences for 601.2 are in the region SHL −6.5 to −5.5, adjacent to the SHL −5 region, where the overtwisted stretch occurs in the structure. This means that the distinction between the original 601 and 601.2 or 601.3 is important.
As expected, flexible pyrimidine–purine steps in the narrow minor groove facing the histone octamer alternate with externally exposed G/C rich grooves along the DNA helical repeat. The undertwisted stretch at SHL ±5 is so stable that crystallisation in the standard lattice requires a 145 bp fragment to replicate terminal base pair stacking normally provided by 147 bp DNA (Vasudevan et al. Citation2010). The stretch is also observed in the RCC1 ternary binding protein, which does not require stacking at all for crystal packing (Makde et al. Citation2010).
This high thermodynamic and structural stability of 601 DNA wrapping has the practical advantage that nucleosomes are readily formed and behave homogeneously, and so these sequences have become near-ubiquitous as an experimental substrate. Interestingly, this stability brings with it the disadvantage that 601 nucleosomes are unlikely to be useful for studying nucleosome dynamics. For example, the thermal energy required to induce sliding on 601 is above 55°C, resulting in the vast majority of nucleosomes being unfolded rather than slid. However, even this extremely strong positioning sequence can be overcome by energy-driven processes in vivo like polymerase passage (Perales et al. Citation2011).
Parallel work by using SELEX for mouse genomic DNA (Widlund et al. Citation1997) identified high-stability native sequences although these have less stability than 601 (Thåström et al. Citation1999).
5S rDNA sequence
The original in vitro nucleosome positioning sequence used for detailed biochemical studies over many years was a 260 bp fragment from the 5S ribosomal RNA gene of Lytechinus variegatus, a spiny sea urchin (Simpson and Stafford Citation1983). It lacks clear periodic TA steps although one arm has the TTAAA motif 2 bp upstream and a TA base step 2 bp downstream of SHL 1.5 ().
The observation of precise nucleosome organising behaviour of this sequence by Simpson in 1983 arose from interest in regulation of large numbers of 5S gene copies during oogenesis in organisms that were amenable to biochemical investigations, such as L. variegatus and the frog model Xenopus. 5S transcription by RNA polymerase III is distinct from the larger rRNA precursor transcript generated by RNA polymerase I, and is directed by an internal control region to which the multiple zinc finger protein TFIIIA binds (Vitolo et al. Citation2004). Since 5S genes are well conserved, L. variegatus, Xenopus borealis, human and mouse 5S genes have all been used as nucleosome positioning sequences.
The 5S positioning sequence was the basis for a series of landmark in vitro nucleosome investigations. A highly repetitive DNaseI digestion pattern was observed by Simpson (Simpson and Stafford Citation1983), which inspired its use in high-resolution hydroxyl radical footprinting to demonstrate the detailed periodicity of DNA wrapping around the nucleosome in solution on the X. borealis 5S sequence (Hayes et al. Citation1990). The simultaneous binding of TFIIIA to its site overlapping the 5S positioned nucleosome, and the potential for this to direct RNA polymerase III or be prevented by H1-driven compaction, provided a paradigm for the active role of nucleosomes in gene regulation (Rhodes Citation1985). Multimerisation of the L. variegatus 5S sequence created a substrate for assembly of defined nucleosomal arrays of 208 bp, which have been the basis for many investigations into the role of higher order chromatin structure (Simpson et al. Citation1985). This repeat was originally reported as 207 bp, but later corrected to 208 bp (Dong and Van Holde Citation1991). The clever use of permuted dimeric 5S sequences also provided the first direct demonstration of thermally induced nucleosome sliding (Meersseman et al. Citation1992).
This dynamic behaviour of the 5S nucleosome is in marked contrast to 601. 5S nucleosomes are very labile and can slide readily during incubations or storage even below 37°C (Meersseman et al. Citation1992; Flaus et al. Citation1996). In fact, the 5S sequence is only slightly more stable than genomic or chemically synthesised bulk random DNA in competitive reconstitution (Widom Citation2001).
MMTV LTR nucleosomes A and B
The in vitro nucleosome positioning sequences for which there is most understanding of in vivo roles are MMTV nucleosomes A and B (). These are the pair most downstream in an array of at least six nucleosomes covering the retroviral long terminal repeat (LTR) (Richard-Foy and Hager Citation1987).
LTRs are duplicated sequences that flank all retroviral genomes, but the MMTV LTR is unusual in having an extended length that encodes a superantigen gene and a hormone-inducible promoter (Ross Citation2010). The MMTV 5′ LTR sits upstream of the viral genome and the promoter drives viral gene transcription, whereas the MMTV 3′ LTR is adjacent to mouse genomic sequences and can influence proto-oncogene transcription (Richard-Foy and Hager Citation1987). In fact, most oncogenic effects are due to insertion of the LTR enhancers near a native promoter to induce inappropriate regulation (enhancer insertion), rather than driving transcription from the LTR promoter itself (promoter insertion) (Callahan and Smith Citation2000; Theodorou et al. Citation2007).
Since the end of the most downstream nucleosome A (nucA) incorporates host genomic sequences, different MMTV isolates have different nucA sequences. Furthermore, the superantigen sequence is hypervariable and this affects sequences across nucB (Brandt-Carlson et al. Citation1993), so care is needed when comparing results from different isolates and this may have led to different nucleosome mapping assignments (Piña et al. Citation1990; Archer et al. Citation1991; Fragoso et al. Citation1995; Flaus and Richmond Citation1998).
MMTV LTR promoter function is an important paradigm for chromatin-mediated gene regulation (Beato and Vicent Citation2011). Nucleosome B encompasses a number of hormone response elements (HREs) that come into close proximity when wrapped on the nucleosome. The HREs can be exposed on the outer surface of the nucleosomal DNA enabling hormone receptors such as progesterone receptor (PR) to bind to these sites cooperatively as dimers and multimers. Recruitment of PR and homologues, possibly assisted by pioneer transcription factors such as FoxA1, causes rapid eviction of heterochromatic factors such as H1 and HP1γ from the nucleosomes assisted by the NURF chromatin remodeller together with post-translational modifications of histones. This in turn leads to subsequent recruitment of the BAF chromatin remodelling complex and histone acetyl transferases (HATs) with further remodelling including eviction of H2A–H2B dimers. The newly opened location around nucA is now accessible for binding by NF1 and the basal transcription machinery. Importantly, the association of PR and other factors is rapid and stochastic, creating a dynamic and responsive induction–repression cycle (Grøntved and Hager Citation2011).
The in vitro positioning of nucA and nucB are dominated by different DNA sequence effects (Flaus and Richmond Citation1998). NucB is significantly AT-rich with only 35% G/C content, and the A/T and G/C dinucleotides are clearly periodic. It is likely that this periodicity leads to non-isotropic bendability of the sequence, which is responsible for strong rotational phasing consistent with the functional requirement to expose HREs. This also means that thermal nucleosome sliding can only be induced at elevated temperatures of more than 50°C (Flaus and Richmond Citation1998). In contrast, nucA has a more typical 57% G/C content and less apparent periodicity and so it can slide at more moderate temperatures around 47°C. The wrapped 147 bp region of nucA is flanked by several homopolymer runs of four to six A/T nucleotides that direct strong positioning. Dinucleosomal nucAB substrates can also be reconstituted in vitro, with loading preferentially to nucA position at low octamer/DNA ratios, consistent with nucA having a role in seeding statistical positioning of the array.
Other in vitro nucleosome positioning sequence
A number of other sequences have been used to direct in vitro nucleosome positioning for functional studies of thermal or ATP-dependent nucleosome sliding and remodelling.
Hsp70 promoter
The Drosophila hsp70 promoter is a well-studied model for regulation by promoter-proximal pausing of polymerases (Saunders et al. Citation2006), and an extended region was assembled into organised chromatin using cell-free extracts in vitro (Tsukiyama et al. Citation1994). A mononucleosomal fragment within this region has been shown to direct approximately five different positions that are distinguishable by native gel electrophoresis, and has been used as a substrate for the study of nucleosome sliding (Hamiche et al. Citation1999).
Ribosomal RNA gene regulatory region
The regulation of mammalian ribosomal RNAs transcribed by RNA polymerase I is of interest because epigenetic mechanisms enable copies to be established in either active or inactive states. A nucleosome adjacent to the mouse TTF-I factor binding site (Längst et al. Citation1998) is differently positioned depending on the activity state of the rDNA copy, under the control of chromatin remodelling activities (Li et al. Citation2006). Like the hsp70 promoter, this sequence can direct the positioning of a nucleosome in vitro to a number of positions including one known to be repressive in vivo, and therefore provides a model for the role of chromatin remodelling (Felle et al. Citation2010).
Histone H4 and trinucleotide repeat genes
Early analysis of histone gene clusters revealed an unusual group of 6 CTG trinucleotide repeats downstream of the Xenopus laevis histone H4 gene (Perry et al. Citation1985). This region showed high affinity in competitive nucleosome reconstitution as well as unique positioning in vitro (Godde and Wolffe Citation1996). Deletion of the CTG repeats and adjacent CGG repeats significantly affected these properties. Unique positioning was also observed on the human DRPLA gene encoding Atrophin 1, which has 10 CTG repeats, and methylation-sensitive affinity and positioning was observed for CGG repeats in the FMR1 gene affecting fragile X mental retardation (Godde et al. Citation1996). Nucleosome stabilisation by a natural CAG repeat containing sequence has also been reported (Thåström et al. Citation1999). In contrast, methylation-modulated nucleosome exclusion was shown for CCG repeats at fragile X sites themselves (Wang and Griffith Citation1996). Surprisingly, this positioning property of trinucleotide repeats has not been used as an in vitro tool or revisited for some time despite medical links with trinucleotide repeat disorders.
Alpha satellite DNA
Alpha satellite DNA is a 171 bp conserved repeat found at the core centromere of primates (Schueler and Sullivan Citation2006). Distributed copies of the repeat contain a 17 bp site for the sequence-specific, non-essential CENP-B binding protein. The alphoid repeats are assembled into nucleosome-like structures, at least some of which contain the centromere-specific H3 variant CENP-A (Vafa and Sullivan Citation1997). In vitro nucleosome mapping shows multiple nucleosome positions on alpha satellite repeats for both H3 and CENP-A containing nucleosomes, which become more organised in the presence of CENP-B (Yoda et al. Citation1998; Tanaka et al. Citation2005). A large majority of nucleosome crystal structures are based on a palindromic 73 bp region from the human alpha satellite repeat (Harp et al. Citation1996). However, the functionally relevant nucleosome positioning is unclear and complicated by active debate over the repeating chromatin unit size, the wrapping topology, and whether relevant centromeric chromatin is wrapped as either H3 and CENP-A containing nucleosomes or both (Tachiwana et al. Citation2011), or possibly even other histone fold structures of centromeric proteins (Nishino et al. Citation2012).
Engineered statistical positioning
Nucleosome positioning has also been engineered by statistical positioning using DNA sequences as barriers to investigate the effects of chromatin context. For example, the N-terminal coding region of the chloramphenicol acetyl transferase (CAT) reporter gene was found to have a strong nucleosome positioning effect and used to direct nucleosome positioning over adjacent sequences (Wolffe and Drew Citation1989). The strategy has also been used to test the consequences of mutual invasion of nucleosome territories by using overlapping 601 positioning sequences (Engeholm et al. Citation2009). 5S rDNA positioning sequence arrays have been generated with the transcription factor and restriction enzyme sites embedded at the centre to test accessibility on the oligo-nucleosomal scale (Owen-Hughes and Workman Citation1996).
Similarly, nucleosomal DNA can be engineered to incorporate tertiary factor binding sites. For example, Widom and colleagues introduced additional restriction enzyme sites within the 5S (Polach and Widom Citation1995) and 601 derived positioning sequences to observe dynamic site exposure. Workman and colleagues used the converse ability of arbitrary sequence mononucleosomal length DNA to be wrapped by embedding Gal4 and other transcription factor binding sites in a circular permutation vector and studied the effect of different rotational phases (Adams and Workman Citation1995).
Designed synthetic positioning sequences
The ultimate test of nucleosome positioning is prediction. Shrader and Crothers developed a series of sequences for nucleosome positioning based on DNA bendability and compared these using a quantitative competitive reconstitution assay where a small amount of labelled DNA was mixed in with bulk unlabelled competitor fragments (Shrader and Crothers Citation1989). This assay reports relative free energies from the resulting equilibrium. Their original ‘TG’ and related sequences were based on simple alternation of A/T and G/C rich stretches out of phase in 10 bp intervals using concatenated 20 bp repeats, and showed high competitive affinity and strong rotational phasing ability. As expected, the sequences do not exhibit strong translational positioning.
However, a more complex specificity was uncovered when later studies varied the exact sequence and placement of stretches within the overall periodic pattern. This is presumably due to the non-uniform path of DNA in the nucleosome, which favours particular deformations at certain locations (summarised in Widom Citation2001). Recent calculations based on conflation of roll parameters around the superhelix with weighting of the SHL ±2 location were able to predict binding energy with good accuracy (Battistini et al. Citation2012), again underlining the significance of this location to nucleosome properties.
Although the influence of DNA structure remains an area of active debate, the use of designed sequences for understanding nucleosome wrapping has been largely superseded by analyses of data from large-scale sequencing (Kaplan et al. Citation2010; Zhang et al. Citation2010) and high-resolution nucleosome crystal structures (Olson and Zhurkin Citation2011; Trifonov Citation2011 and accompanying critiques). These are leading to careful reconsideration of models for both chromatin organisation and DNA structure, which may hopefully allow renewed opportunities for design and prediction.
Experimental nucleosome techniques in vitro
Much of the exquisite biochemical detail now accumulating about nucleosome properties builds on techniques for assembling and assaying nucleosomes that reach back several decades.
Histone purification
Originally, histones were purified from nuclei extracted from biological tissue, with the most convenient source being chicken blood because, unlike mammals, other vertebrate erythrocytes retain their nuclei in a highly compact form after pyknosis (Ji et al. Citation2011). Histone octamer complexes could therefore be readily isolated from nuclei preparations by hydroxyapatite chromatography. This has been superseded by recombinant production of histones, which can be efficiently expressed as insoluble inclusion bodies in Escherichia coli (Luger, Rechsteiner, et al. 1997), and purified under denaturing conditions by gel filtration or cation exchange chromatography. Recombinant expression in bacteria enables engineering of mutants and avoids post-translational modifications. Individual histones are lyophilised with yields of 20–200 mg l−1 culture.
Although human sequence core histones can be produced ( and nucleosome containing these histones has been crystallised (PDB 2CV5, Tsunaka et al. Citation2005), histones derived from X. laevis sequences ( have been the dominant recombinant proteins used for in vitro nucleosome studies. The most widely used reference crystal structure solved at 1.9 Å resolution contains X. laevis sequences (PDB 1KX5; Davey et al. Citation2002).
Figure 6. Core histone sequences. (a) Human core histone sequences from PDB structure 2CV5 aligned by α1–α2–α3 histone fold domain regions as grey boxes. H3 αN, H2B αC and short H2A αC regions are identified as open boxes. (b) Xenopus laevis core histone sequences from PDB 1KX5 as in (a). N-terminal methionine and initial tripeptide PEP in human H2B are not numbered.

It is also possible to express and purify histones in a soluble form by co-expression of histone fold dimer pairs (Hayes and Lee Citation1997; Black et al. Citation2004).
Histone octamer assembly
Histone octamers are readily assembled by refolding from equimolar histone mixtures, starting from a completely unfolded state in guanidinium chaotrope and dialysing into 2 M NaCl (Luger, Rechsteiner, et al. 1997; Luger et al. Citation1999). (H3–H4)2 tetramer and H2A–H2B dimer complexes can also be assembled at 1 M NaCl. The simple histone alpha helical structure and specific histone fold pairings spontaneously reform, while charge–charge interactions are shielded in the highly ionic buffers. These complexes are purified by gel filtration chromatography to ensure correct stoichiometric ratios on a scale of 0.5–10 mg.
Nucleosomal DNA preparation
DNA fragments of nucleosomal length with defined sequence cannot yet be made economically by direct chemical synthesis, but are conveniently prepared by PCR in quantities up to 1 nmol (Bruno et al. Citation2004). This has the additional advantage that primer oligonucleotides can be pre-labelled on their termini with fluorescent dyes or radionucleotides. PCR products are purified by gel electrophoresis, silica-based kit or anion exchange chromatography. For the even larger scales needed for crystallisation, DNA fragments are cloned into plasmids as direct repeats of 16 or more copies separated by restriction sites (Palmer et al. Citation1995; Dyer et al. Citation2004). Large-scale plasmid purification is followed by release of fragments by restriction enzyme digestion and self-ligation.
Nucleosome assembly or reconstitution
Nucleosome assembly is frequently referred to as ‘reconstitution’ because histone octamers from native sources were originally being reassembled into a nucleosome. Since this is not strictly true for recombinant histones, the term ‘assembly’ is more correct.
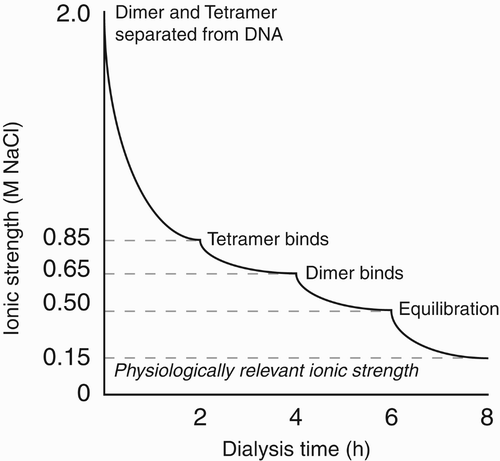
The dominant method of nucleosome assembly is stepwise salt gradient dialysis, which relies on the fact that (H3–H4)2 tetramers and H2A–H2B dimers equilibrate their binding to DNA at 0.85 M and 0.65 M salt, respectively (Oudet et al. Citation1975; Germond et al. Citation1976). Equimolar amounts of histone octamer and DNA are mixed at 1.6–2.0 M NaCl and dialysed sequentially at 0.85, 0.65, 0.5 M and finally at a physiologically relevant salt concentration (; Rhodes and Laskey Citation1989; Luger et al. Citation1999). Although preparative gel electrophoresis or ion exchange chromatography are possible, there is usually no purification step after nucleosome assembly so inaccurate histone/DNA ratios can lead to contaminating complexes. Nucleosomes can be prepared in volumes of 10–1000 μl at 1–10 μM concentration.
Figure 7. Salt gradient dialysis assembly of nucleosomes. Histone octamer comprising (H3–H4)2 tetramer and H2A–H2B dimer are mixed with DNA at high ionic strength (2.0 M NaCl) and then progressively dialysed through specific reducing salt concentrations (0.85 M, 0.65 M and 0.5 M NaCl), leading first to tetramer-DNA then dimer-DNA association for stable nucleosome assembly at physiological buffer conditions.
There are a number of alternatives to stepwise salt dialysis assembly of nucleosomes. An equivalent salt gradient can be achieved by a continuous exponential dialysis using a pump to pass through the same ionic strengths at a similar rate (; Luger et al. Citation1999). Simple stepwise dilution can also be used, although it is important to note that nucleosomes are unstable at dilutions in the 10–100 nM range (Godde and Wolffe Citation1995). Salt–urea dialysis combines histone assembly and DNA association in a simultaneous refolding and assembly, although the disadvantage of this method is that it depends on exact equimolar mixing of all components.
Histone exchange is a true reconstitution where donor nucleosomes are mixed with free target DNA then either high-salt dilution is carried out or a polyanion such as polyglutamate is used as a transfer agent at low ionic strength (Stein et al. Citation1979). This results in mixed populations of histone octamer on both donor and acceptor DNAs. Histone chaperones such as nucleoplasmin (Earnshaw et al. Citation1980) and Nap1 (Ishimi and Kikuchi Citation1991) have also been shown to act as transfer agents under physiologically relevant salt conditions, and Nap1 has been proposed as route to measure nucleosome affinities (Andrews et al. Citation2010).
Methods for in vitro assembly using cell extracts have also been developed, which recapitulate native nucleosome arrangements (Tsukiyama et al. Citation1994; Korber and Hörz Citation2004) presumably by involving native chaperone and remodeller activities. These are limited in scale and introduce potential contaminants in downstream assays.
Mapping nucleosome positions
Nucleosome mapping uses biophysical properties, footprinting or direct observation to determine the location of nucleosome positioning.
Native gel mobility of nucleosomes
One of the most popular methods for observing nucleosome positions is by native (non-denaturing) electrophoresis in low-density native polyacrylamide (PAGE) or mixed agarose–polyacrylamide gels. Under gentle thermal conditions, in low-salt buffers, it is possible to resolve small conformational differences using native PAGE. Bradbury and colleagues showed that different nucleosome positions on an extended DNA fragment can be resolved (Pennings et al. Citation1991), and used this in the first direct demonstrations of nucleosome sliding (Pennings et al. Citation1991; Meersseman et al. Citation1992). In general, nucleosomes with more central positions on a specific DNA fragment will migrate more slowly than those at terminal positions because centrally positioned nucleosomes have DNA projections from both sides of the histone octamer wrapped region and occupy a larger effective hydrodynamic radius.
However, some caution is needed when interpreting nucleosome positioning in native gels. Firstly, flexibility of entry/exit DNA angles in the nucleosome is large and variable (Furrer et al. Citation1995; Li and Widom Citation2004) and therefore even nucleosomes at equivalent locations on opposite ends of mono-nucleosomal DNA fragments can migrate differently (Flaus et al. Citation1996). Sub-nucleosomal structures such as hexasomes lacking one H2A–H2B dimer or tetrasomes lacking both H2A–H2B dimers run in a similar region of the gel to positioned nucleosomes (Bruno et al. Citation2003). Finally, intercalating DNA stains such as ethidium bromide bind more readily to free than wrapped DNA, so will be sensitive to the relative length of exposed DNA and confound interpretations of occupancy.
Footprinting by restriction enzymes
A second popular method of nucleosome mapping is to compare the DNA accessibility for restriction enzymes, which is reduced by wrapping in a nucleosome (Pfeiffer et al. Citation1975). As demonstrated by Widom and colleagues, there is a gradient of accessibility for DNA sites that is maximal at the entry/exit region and minimal near the dyad axis due to dynamic unpeeling of DNA from the outer edge of the structure (Polach and Widom Citation1995; Li and Widom Citation2004). This means that almost all sites will be accessible to some degree. Single molecule measurements suggest this access has an initial unpeeling event frequency of 4 s−1 with rapid rebinding of DNA in 10–50 ms (Li et al. Citation2005).
Footprinting by non-specific nucleases
The relative protection of wrapped DNA also enables the use of non-specific nucleases to report positioning as a footprint (Hampshire et al. Citation2007). An early in vitro footprinting strategy used methidium propyl EDTA (MPE), which fused an intercalating dye to an iron ion chelating group (Hertzberg and Dervan Citation1982). Because intercalation is strongly favoured in undistorted linker DNA and the localised Fe2+ ions could generate hydroxy radicals by the Fenton reaction, MPE acts as a nuclease with specificity for free DNA. However, binding preference is not absolute and the diffusible hydroxy radicals limited resolution.
Another footprinting strategy uses E. coli exonuclease III (ExoIII), a single-stranded 3′ to 5′ exonuclease that requires a free 3′ terminal on which to initiate ( and ; Rogers and Weiss Citation1980). This can map nucleosomes at high resolution on DNA fragments (Riley and Weintraub Citation1978), either with detection of single-stranded protected regions or as double-stranded regions after subsequent S1 nuclease treatment to remove the exposed 5′ single-stranded regions left by ExoIII (Archer and Ricci Citation1999). ExoIII has some sequence bias and an inherent processivity (Linxweiler and Hörz Citation1982), meaning that over digestion allows it to progressively invade the labile outer turns of DNA on the nucleosome.
Figure 8. Nucleosome mapping by non-specific nucleases. Schematic showing respectively exonuclease III (ExoIII) digestion of linker DNA from 3’ hydroxyl termini, micrococcal nuclease (MNase) endonucleolytic activity in linker DNA, Fe2+/EDTA generated small hydroxy radicals cleaving regularly at exposed strands within nucleosome and throughout linkers, histone tethered Fe2+/EDTA by hydroxy radicals at specific symmetric points within the nucleosome, and larger deoxynuclease I (DNaseI) at exposed minor grooves within the nucleosome and throughout linkers.
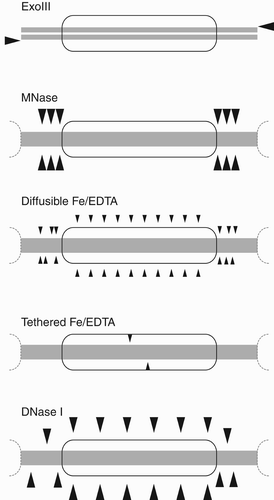
Table 4. Properties of important footprinting nucleases.
The most commonly used chromatin footprinting nuclease is Staphylococcus aureus micrococcal nuclease (MNase), which is an endonuclease that recognises and cuts single DNA strands either alone or within a duplex ( and ). MNase cleaves with strong bias for the linker DNA over nucleosomes (Lutter Citation1978; Cockell et al. Citation1983), although there is a small fraction of nicking within the nucleosome core. MNase also shows a very strong bias for cutting 5′ of A and T nucleotides (Sulkowski and Laskowski Citation1962), so it is likely that initial single strand cuts will occur at A/T nucleotides in linkers. Nevertheless, prolonged digestion generates exonucleolytic-like double strand cutting up to the protected edge of the nucleosome (Cockell et al. Citation1983). Controls are required to ensure that the MNase sequence bias does not confound results by failing to reach completion in the linkers (Dingwall et al. Citation1981) or by generation of biased single stranded cuts at A/T bases in the core (McGhee and Felsenfeld Citation1983; Allan et al. Citation2012).
The use of MNase to generate mononucleosome length DNA fragments that can be identified by high throughput sequencing and mapped back onto genome scaffolds underlies the current in vivo nucleosome positioning revolution, but can also be used for in vitro mapping (Fraser et al. Citation2009; Allan et al. Citation2012).
Direct histone octamer localisation by site-directed mapping
An early example of direct determination of nucleosome positioning through histone–DNA interactions was the site-specific crosslinking of Mirzabekov and colleagues (Mirzabekov et al. Citation1978). However, this is a difficult technique to perform successfully. An alternative strategy is to tether a small chemical nuclease to the histone octamer and detect the sites of DNA strand cleavage (). This has been achieved using a thiol-reactive EDTA derivative attached to engineered cysteine residues at locations in histone sequences close to DNA (Flaus et al. Citation1996). Loading of Fe2+ ions and induction of the Fenton reaction generates hydroxy radicals that are able to diffuse up to 10 Å (Ebright et al. Citation1992) and can report the nucleosome position at base pair resolution.
Internal nucleosome structure by non-specific nucleases
Non-specific endonucleases can also be used to reveal details of the DNA wrapping within the nucleosome. Because the outward facing sections of the DNA backbone are much more accessible for cleavage than those associated with the histone octamer surface, the periodic pattern of cleavages reports the exact helical pitch of DNA as well as irregular features.
Freely diffusing Fe2+ ions chelated by EDTA have been used with the Fenton reaction to generate hydroxy radicals for internal footprinting (Hayes et al. Citation1990). The hydroxy radicals are small and typically provide a fine footprint of DNA exposure in solution ( and ). Chelation is required to avoid bias that would be caused if Fe2+ ions were localised to DNA phosphates as counter ions, but radical-induced DNA backbone cleavage rates can still be affected to some extent by local stereochemistry.
Bovine pancreatic Deoxynuclease I (DNaseI) is a non-sequence specific endonuclease that binds in the minor groove of double stranded DNA to cut accessible strands, including within the nucleosome ( and ; Lutter Citation1979). It has strong structural specificity for the geometry of the minor groove, and gives staggered cuts on each strand by a ‘double hit’ mechanism (Suck and Oefner Citation1986). Because it is a large enzyme of 30 kDa, and may be glycosylated, DNaseI only cleaves the most accessible DNA regions. It is also popular for in vivo studies of genome accessibility, where DNaseI hypersensitivity correlates with open regions of transcription factor access in chromatin (Cockerill Citation2011).
These internal footprinting agents reflect the DNA helix parameters in what is known as the ‘local frame’ of reference on the nucleosome. If wrapped DNA is removed from its superhelical path and made linear, then the same footprinted points will have a different apparent twist known as the ‘laboratory frame’. The typical change of the period from 10.17 to 10.5 bp/turn sums to a difference of half a turn of helicity, and gave rise to a confusion known as the ‘linking number paradox’ because early analyses did not take the change of frame into account (Travers and Klug Citation1987). The ‘paradox’ has been resolved but the term sometimes appears in discussions of the local helical winding parameter.
Single molecule imaging
Despite the origins of nucleosomes as ‘beads on a string’, observation using direct imaging techniques is sensitive to artefacts caused by aggressive sample treatments, disruptive interactions with solid supports and unfolding at high dilutions. Awareness of these features has led to direct methods for measuring nucleosome positioning and dynamics, which are of increasing value.
Atomic force microscopy (AFM) uses physical tapping of a probe at high spatial resolution over a surface and is capable of nanometer spatial resolution or up to 100 ms time resolution (Lyubchenko et al. Citation2011). Nucleosomes are measured by depositing them on a derivative mica surface under a thin layer of buffer (Lyubchenko Citation2011), so AFM has been used to measure the contour length of linker DNA to infer positioning, and the geometry of entry/exit DNA and proximity of histone complexes to understand inter-nucleosomal packing (Engeholm et al. Citation2009; Filenko et al. Citation2012). It has also recently been used to observe nucleosome dynamics (Miyagi et al. Citation2011) and chromatin remodelling (van Vugt et al. Citation2009; Shukla et al. Citation2010; Montel et al. Citation2011).
Electron microscopy at cryogenic temperatures (CryoEM) below −150°C has also been used for direct imaging nucleosomes (Bednar and Woodcock Citation1999; Leschziner Citation2011), such as the broad spread of entry/exit DNA splayed at angles of 30°C relative to the dyad axis (Furrer et al. Citation1995).
Single-pair FRET (spFRET) methods use through-space energy transfer between a specific pair of donor and acceptor dyes to measure spatial separation when tethered to locations on the nucleosome (Buning and van Noort Citation2010). This FRET response is highly sensitive over a window around the characteristic Förster radius of the dye pair, which is typically in the range 10–100 Å. spFRET can be performed in solution (Tóth et al. Citation2001), or particles can be spread on an imaging surface and the relative signal of the FRET dyes observed for each single molecule spot simultaneously for thousands of spots with high time resolution (Koopmans et al. Citation2007). spFRET revealed rates of site exposure of DNA from the edge of the nucleosome (Li et al. Citation2005) and quantitation of nucleosome sliding (Racki et al. Citation2009).
Conclusion
As the universal packaging system of eukaryotic genomes, chromatin is composed of fundamental nucleosome units organised by highly conserved histone proteins. Understanding their behaviour is key to explaining the eukaryotic genome and its transactions. Nucleosome positioning is the stable location of DNA relative to the histone octamer and is an important feature of chromatin behaviour.
Biochemical investigations into nucleosome positioning and dynamics have led to three general principles: (1) DNA sequence accessibility depends on its position within the nucleosome structure, (2) DNA sequences can influence their nucleosomal location and (3) the structural properties of DNA and of the histone octamer control the potential for the nucleosome to undergo structural changes. These depend on the feature-rich spiral of histone fold dimers that present a path for combinatorial histone–DNA interactions in the nucleosome.
Nucleosome positioning is an indirect readout of DNA sequence properties onto this histone surface through the DNA backbone conformation. Since the 147 bp of bound DNA includes a considerable number of interactions, cooperative effects can be built up in phase and a number of DNA sequences that form very stable interactions have been identified for use in in vitro studies of nucleosome positioning. These include the artificially selected Widom 601 DNA, the 5S rRNA gene from various organisms and the MMTV 3′ LTR isolates.
A battery of techniques have been perfected over several decades to enable the preparation of histones and DNA, their assembly into nucleosomes, and the footprinting and mapping of nucleosome positions. Being able to assemble homogenous defined nucleosome substrates is necessary for biochemical assays of nucleosome dynamics and chromatin active enzymes. Although the stable nucleosomes reflect high histone–DNA affinity in vitro and contribute to in vivo properties, DNA sequence effects do not appear to dominate the complex forces that direct nucleosome positioning in the genome. Nevertheless, the principles and practical aspects of experimental investigations using positioned nucleosomes in vitro provide important tools and insights into the structure and function of chromatin at the molecular level.
Acknowledgements
The author is funded by a Science Foundation Ireland Principal Investigator award and Irish Health Research Board research project grants.
References
- Adams , C C and Workman , J L . 1995 . Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative . Mol Cell Biol , 15 ( 3 ) : 1405 – 1421 .
- Allan , J , Fraser , R M , Owen-Hughes , T A and Keszenman-Pereyra , D . 2012 . Micrococcal nuclease does not substantially bias nucleosome mapping . J Mol Biol , 417 ( 3 ) : 152 – 164 .
- Anderson , J D and Widom , J . 2000 . Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites . J Mol Biol , 296 ( 4 ) : 979 – 987 .
- Anderson , J D and Widom , J . 2001 . Poly(dA-dT) promoter elements increase the equilibrium accessibility of nucleosomal DNA target sites . Mol Cell Biol , 21 ( 11 ) : 3830 – 3839 .
- Anderson , J D , Thåström , A and Widom , J . 2002 . Spontaneous access of proteins to buried nucleosomal DNA target sites occurs via a mechanism that is distinct from nucleosome translocation . Mol Cell Biol , 22 ( 20 ) : 7147 – 7157 .
- Andrews , A J , Chen , X , Zevin , A , Stargell , L A and Luger , K . 2010 . The histone chaperone Nap1 promotes nucleosome assembly by eliminating nonnucleosomal histone DNA interactions . Mol Cell , 37 ( 6 ) : 834 – 842 .
- Archer , T K , Cordingley , M G , Wolford , R G and Hager , G L . 1991 . Transcription factor access is mediated by accurately positioned nucleosomes on the mouse mammary tumor virus promoter . Mol Cell Biol , 11 ( 2 ) : 688 – 698 .
- Archer , T K and Ricci , A R . 1999 . Exonuclease III as a probe of chromatin structure in vivo . Methods Enzymol , 304 : 584 – 599 .
- Battistini , F , Hunter , C A , Moore , I K and Widom , J . 2012 . Structure-based identification of new high affinity nucleosome binding sequences . J Mol Biol , 420 ( 1–2 ) : 8 – 16 .
- Beato , M and Vicent , G P . 2011 . Impact of chromatin structure and dynamics on PR signaling . The initial steps in hormonal gene regulation. Mol Cell Endocrinol , 357 ( 1–2 ) : 37 – 42 .
- Bednar , J and Woodcock , C L . 1999 . Cryoelectron microscopic analysis of nucleosomes and chromatin . Methods Enzymol , 304 : 191 – 213 .
- Black , B E , Foltz , D R , Chakravarthy , S , Luger , K , Woods , V L and Cleveland , D W . 2004 . Structural determinants for generating centromeric chromatin . Nature , 430 ( 6999 ) : 578 – 582 .
- Blacketer , M J , Feely , S J and Shogren-Knaak , M A . 2010 . Nucleosome interactions and stability in an ordered nucleosome array model system . J Biol Chem , 285 ( 45 ) : 34597 – 34607 .
- Brandt-Carlson , C , Butel , J S and Wheeler , D . 1993 . Phylogenetic and structural analyses of MMTV LTR ORF sequences of exogenous and endogenous origins . Virology , 193 ( 1 ) : 171 – 185 .
- Bruno , M , Flaus , A and Owen-Hughes , T A . 2004 . Site-specific attachment of reporter compounds to recombinant histones . Methods Enzymol , 375 : 211 – 228 .
- Bruno , M , Flaus , A , Stockdale , C , Rencurel , C , Ferreira , H and Owen-Hughes , T A . 2003 . Histone H2A/H2B dimer exchange by ATP-dependent chromatin remodeling activities . Mol Cell , 12 ( 6 ) : 1599 – 1606 .
- Buning , R and van Noort , J . 2010 . Single-pair FRET experiments on nucleosome conformational dynamics . Biochimie , 92 ( 12 ) : 1729 – 1740 .
- Callahan , R and Smith , G H . 2000 . MMTV-induced mammary tumorigenesis: gene discovery, progression to malignancy and cellular pathways . Oncogene , 19 ( 8 ) : 992 – 1001 .
- Chua , EY D , Vasudevan , D , Davey , G E , Wu , B and Davey , C A . 2012 . The mechanics behind DNA sequence-dependent properties of the nucleosome . Nucleic Acids Res , 40 ( 13 ) : 6338 – 6352 .
- Cockell , M , Rhodes , D and Klug , A . 1983 . Location of the primary sites of micrococcal nuclease cleavage on the nucleosome core . J Mol Biol , 170 ( 2 ) : 423 – 446 .
- Cockerill , P N . 2011 . Structure and function of active chromatin and DNase I hypersensitive sites . FEBS J , 278 ( 13 ) : 2182 – 2210 .
- D'Anna , J A and Isenberg , I . 1974 . A histone cross-complexing pattern . Biochemistry , 13 ( 24 ) : 4992 – 4997 .
- Davey , C A , Sargent , D F , Luger , K , Maeder , A W and Richmond , T J . 2002 . Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution . J Mol Biol , 319 ( 5 ) : 1097 – 1113 .
- Dechassa , M L and Luger , K . 2012 . “ Nucleosomes as control elements for accessing the genome ” . In Genome organization and function in the cell nucleus , Edited by: Rippe , K . 55 – 87 . Weinheim (Germany) : Wiley-VCH .
- Dingwall , C , Lomonossoff , G P and Laskey , R A . 1981 . High sequence specificity of micrococcal nuclease . Nucleic Acids Res , 9 ( 12 ) : 2659 – 2673 .
- Donehower , L A , Huang , A L and Hager , G L . 1981 . Regulatory and coding potential of the mouse mammary tumor virus long terminal redundancy . J Virol , 37 ( 1 ) : 226 – 238 .
- Dong , F and Van Holde , K E . 1991 . Nucleosome positioning is determined by the (H3-H4)2 tetramer . Proc Natl Acad Sci USA , 88 ( 23 ) : 10596 – 10600 .
- Drew , H R . 1991 . Can one measure the free energy of binding of the histone octamer to different DNA sequences by salt-dependent reconstitution? . J Mol Biol , 219 ( 3 ) : 391 – 392 .
- Drew , H R and Travers , A A . 1985 . DNA bending and its relation to nucleosome positioning . J Mol Biol , 186 ( 4 ) : 773 – 790 .
- Dyer , P N , Edayathumangalam , R S , White , C L , Bao , Y , Chakravarthy , S , Muthurajan , U M and Luger , K . 2004 . Reconstitution of nucleosome core particles from recombinant histones and DNA . Methods Enzymol , 375 : 23 – 44 .
- Earnshaw , W C , Honda , B M , Laskey , R A and Thomas , J O . 1980 . Assembly of nucleosomes: the reaction involving X. laevis . nucleoplasmin. Cell , 21 ( 2 ) : 373 – 383 .
- Ebright , Y W , Chen , Y , Pendergras , P S and Ebright , R H . 1992 . Incorporation of an EDTA-metal complex at a rationally selected site within a protein: application to EDTA-iron DNA affinity cleaving with catabolite gene activator protein (CAP) and Cro . Biochemistry , 31 ( 44 ) : 10664 – 10670 .
- Edayathumangalam , R S , Weyermann , P , Dervan , P B , Gottesfeld , J M and Luger , K . 2005 . Nucleosomes in solution exist as a mixture of twist-defect states . J Mol Biol , 345 ( 1 ) : 103 – 114 .
- Edayathumangalam , R S , Weyermann , P , Gottesfeld , J M , Dervan , P B and Luger , K . 2004 . Molecular recognition of the nucleosomal “supergroove” . Proc Natl Acad Sci USA , 101 ( 18 ) : 6864 – 6869 .
- Eickbush , T H and Moudrianakis , E N . 1978 . The histone core complex: an octamer assembled by two sets of protein-protein interactions . Biochemistry , 17 ( 23 ) : 4955 – 4964 .
- Engeholm , M , de Jager , M , Flaus , A , Brenk , R , van Noort , J and Owen-Hughes , T A . 2009 . Nucleosomes can invade DNA territories occupied by their neighbors . Nature Struct Mol Biol , 16 ( 2 ) : 151 – 158 .
- Felle , M , Exler , J H , Merkl , R , Dachauer , K , Brehm , A , Grummt , I and Längst , G . 2010 . DNA sequence encoded repression of rRNA gene transcription in chromatin . Nucleic Acids Res , 38 ( 16 ) : 5304 – 5314 .
- Fernandez , A G and Anderson , J N . 2007 . Nucleosome positioning determinants . J Mol Biol , 371 ( 3 ) : 649 – 668 .
- Ferreira , H , Somers , J , Webster , R , Flaus , A and Owen-Hughes , T A . 2007 . Histone tails and the H3 alphaN helix regulate nucleosome mobility and stability . Mol Cell Biol , 27 ( 11 ) : 4037 – 4048 .
- Filenko , N A , Palets , D B and Lyubchenko , Y L . 2012 . Structure and dynamics of dinucleosomes assessed by atomic force microscopy . J Amino Acids , : 650840
- Fitzgerald , D J and Anderson , J N . 1999 . DNA distortion as a factor in nucleosome positioning . J Mol Biol , 293 ( 3 ) : 477 – 491 .
- Flaus , A and Richmond , T J . 1998 . Positioning and stability of nucleosomes on MMTV 3’LTR sequences . J Mol Biol , 275 ( 3 ) : 427 – 441 .
- Flaus , A , Luger , K , Tan , S and Richmond , T J . 1996 . Mapping nucleosome position at single base-pair resolution by using site-directed hydroxyl radicals . Proc Natl Acad Sci USA , 93 ( 4 ) : 1370 – 1375 .
- Fragoso , G , John , S , Roberts , M S and Hager , G L . 1995 . Nucleosome positioning on the MMTV LTR results from the frequency-biased occupancy of multiple frames. Genes Dev , 9 ( 15 ) : 1933 – 1947 .
- Fraser , R , Keszenman-Pereyra , D , Simmen , M and Allan , J . 2009 . High-resolution mapping of sequence-directed nucleosome positioning on genomic DNA . J Mol Biol , 390 ( 2 ) : 292 – 305 .
- Furrer , P , Bednar , J , Dubochet , J , Hamiche , A and Prunell , A . 1995 . DNA at the entry-exit of the nucleosome observed by cryoelectron microscopy . J Struct Biol , 114 ( 3 ) : 177 – 183 .
- Germond , J E , Bellard , M , Oudet , P and Chambon , P . 1976 . Stability of nucleosomes in native and reconstituted chromatins . Nucleic Acids Res , 3 ( 11 ) : 3173 – 3192 .
- Gkikopoulos , T , Schofield , P , Singh , V , Pinskaya , M , Mellor , J , Smolle , M , Workman , J L , Barton , G J and Owen-Hughes , T A . 2011 . A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization . Science , 333 ( 6050 ) : 1758 – 1760 .
- Godde , J S and Wolffe , A P . 1995 . Disruption of reconstituted nucleosomes . The effect of particle concentration, MgCl2 and KCl concentration, the histone tails, and temperature. J Biol Chem , 270 ( 46 ) : 27399 – 27402 .
- Godde , J S and Wolffe , A P . 1996 . Nucleosome assembly on CTG triplet repeats . J Biol Chem , 271 ( 25 ) : 15222 – 15229 .
- Godde , J S , Kass , S U , Hirst , M C and Wolffe , A P . 1996 . Nucleosome assembly on methylated CGG triplet repeats in the fragile X mental retardation gene 1 promoter . J Biol Chem , 271 ( 40 ) : 24325 – 24328 .
- Grigoryev , S A and Woodcock , C L . 2012 . Chromatin organization – The 30nm fiber . Exp Cell Res , (318) : 1448 – 1455 .
- Grøntved , L and Hager , G L . 2011 . Impact of chromatin structure on PR signaling: Transition from local to global analysis . Mol Cell Endocrinol , 357 ( 1/2 ) : 30 – 36 .
- Hall , M A , Shundrovsky , A , Bai , L , Fulbright , R M , Lis , J T and Wang , M D . 2009 . High-resolution dynamic mapping of histone-DNA interactions in a nucleosome . Nature Struct Mol Biol , 16 ( 2 ) : 124 – 129 .
- Hamiche , A , Sandaltzopoulos , R , Gdula , D A and Wu , C . 1999 . ATP-dependent histone octamer sliding mediated by the chromatin remodeling complex NURF . Cell , 97 ( 7 ) : 833 – 842 .
- Hampshire , A J , Rusling , D A , Broughton-Head , V J and Fox , K R . 2007 . Footprinting: a method for determining the sequence selectivity, affinity and kinetics of DNA-binding ligands . Methods , 42 ( 2 ) : 128 – 140 .
- Harp , J M , Uberbacher , E C , Roberson , A E , Palmer , E L , Gewiess , A and Bunick , G J . 1996 . X-ray diffraction analysis of crystals containing twofold symmetric nucleosome core particles . Acta Crystallogr D Biol Crystallogr , 52 ( Pt 2 ) : 283 – 288 .
- Hartlepp , K F , Fernández-Tornero , C , Eberharter , A , Grüne , T , Müller , C W and Becker , P B . 2005 . The histone fold subunits of Drosophila CHRAC facilitate nucleosome sliding through dynamic DNA interactions . Mol Cell Biol , 25 ( 22 ) : 9886 – 9896 .
- Hayes , J J , Clark , D J and Wolffe , A P . 1991 . Histone contributions to the structure of DNA in the nucleosome . Proc Natl Acad Sci USA , 88 ( 15 ) : 6829 – 6833 .
- Hayes , J J and Lee , K M . 1997 . In vitro reconstitution and analysis of mononucleosomes containing defined DNAs and proteins . Methods , 12 ( 1 ) : 2 – 9 .
- Hayes , J J , Tullius , T D and Wolffe , A P . 1990 . The structure of DNA in a nucleosome . Proc Natl Acad Sci USA , 87 ( 19 ) : 7405 – 7409 .
- Hertzberg , R P and Dervan , P B . 1982 . Cleavage of double helical DNA by methidium-propyl-EDTA-iron(II) . J Am Chem Soc , 104 ( 1 ) : 313 – 315 .
- Hughes , A and Rando , O J . 2009 . Chromatin “programming” by sequence–is there more to the nucleosome code than %GC? . J Biol , 8 ( 11 ) : 96
- Ishimi , Y and Kikuchi , A . 1991 . Identification and molecular cloning of yeast homolog of nucleosome assembly protein I which facilitates nucleosome assembly in vitro . J Biol Chem , 266 ( 11 ) : 7025 – 7029 .
- Iyer , V R . 2012 . Nucleosome positioning: bringing order to the eukaryotic genome . Trends Cell Biol , 22 ( 5 ) : 250 – 256 .
- Ji , P , Murata-Hori , M and Lodish , H F . 2011 . Formation of mammalian erythrocytes: chromatin condensation and enucleation . Trends Cell Biol , 21 ( 7 ) : 409 – 415 .
- Jorcano , J L and Ruiz-Carrillo , A . 1979 . H3.H4 tetramer directs DNA and core histone octamer assembly in the nucleosome core particle . Biochemistry , 18 ( 5 ) : 768 – 774 .
- Kamada , K , Shu , F , Chen , H , Malik , S , Stelzer , G , Roeder , R G , Meisterernst , M and Burley , S K . 2001 . Crystal structure of negative cofactor 2 recognizing the TBP-DNA transcription complex . Cell , 106 ( 1 ) : 71 – 81 .
- Kaplan , N , Moore , I , Fondufe-Mittendorf , Y , Gossett , A J , Tillo , D , Field , Y , Hughes , T R , Lieb , J D , Widom , J and Segal , E . 2010 . Nucleosome sequence preferences influence in vivo nucleosome organization . Nature Struct Mol Biol , 17 ( 8 ) : 918 – 920; . author reply 920–922.
- Klug , A , Rhodes , D , Smith , J , Finch , J T and Thomas , J O . 1980 . A low resolution structure for the histone core of the nucleosome . Nature , 287 ( 5782 ) : 509 – 516 .
- Koopmans , WJ A , Brehm , A , Logie , C , Schmidt , T and van Noort , J . 2007 . Single-pair FRET microscopy reveals mononucleosome dynamics . J Fluoresc , 17 ( 6 ) : 785 – 795 .
- Korber , P and Hörz , W . 2004 . In vitro assembly of the characteristic chromatin organization at the yeast PHO5 promoter by a replication-independent extract system . J Biol Chem , 279 ( 33 ) : 35113 – 35120 .
- Kornberg , R D . 1974 . Chromatin structure: a repeating unit of histones and DNA . Science , 184 ( 4139 ) : 868 – 871 .
- Kornberg , R D and Lorch , Y . 2007 . Chromatin rules . Nature Struct Mol Biol , 14 ( 11 ) : 986 – 988 .
- Kornberg , R D and Stryer , L . 1988 . Statistical distributions of nucleosomes: nonrandom locations by a stochastic mechanism . Nucleic Acids Res , 16 ( 14A ) : 6677 – 6690 .
- Längst , G , Becker , P B and Grummt , I . 1998 . TTF-I determines the chromatin architecture of the active rDNA promoter . EMBO J , 17 ( 11 ) : 3135 – 3145 .
- Leschziner , A E . 2011 . Electron microscopy studies of nucleosome remodelers . Curr Opin Struct Biol , 21 ( 6 ) : 709 – 718 .
- Li , G , Levitus , M , Bustamante , C and Widom , J . 2005 . Rapid spontaneous accessibility of nucleosomal DNA . Nature Struct Mol Biol , 12 ( 1 ) : 46 – 53 .
- Li , G and Widom , J . 2004 . Nucleosomes facilitate their own invasion . Nature Struct Mol Biol , 11 ( 8 ) : 763 – 769 .
- Li , J , Längst , G and Grummt , I . 2006 . NoRC-dependent nucleosome positioning silences rRNA genes . EMBO J , 25 ( 24 ) : 5735 – 5741 .
- Linxweiler , W and Hörz , W . 1982 . Sequence specificity of exonuclease III from E. coli . Nucleic Acids Res , 10 ( 16 ) : 4845 – 4859 .
- Lohr , D , Tatchell , K and Van Holde , K E . 1977 . On the occurrence of nucleosome phasing in chromatin . Cell , 12 ( 3 ) : 829 – 836 .
- Lowary , P T and Widom , J . 1998 . New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning . J Mol Biol , 276 ( 1 ) : 19 – 42 .
- Luger , K and Richmond , T J . 1998a . DNA binding within the nucleosome core . Curr Opin Struct Biol , 8 ( 1 ) : 33 – 40 .
- Luger , K and Richmond , T J . 1998b . The histone tails of the nucleosome . Curr Opin Genet Dev , 8 ( 2 ) : 140 – 146 .
- Luger , K , Mäder , A W , Richmond , R K , Sargent , D F and Richmond , T J . 1997 . Crystal structure of the nucleosome core particle at 2.8 A resolution . Nature , 389 ( 6648 ) : 251 – 260 .
- Luger , K , Rechsteiner , T J , Flaus , A , Waye , M M and Richmond , T J . 1997 . Characterization of nucleosome core particles containing histone proteins made in bacteria . J Mol Biol , 272 ( 3 ) : 301 – 311 .
- Luger , K , Rechsteiner , T J and Richmond , T J . 1999 . Preparation of nucleosome core particle from recombinant histones . Methods Enzymol , 304 : 3 – 19 .
- Lutter , L C . 1978 . Kinetic analysis of deoxyribonuclease I cleavages in the nucleosome core: evidence for a DNA superhelix . J Mol Biol , 124 ( 2 ) : 391 – 420 .
- Lutter , L C . 1979 . Precise location of DNase I cutting sites in the nucleosome core determined by high resolution gel electrophoresis . Nucleic Acids Res , 6 ( 1 ) : 41 – 56 .
- Lyubchenko , Y L . 2011 . Preparation of DNA and nucleoprotein samples for AFM imaging . Micron , 42 ( 2 ) : 196 – 206 .
- Lyubchenko , Y L , Shlyakhtenko , L S and Ando , T . 2011 . Imaging of nucleic acids with atomic force microscopy . Methods , 54 ( 2 ) : 274 – 283 .
- Makde , R D , England , J R , Yennawar , H P and Tan , S . 2010 . Structure of RCC1 chromatin factor bound to the nucleosome core particle . Nature , 467 ( 7315 ) : 562 – 566 .
- Mariño-Ramírez , L , Kann , M G , Shoemaker , B A and Landsman , D . 2005 . Histone structure and nucleosome stability . Expert Rev Proteomic , 2 ( 5 ) : 719 – 729 .
- Mavrich , T N , Ioshikhes , I P , Venters , B J , Jiang , C , Tomsho , L P , Qi , J , Schuster , S C , Albert , I and Pugh , B F . 2008 . A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome . Genome Res , 18 ( 7 ) : 1073 – 1083 .
- McGhee , J D and Felsenfeld , G . 1983 . Another potential artifact in the study of nucleosome phasing by chromatin digestion with micrococcal nuclease . Cell , 32 ( 4 ) : 1205 – 1215 .
- Meersseman , G , Pennings , S and Bradbury , E M . 1992 . Mobile nucleosomes – a general behavior . EMBO J , 11 ( 8 ) : 2951 – 2959 .
- Mills , J B , Cooper , J P and Hagerman , P J . 1994 . Electrophoretic evidence that single-stranded regions of one or more nucleotides dramatically increase the flexibility of DNA . Biochemistry , 33 ( 7 ) : 1797 – 1803 .
- Mirzabekov , A D , Shick , V V , Belyavsky , A V and Bavykin , S G . 1978 . Primary organization of nucleosome core particle of chromatin: sequence of histone arrangement along DNA . Proc Natl Acad Sci USA , 75 ( 9 ) : 4184 – 4188 .
- Miyagi , A , Ando , T and Lyubchenko , Y L . 2011 . Dynamics of nucleosomes assessed with time-lapse high-speed atomic force microscopy . Biochemistry , 50 ( 37 ) : 7901 – 7908 .
- Montel , F , Castelnovo , M , Menoni , H , Angelov , D , Dimitrov , S and Faivre-Moskalenko , C . 2011 . RSC remodeling of oligo-nucleosomes: an atomic force microscopy study . Nucleic Acids Res , 39 ( 7 ) : 2571 – 2579 .
- Negri , R , Buttinelli , M , Panetta , G , De Arcangelis , V , Di Mauro , E and Travers , A A . 2001 . Sequence dependence of translational positioning of core nucleosomes . J Mol Biol , 307 ( 4 ) : 987 – 999 .
- Nelson , H C , Finch , J T , Luisi , B F and Klug , A . 1987 . The structure of an oligo(dA).oligo(dT) tract and its biological implications . Nature , 330 ( 6145 ) : 221 – 226 .
- Nishino , T , Takeuchi , K , Gascoigne , K E , Suzuki , A , Hori , T , Oyama , T , Morikawa , K , Cheeseman , I M and Fukagawa , T . 2012 . CENP-T-W-S-X forms a unique centromeric chromatin structure with a histone-like fold . Cell , 148 ( 3 ) : 487 – 501 .
- Olins , AL L and Olins , D E . 1974 . Spheroid chromatin units (v bodies) . Science , 183 ( 4122 ) : 330 – 332 .
- Olson , W K and Zhurkin , V B . 2011 . Working the kinks out of nucleosomal DNA . Curr Opin Struct Biol , 21 ( 3 ) : 348 – 357 .
- Oudet , P , Gross-Bellard , M and Chambon , P . 1975 . Electron microscopic and biochemical evidence that chromatin structure is a repeating unit . Cell , 4 ( 4 ) : 281 – 300 .
- Owen-Hughes , T A and Workman , J L . 1996 . Remodeling the chromatin structure of a nucleosome array by transcription factor-targeted trans-displacement of histones . EMBO J , 15 ( 17 ) : 4702 – 4712 .
- Palmer , E L , Gewiess , A , Harp , J M , York , M H and Bunick , G J . 1995 . Large-scale production of palindrome DNA fragments . Anal Biochem , 231 ( 1 ) : 109 – 114 .
- Pennings , S , Meersseman , G and Bradbury , E M . 1991 . Mobility of positioned nucleosomes on 5 S rDNA . J Mol Biol , 220 ( 1 ) : 101 – 110 .
- Perales , R , Zhang , L and Bentley , D . 2011 . Histone occupancy in vivo at the 601 nucleosome binding element is determined by transcriptional history . Mol Cell Biol , 31 ( 16 ) : 3485 – 3496 .
- Perry , M , Thomsen , G H and Roeder , R G . 1985 . Genomic organization and nucleotide sequence of two distinct histone gene clusters from Xenopus laevis . Identification of novel conserved upstream sequence elements. J Mol Biol , 185 ( 3 ) : 479 – 499 .
- Peters , J P and Maher , L J . 2010 . DNA curvature and flexibility in vitro and in vivo . Q Rev Biophys , 43 ( 1 ) : 23 – 63 .
- Pfeiffer , W , Hörz , W , Igo-Kemenes , T and Zachau , H G . 1975 . Restriction nucleases as probes of chromatin structure . Nature , 258 ( 5534 ) : 450 – 452 .
- Piña , B , Barettino , D , Truss , M and Beato , M . 1990 . Structural features of a regulatory nucleosome . J Mol Biol , 216 ( 4 ) : 975 – 990 .
- Polach , K J and Widom , J . 1995 . Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation . J Mol Biol , 254 ( 2 ) : 130 – 149 .
- Portugal , J and Waring , M J . 1986 . Antibiotics which can alter the rotational orientation of nucleosome core DNA . Nucleic Acids Res , 14 ( 22 ) : 8735 – 8754 .
- Portugal , J and Waring , M J . 1987 . Analysis of the effects of antibiotics on the structure of nucleosome core particles determined by DNAase I cleavage . Biochimie , 69 ( 8 ) : 825 – 840 .
- Puhl , H L and Behe , M J . 1995 . Poly(dA).poly(dT) forms very stable nucleosomes at higher temperatures . J Mol Biol , 245 ( 5 ) : 559 – 567 .
- Puhl , H L , Gudibande , S R and Behe , M J . 1991 . Poly[d(A.T)] and other synthetic polydeoxynucleotides containing oligoadenosine tracts form nucleosomes easily . J Mol Biol , 222 ( 4 ) : 1149 – 1160 .
- Racki , L R , Yang , J G , Naber , N , Partensky , P D , Acevedo , A , Purcell , T J , Cooke , R , Cheng , Y and Narlikar , G J . 2009 . The chromatin remodeller ACF acts as a dimeric motor to space nucleosomes . Nature , 462 ( 7276 ) : 1016 – 1021 .
- Radman-Livaja , M and Rando , O J . 2010 . Nucleosome positioning: how is it established, and why does it matter? . Dev Biol , 339 ( 2 ) : 258 – 266 .
- Rhodes , D . 1979 . Nucleosome cores reconstituted from poly (dA-dT) and the octamer of histones . Nucleic Acids Res , 6 ( 5 ) : 1805 – 1816 .
- Rhodes , D . 1985 . Structural analysis of a triple complex between the histone octamer, a Xenopus gene for 5S RNA and transcription factor IIIA . EMBO J , 4 ( 13A ) : 3473 – 3482 .
- Rhodes , D and Laskey , R A . 1989 . Assembly of nucleosomes and chromatin in vitro . Methods Enzymol , 170 : 575 – 585 .
- Richard-Foy , H and Hager , G L . 1987 . Sequence-specific positioning of nucleosomes over the steroid-inducible MMTV promoter . EMBO J , 6 ( 8 ) : 2321 – 2328 .
- Richmond , T J and Davey , C A . 2003 . The structure of DNA in the nucleosome core . Nature , 423 ( 6936 ) : 145 – 150 .
- Riley , D and Weintraub , H . 1978 . Nucleosomal DNA is digested to repeats of 10 bases by exonuclease III . Cell , 13 ( 2 ) : 281 – 293 .
- Rogers , S G and Weiss , B . 1980 . Exonuclease III of Escherichia coli K-12, an AP endonuclease . Methods Enzymol , 65 ( 1 ) : 201 – 211 .
- Ross , S R . 2010 . Mouse mammary tumor virus molecular biology and oncogenesis. Viruses , 2 ( 9 ) : 2000 – 2012 .
- Sakaue , T , Yoshikawa , K , Yoshimura , S H and Takeyasu , K . 2001 . Histone core slips along DNA and prefers positioning at the chain end . Phys Rev Lett , 87 ( 7 ) : 078105
- Sambrook , J and Russell , D W . 2001 . Molecular cloning: a laboratory manual , 3 , Cold Spring Harbor (NY) : Cold Spring Harbor Laboratory Press .
- Satchwell , S C , Drew , H R and Travers , A A . 1986 . Sequence periodicities in chicken nucleosome core DNA . J Mol Biol , 191 ( 4 ) : 659 – 675 .
- Saunders , A , Core , L J and Lis , J T . 2006 . Breaking barriers to transcription elongation . Nat Rev Mol Cell Biol , 7 ( 8 ) : 557 – 567 .
- Schones , D E , Cui , K , Cuddapah , S , Roh , T- Y , Barski , A , Wang , Z , Wei , G and Zhao , K . 2008 . Dynamic regulation of nucleosome positioning in the human genome . Cell , 132 ( 5 ) : 887 – 898 .
- Schueler , M G and Sullivan , B A . 2006 . Structural and functional dynamics of human centromeric chromatin . Annu Rev Genomics Human Genet , 7 : 301 – 313 .
- Segal , E and Widom , J . 2009a . What controls nucleosome positions? . Trends Genet , 25 ( 8 ) : 335 – 343 .
- Segal , E and Widom , J . 2009b . Poly(dA:dT) tracts: major determinants of nucleosome organization . Curr Opin Struct Biol , 19 ( 1 ) : 65 – 71 .
- Shrader , T E and Crothers , D M . 1989 . Artificial nucleosome positioning sequences . Proc Natl Acad Sci USA , 86 ( 19 ) : 7418 – 7422 .
- Shukla , M S , Syed , S H , Montel , F , Faivre-Moskalenko , C , Bednar , J , Travers , A A , Angelov , D and Dimitrov , S . 2010 . Remosomes: RSC generated non-mobilized particles with approximately 180 bp DNA loosely associated with the histone octamer. Proc Natl Acad Sci USA , 107 ( 5 ) : 1936 – 1941 .
- Simpson , R T . 1978 . Structure of the chromatosome, a chromatin particle containing 160 base pairs of DNA and all the histones . Biochemistry , 17 ( 25 ) : 5524 – 5531 .
- Simpson , R T and Shindo , H . 1979 . Conformation of DNA in chromatin core particles containing poly(dAdT)-poly(dAdT) studied by 31 P NMR spectroscopy . Nucleic Acids Res , 7 ( 2 ) : 481 – 492 .
- Simpson , R T and Stafford , D W . 1983 . Structural features of a phased nucleosome core particle . Proc Natl Acad Sci USA , 80 ( 1 ) : 51 – 55 .
- Simpson , R T , Thoma , F and Brubaker , J M . 1985 . Chromatin reconstituted from tandemly repeated cloned DNA fragments and core histones: a model system for study of higher order structure . Cell , 42 ( 3 ) : 799 – 808 .
- Stein , A , Whitlock , J P and Bina , M . 1979 . Acidic polypeptides can assemble both histones and chromatin in vitro at physiological ionic strength . Proc Natl Acad Sci USA , 76 ( 10 ) : 5000 – 5004 .
- Strauss , J K and Maher , L J . 1994 . DNA bending by asymmetric phosphate neutralization . Science , 266 ( 5192 ) : 1829 – 1834 .
- Suck , D and Oefner , C . 1986 . Structure of DNase I at 2.0 A resolution suggests a mechanism for binding to and cutting DNA . Nature , 321 ( 6070 ) : 620 – 625 .
- Sulkowski , E and Laskowski , M . 1962 . Mechanism of action of micrococcal nuclease on deoxyribonucleic acid . J Biol Chem , 237 : 2620 – 2625 .
- Tachiwana , H , Kagawa , W , Shiga , T , Osakabe , A , Miya , Y , Saito , K , Hayashi-Takanaka , Y , Oda , T , Sato , M Park , S- Y . 2011 . Crystal structure of the human centromeric nucleosome containing CENP-A . Nature , 476 ( 7359 ) : 232 – 235 .
- Tan , S and Davey , C A . 2010 . Nucleosome structural studies . Curr Opin Struct Biol , 21 ( 1 ) : 128 – 136 .
- Tanaka , Y , Tachiwana , H , Yoda , K , Masumoto , H , Okazaki , T , Kurumizaka , H and Yokoyama , S . 2005 . Human centromere protein B induces translational positioning of nucleosomes on alpha-satellite sequences . J Biol Chem , 280 ( 50 ) : 41609 – 41618 .
- Thåström , A , Gottesfeld , J M , Luger , K and Widom , J . 2004 . Histone-DNA binding free energy cannot be measured in dilution-driven dissociation experiments . Biochemistry , 43 ( 3 ) : 736 – 741 .
- Thåström , A , Lowary , P T , Widlund , H R , Cao , H , Kubista , M and Widom , J . 1999 . Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences . J Mol Biol , 288 ( 2 ) : 213 – 229 .
- Theodorou , V , Kimm , M A , Boer , M , Wessels , L , Theelen , W , Jonkers , J and Hilkens , J . 2007 . MMTV insertional mutagenesis identifies genes, gene families and pathways involved in mammary cancer . Nature Genet , 39 ( 6 ) : 759 – 769 .
- Thoma . 1992 . Nucleosome positioning . Biochim Biophys Acta , 1130 ( 1 ) : 1 – 19 .
- Tillo , D and Hughes , T R . 2009 . G+C content dominates intrinsic nucleosome occupancy . BMC Bioinformatics , 10 : 442
- Tóth , K , Brun , N and Langowski , J . 2001 . Trajectory of nucleosomal linker DNA studied by fluorescence resonance energy transfer . Biochemistry , 40 ( 23 ) : 6921 – 6928 .
- Travers , A A and Klug , A . 1987 . The bending of DNA in nucleosomes and its wider implications . Philos Trans R Soc Lond B Biol Sci , 317 ( 1187 ) : 537 – 561 .
- Travers , A A , Caserta , M , Churcher , M , Hiriart , E and Di Mauro , E . 2009 . Nucleosome positioning – what do we really know? . Mol Biosyst , 5 ( 12 ) : 1582 – 1592 .
- Trifonov , E N . 2011 . Cracking the chromatin code: precise rule of nucleosome positioning . Phys Life Rev , 8 ( 1 ) : 39 – 50 .
- Tsukiyama , T , Becker , P B and Wu , C . 1994 . ATP-dependent nucleosome disruption at a heat-shock promoter mediated by binding of GAGA transcription factor . Nature , 367 ( 6463 ) : 525 – 532 .
- Tsunaka , Y , Kajimura , N , Tate , S- I and Morikawa , K . 2005 . Alteration of the nucleosomal DNA path in the crystal structure of a human nucleosome core particle . Nucleic Acids Res , 33 ( 10 ) : 3424 – 3434 .
- Vafa , O and Sullivan , K F . 1997 . Chromatin containing CENP-A and alpha-satellite DNA is a major component of the inner kinetochore plate . Curr Biol , 7 ( 11 ) : 897 – 900 .
- Van Holde , K E . 1989 . “ Chromat ” . In Springer Series in Molecular Biology , New York : Springer-Verlag .
- van Vugt , JJF A , de Jager , M , Murawska , M , Brehm , A , van Noort , J and Logie , C . 2009 . Multiple aspects of ATP-dependent nucleosome translocation by RSC and Mi-2 are directed by the underlying DNA sequence . PloS one , 4 ( 7 ) : e6345
- Vasudevan , D , Chua , EY D and Davey , C A . 2010 . Crystal structures of nucleosome core particles containing the “601” strong positioning sequence . J Mol Biol , 403 ( 1 ) : 1 – 10 .
- Vitolo , J M , Yang , Z , Basavappa , R and Hayes , J J . 2004 . Structural features of transcription factor IIIA bound to a nucleosome in solution . Mol Cell Biol , 24 ( 2 ) : 697 – 707 .
- Wang , Y H and Griffith , J . 1996 . Methylation of expanded CCG triplet repeat DNA from fragile X syndrome patients enhances nucleosome exclusion . J Biol Chem , 271 ( 38 ) : 22937 – 22940 .
- Weintraub , H , Worcel , A and Alberts , B . 1976 . A model for chromatin based upon two symmetrically paired half-nucleosomes . Cell , 9 ( 3 ) : 409 – 417 .
- White , C L , Suto , R K and Luger , K . 2001 . Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions . EMBO J , 20 ( 18 ) : 5207 – 5218 .
- Whitehouse , I. , Rando , O J , Delrow , J. and Tsukiyama , T. 2007 . Chromatin remodelling at promoters suppresses antisense transcription . Nature , 450 ( 7172 ) : 1031 – 1035 .
- Widlund , H R , Cao , H , Simonsson , S , Magnusson , E , Simonsson , T , Nielsen , P E , Kahn , J D , Crothers , D M and Kubista , M . 1997 . Identification and characterization of genomic nucleosome-positioning sequences . J Mol Biol , 267 ( 4 ) : 807 – 817 .
- Widom , J . 1992 . A relationship between the helical twist of DNA and the ordered positioning of nucleosomes in all eukaryotic cells . Proc Natl Acad Sci USA , 89 ( 3 ) : 1095 – 1099 .
- Widom , J . 2001 . Role of DNA sequence in nucleosome stability and dynamics . Q Rev Biophys , 34 ( 3 ) : 269 – 324 .
- Wolffe , A P and Drew , H R . 1989 . Initiation of transcription on nucleosomal templates . Proc Natl Acad Sci USA , 86 ( 24 ) : 9817 – 9821 .
- Woods , K K , Maehigashi , T , Howerton , S B , Sines , C C , Tannenbaum , S and Williams , L D . 2004 . High-resolution structure of an extended A-tract: [d(CGCAAATTTGCG)]2 . J Am Chem Soc , 126 ( 47 ) : 15330 – 15331 .
- Wu , B , Mohideen , K , Vasudevan , D and Davey , C A . 2010 . Structural insight into the sequence dependence of nucleosome positioning . Structure , 18 ( 4 ) : 528 – 536 .
- Xie , X , Kokubo , T , Cohen , S L , Mirza , U A , Hoffmann , A , Chait , B T , Roeder , R G , Nakatani , Y and Burley , S K . 1996 . Structural similarity between TAFs and the heterotetrameric core of the histone octamer . Nature , 380 ( 6572 ) : 316 – 322 .
- Yakovchuk , P , Protozanova , E and Frank-Kamenetskii , M D . 2006 . Base-stacking and base-pairing contributions into thermal stability of the DNA double helix . Nucleic Acids Res , 34 ( 2 ) : 564 – 574 .
- Yoda , K , Ando , S , Okuda , A , Kikuchi , A and Okazaki , T . 1998 . In vitro assembly of the CENP-B/alpha-satellite DNA/core histone complex: CENP-B causes nucleosome positioning . Gene Cells , 3 ( 8 ) : 533 – 548 .
- Yodh , J G , Woodbury , N , Shlyakhtenko , L S , Lyubchenko , Y L and Lohr , D . 2002 . Mapping nucleosome locations on the 208-12 by AFM provides clear evidence for cooperativity in array occupation . Biochemistry , 41 ( 11 ) : 3565 – 3574 .
- Yuan , G- C , Liu , Y- J , Dion , M F , Slack , M D , Wu , L F , Altschuler , S J and Rando , O J . 2005 . Genome-scale identification of nucleosome positions in S. cerevisiae. . Science , 309 ( 5734 ) : 626 – 630 .
- Zhang , Y , Moqtaderi , Z , Rattner , B P , Euskirchen , G , Snyder , M , Kadonaga , J T and Struhl , XSL K . 2010 . Evidence against a genomic code for nucleosome positioning . Nature Struct Mol Biol , 17 ( 8 ) : 920 – 922 .
- Zhang , Z , Wippo , C J , Wal , M , Ward , E , Korber , P and Pugh , B F . 2011 . A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome . Science , 332 ( 6032 ) : 977 – 980 .
- Zhurkin , V B . 2011 . The first thirty years of nucleosome positioning . Phys Life Rev , 8 ( 1 ) : 64 – 66; . discussion 69–72.