
Abstract
Culture-dependent methods and molecular techniques were used to simultaneously investigate the cultivable bacterial diversity and amylase production in two typical light-flavor Daqus of Chinese spirits. Eight bacteria were identified from Niulanshan Daqu (Bacillus licheniformis, Bacillus subtilis, Bacillus cereus, Bacillus sonorensis, Streptomyces albus, Bacillus atrophaeus, Bacillus tequilensis and Bacillus megaterium) and eight from Hongxing Daqu (B. licheniformis, B. subtilis, B. cereus, Bacillus thuringiensis, Bacillus altitudinis, Bacillus pumilus, Geobacillus stearothermophilus and Bacillus amyloliquefaciens). All of the bacterial isolates from the Hongxing Daqu could produce extracellular α-amylase with a maximum yield of 25.3 U/ml by B. cereus H17, whereas B. licheniformis H55 could produce a maximum glucoamylase yield of 41.6 U/ml. Some of the bacterial isolates from the Niulanshan Daqu could also produce extracellular α-amylase and glucoamylase. The maximum yield of 27.6 U/ml α-amylase was achieved by B. subtilis N3, and the maximum yield of 58.1 U/ml glucoamylase was achieved by B. cereus N25. Bacillus licheniformis, B. subtilis and B. cereus were not only the dominant bacteria, but also possessed high α-amylase and glucoamylase activities, which may play very important roles during fermentation.
Introduction
Chinese spirits are commonly known as ‘Chinese liquor' and are very popular in China. There are three main types of Chinese spirit: sauce-flavor liquor, intense-flavor liquor and light-flavor liquor (Li et al. Citation2013). Chinese spirits are typically obtained from cereals such as sorghum and wheat by complex fermentation processes using natural mixed culture starters (i.e. ‘Daqu'). Daqu, which is made from raw wheat, barley and/or peas, is associated with the degradation of starch and the production of alcohol, and is also the determinative factor for liquor flavoring (Liu et al. Citation2012). Daqu contains microbes and enzymes, and both glucoamylase and α-amylase are two important amylases for starch hydrolysis. In the brewing industry, the hydrolyzates are used as nutrients in microbial fermentation for ethanol production after enzymatic hydrolysis. However, amylase production of bacteria from Daqus is poorly understood. During fermentation, the degradation of biopolymers such as starch and the formation of aromatic compounds are usually performed by microbes (Wang et al. Citation2008). Therefore, it is important to check the exact composition of the natural product's microflora. However, very little is known about the specific microbes associated with Daqus, despite the fact that the subject has been studied by microbiologists (Wang et al. Citation2008; Zheng et al. Citation2012).
Traditional microbiological methods, such as isolation and enumeration in selective media, are effective in qualitative and quantitative analysis (Shi et al. Citation2009; Zheng et al. Citation2012). Sequencing analysis of the 16S rRNA gene has been used for rapid bacterial identification (Jeyaram et al. Citation2010). In this study, reports on an investigation of the cultivable bacterial diversity and amylase production from two typical light-flavor Daqus in Chinese spirits.
Materials and methods
The two Daqus were obtained from two major Chinese spirit factories in Beijing, which are well known in China. The sampling data are shown in . Three parallel samples were collected from the top, middle and bottom in the store room. After being milled and mixed, samples were transferred to sterile bags, sealed and stored at 4°C for further use.
Table 1. Samples of two typical light-flavor Daqus of Chinese spirits.
Ten grams of Daqu powder was homogenized in 90 ml of a sterile 0.9% NaCl solution in a rocking incubator for 30 min at 180 rpm. Samples (1.0 ml) of the homogenate were serially diluted 10-fold in a sterile 0.9% NaCl solution, from which aliquots (0.1 ml) were plated on beef extract peptone agar plates with the following components (w/v): 0.5% beef extract, 1.0% peptone and 0.5% NaCl. Inoculated plates were incubated at 37°C for 24–48 h for isolation and screening on the basis of different colony morphologies (diameter, shape, color, surface and spores).
The genomic DNAs of the isolates were extracted, amplified and sequenced according to the methods described by Li et al. (Citation2012). The 16S rDNA sequence data of the isolates reported here have been submitted to GenBank nucleotide sequence databases with the accession numbers shown in Tables and .
Table 2. Bacterial diversity and amylase production in Niulanshan Daqu.
Table 3. Bacterial diversity and amylase production in Hongxing Daqu.
The isolates from the Daqus were diluted in a sterile 0.9% NaCl solution and then plated on to starch agar plates containing 2.0% soluble starch, 1.0% peptone and 0.5% NaCl. Inoculated plates were incubated for 1–3 days at 37°C to obtain colonial growth. The incubated plates were stained with iodine solution, and the colonies with clear zones formed by the hydrolysis of starch were evaluated as amylase producers. The components (w/v) of the medium used for liquid inoculum culture were as follows: 0.3% beef extract, 1.0% peptone and 0.5% NaCl, pH 7.2. The medium was sterilized by autoclaving at 121°C for 20 min. Medium (20 ml) in 150 ml Erlenmeyer flasks was inoculated and cultivated at 37°C by shaking at 180 rpm for 24 h. The medium in a 500 ml Erlenmeyer flask contained (w/v): soluble starch 2.0%, beef extract 0.5%, peptone 1.0% and NaCl 0.5%, pH 7.2. After sterilization, the medium (100 ml) was inoculated with 6 ml of a 24-h-old inoculum culture, and cultivated at 37°C by shaking at 180 rpm for 48 h, then centrifuged at 8000 rpm at 4°C for 15 min to remove the cells and debris. The supernatant was designated as the crude enzyme for future enzyme activity assays (Qureshi et al. Citation2015).
For α-amylase assays, the starch–iodine method was used (Shankar et al. Citation2009). One unit of α-amylase activity was defined as the amount of enzyme that hydrolyzed 1 mg of starch per hour under the assay conditions. Glucoamylase activity was determined by measuring released reducing sugars using the dinitrosalicylic acid method (Kumar & Satyanarayana Citation2009). One unit of glucoamylase activity was defined as the amount of enzyme that liberated 1 mg of reducing sugar as glucose per hour under the assay conditions.
Results and discussion
In total, 16 and 15 bacterial strains were randomly selected and identified by sequencing the 16S rDNA from the Niulanshan and Hongxing Daqus, respectively. The sequences have been submitted to the GenBank database with accession numbers (Tables and ). Eight species were encountered in each of the Niulanshan and Hongxing Daqus. The results are presented in Tables and . The Bacillus genus was dominant in the two light-flavor style Daqus. This observation is consistent with that of previous studies (Kumar & Satyanarayana Citation2009; Zheng et al. Citation2012). In particular, B. licheniformis, B. subtilis and B. cereus together represented approximately 68.8% and 66.7% of the bacteria isolated from the Niulanshan and Hongxing Daqus, with approximately 37.5% and 20.0% of the sequences corresponding to B. licheniformis. In addition, B. sonorensis, Streptomyces albus, B. atrophaeus, B. tequilensis and B. megaterium were observed in the Niulanshan Daqu, while B. thuringiensis, B. altitudinis, B. pumilus, Geobacillus stearothermophilus and B. amyloliquefaciens were found in the Hongxing Daqu (Tabassum et al. Citation2014).
To understand the phylogenetic position of these strains from the two typical light-flavor Daqus of Chinese spirits, phylogenetic trees were constructed based on comparison of the 16S rDNA sequences of the strains from the two typical light-flavor Daqus and those of reference bacteria (Figures and ).
Figure 1. Phylogenetic relationships among bacterial 16S rDNA sequences in the Daqu of Niulanshan liquor and with previously reported sequences. The number on each branch indicates the percentage of 1000 replicates that are included in the branch. Sequences determined in this study are shown in bold, and GenBank accession numbers are shown in parentheses for all of the related sequences. The scale bar of 0.02 represents a 2% nucleotide substitution rate according to the Jukes–Cantor evolutionary distance (Surhio et al. Citation2014).
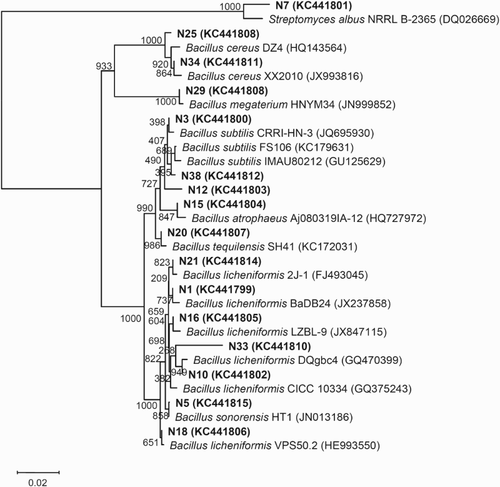
Figure 2. Phylogenetic relationships among bacterial 16S rDNA sequences in the Daqu of Hongxing liquor and with previously reported sequences. The number on each branch indicates the number out of 1000 replicates that are included in the branch. Sequences determined in this study are shown in bold, and GenBank accession numbers are shown in parentheses for all of the related sequences. The scale bar of 0.02 represents a 2% nucleotide substitution rate according to the Jukes–Cantor evolutionary distance.
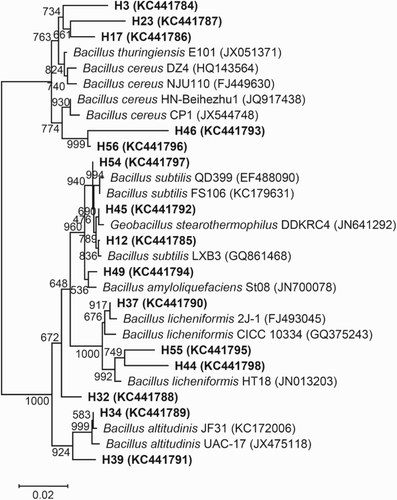
Several factors, including the regional climate, water used in production and microorganisms in the air jointly influenced the microorganism composition in Daqus. Nevertheless, temperature was an important technological parameter, which would affect the growth and death of microorganisms and finally determine the microorganism community structure in Daqus (Kumar & Satyanarayana Citation2009; Zheng et al. Citation2012). Both Niulanshan Daqu and Hongxing Daqu are representatives of the light-flavor Daqus. During its manufacture by solid-state fermentation, the temperature in the center of the Daqu blocks during fermentation does not exceed 50°C (). Most bacteria tolerate these temperatures and, therefore, a wide range of bacterial species was observed in the two light-flavor Daqus (Figures and ). Although both the Niulanshan and Hongxing Daqus are light-flavor Daqus, the community structures of bacteria were obviously different between them. Bacillus species were the most frequently isolated bacteria from the two light-flavor Daqus. Bacillus bacteria have a better ability than other bacteria to survive under conditions of low moisture content and high temperature (Wang et al. Citation2008). Bacillus licheniformis, B. subtilis and B. cereus were dominant in the Daqus (Figures and ). Other species, such as B. atrophaeus, B. sonorensis, B. megaterium, Streptomyces albus and Bacillus tequilensis, were observed only in the Niulanshan Daqu, whereas B. amyloliquefaciens, B. pumilus, B. altitudinis, B. thuringiensis and Geobacillus stearothermophilus were all observed in the Hongxing Daqu. In comparison, both B. licheniformis and B. subtilis, which were detected by a culture-dependent method, were predominant in Fen Daqu, which is a light-flavor Daqu from the Shanxi Province (Kumar & Satyanarayana Citation2009; Zheng et al. Citation2012). The present results are in agreement with these findings. Bacillus licheniformis, a facultative anaerobe, is a bacterium that can grow in adverse ecological niches (Shi et al. Citation2009). It has been found that B. pumilus exists in Hongxing Daqu and Fen Daqu. However, B. cereus was not the dominant species in Fen Daqu and was found only in the outer part of Fen Daqu. In addition, Brevibacterium sp., Enterococcus faecalis, Lactobacillus plantarum, Pediococcus pentosaceus, Salmonella enterica, Leuconostoc citreum, Micrococcus luteus, Pseudomonas aeruginosa and Escherichia coli were encountered in Fen Daqu (Zheng et al. Citation2012); nevertheless, none of them was observed in either Niulanshan Daqu or Hongxing Daqu. Therefore, the bacterial community structures in both Niulanshan and Hongxing Daqus were obviously different from those in Fen Daqu. Many intrinsic and extrinsic factors of Daqu production influence the richness and structure of the microbial community, including the raw material variety, moisture content, temperature control, geographical location, climate and water (Wang et al. Citation2011).
Following staining with iodine solution, the isolates from the two Daqus that exhibited visible clear zones around the colonies on agar plates with starch as the sole carbon source were selected as good producers of amylase (Tables and ). All 15 bacterial isolates from the Hongxing Daqu could produce extracellular α-amylase at 37°C after 48 h of incubation, with a maximum yield of 25.3 U/ml by B. cereus H17, whereas B. licheniformis H55 could produce a maximum glucoamylase yield of 41.6 U/ml among the nine bacterial isolates that could produce glucoamylase at 37°C after 48 h of incubation (). Of the total enzyme, the activity of α-amylase and glucoamylase produced by B. licheniformis, B. subtilis and B. cereus accounted for 27.5% and 52.2%, 18.1% and 2.5%, and 33.7% and 38.0%, respectively. Furthermore, B. amyloliquefaciens could produce glucoamylase.
In total, eight and eight bacterial isolates from the Niulanshan Daqu could produce extracellular α-amylase and glucoamylase, respectively. The maximum yield of 27.6 U/ml α-amylase was achieved by B. subtilis N3 and the maximum yield of 58.1 U/ml glucoamylase was achieved by B. cereus N25 (). Bacillus licheniformis, B. subtilis and B. cereus were the dominant producers of both α-amylase and glucoamylase. Of the total enzyme, the activity of α-amylase and glucoamylase produced by these bacteria accounted for 14.6% and 23.4%, 33.1% and 11.1%, and 20.6% and 38.3%, respectively. In addition, B. atrophaeus and B. tequilensis could produce α-amylase and glucoamylase (Khaskheli et al. Citation2015).
The variety of bacteria found in the Daqus produced complicated enzyme systems that converted the Daqus into unique glycation and fermented substances, which are useful for subsequent reactions leading to flavor production (Shi et al. Citation2009). Amylases are one of the most important enzymes and are of great significance in today's biotechnology industry. They have several different industrial applications, including foods, pharmaceuticals, detergents and animal feed. The variety of bacteria found in the two Daqus could produce amylase, but the compositions of bacteria producing amylase obviously differed between them, although they all belonged to light-flavor Daqus (Tables and ). As the dominant bacterial species in the two typical Daqus, B. licheniformis and B. subtilis also possessed high α-amylase and glucoamylase activities, which enabled them to utilize raw starch as a carbon source and helped them to play a very important role during fermentation. Of the total enzyme from the Hongxing and Niulanshan Daqus, the activity of α-amylase secreted by B. licheniformis and B. subtilis accounted for 27.5% and 18.1%, and 14.6% and 33.1%, respectively. In contrast, the total activity of glucoamylase accounted for 52.2% and 2.5%, and 23.4% and 11.1%. Moreover, B. cereus was a chief bacterial species that produced amylase in the Hongxing and Niulanshan Daqus, with the total activity of α-amylase and glucoamylase produced by B. cereus accounting for 33.7% and 38.0%, and 20.6% and 38.3%, respectively.
Comparison of the activities of α-amylase and glucoamylase in the Hongxing Daqu with those in the Niulanshan Daqu indicated that the predominant bacterial genus and species secreting both α-amylase and glucoamylase were similar, including the Bacillus genus and species of B. licheniformis, B. subtilis and B. cereus. Bacillus species are important sources of amylases and proteases. The hydrolytic capabilities of these microorganisms can result in a precursor-rich environment, which is useful for subsequent reactions leading to flavor production (Wang et al. Citation2008). Bacillus subtilis and B. licheniformis may facilitate the conversion of starch into fermentable carbohydrates owing to their amylolytic activity, thus generating a suitable substrate for the second stage of liquor production, i.e. alcoholic fermentation (Zheng et al. Citation2012). Other bacterial species, such as B. amyloliquefaciens, could produce both α-amylase and glucoamylase only in the Hongxing Daqu. Bacillus amyloliquefaciens, which is one of the most widely used species for the bulk production of amylase, is a potent producer of amylase and other industrial exoenzymes (Liu et al. Citation2012). Bacillus atrophaeus and B. tequilensis could produce α-amylase as well as glucoamylase only in the Niulanshan Daqu. It was found that different maximum yields of α-amylase and glucoamylase were produced by different bacterial species in the Daqus. The maximum α-amylase yields of 25.3 U/ml and 27.6 U/ml, which were produced by B. cereus H17 from the Hongxing Daqu and B. subtilis N3 from the Niulanshan Daqu, are similar. However, the maximum glucoamylase yield by B. cereus N25 from the Niulanshan Daqu was 1.4-fold higher than that by B. licheniformis H55 from the Hongxing Daqu. The activity of α-amylase and glucoamylase in Daqus is a key element in the production of Chinese spirits, and these enzymes act to decompose starch in the raw materials (Kayani et al. Citation2014).
In conclusion, 16 and 15 bacteria strains were randomly selected and identified by sequencing of the 16S rDNA from Niulanshan and Hongxing Daqus, respectively. The indigenous bacterial communities in the two Daqus were complex and consisted of a wide variety of bacteria. Both the bacterial community structures and characteristics of amylase production were obviously different. However, B. licheniformis, B. subtilis and B. cereus were not only the dominant bacteria but also possessed high α-amylase and glucoamylase activities that enabled them to utilize raw starch as a carbon source, and which may help them to play a very important role during fermentation. Further investigation needs to be carried out to obtain more detailed information on the specific function of each individual microorganism and its contribution to the final formation of the unique aroma in Chinese spirits.
Acknowledgements
The authors are thankful to those who sent the Daqu samples for this research.
Funding
This research was financially supported by the National Key Technology R&D Program of China [no. 2012BAK17B11] and Beijing Municipal Commission of Education and Subsidy for Outstanding People of Beijing [no. KM201311417007; PXM2013_014209_07_000082; 2011A005022000004].
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Jeyaram K, Romi W, Singh TA, Devi AR, Devi SS. 2010. Bacterial species associated with traditional starter cultures used for fermented bamboo shoot production in Manipur state of India. Int J Food Microbiol. 143:1–8.
- Kayani S, Ahmad M, Zafar M, Sultana S, Khan MPZ, Ashraf MA, Hussain J, Yaseen G. 2014. Ethnobotanical uses of medicinal plants for respiratory disorders among the inhabitants of Gallies-Abbottabad, Northern Pakistan. J Ethnopharmacol. doi:https://doi.org/10.1016/j.jep.2014.08.005.
- Khaskheli AA, Talpur FN, Ashraf MA, Cebeci A, Jawaid S, Afridi HI. 2015. Monitoring the Rhizopus oryzae lipase catalyzed hydrolysis of castor oil by ATR-FTIR spectroscopy. J Mol Catal B: Enzym. 113(1):56–61. https://doi.org/10.1016/j.molcatb.2015.01.002.
- Kumar P, Satyanarayana T. 2009. Overproduction of glucoamylase by a deregulated mutant of a thermophilic mould thermomucor indicae-seudaticae. Appl Biochem Biotechnol. 158:113–25.
- Li K, Zhang Q, Zhong XT, Jia BH, Yuan CH, Liu S, Che ZM, Xiang WL. 2013. Microbial diversity and succession in the Chinese Luzhou-Flavor Liquor fermenting cover lees as evaluated by SSU rRNA profiles. Indian J Microbiol. 53(4):425–31.
- Li ZM, Bai ZH, Zhang BG, Li BJ, Jin B, Zhang M, Lin F, Zhang HX. 2012. Purification and characterization of alkaline pectin lyase from a newly isolated Bacillus clausii and its application in elicitation of plant disease resistance. Appl Biochem Biotechnol. 167:2241–56.
- Liu X, Guo KL, Zhang HX. 2012. Determination of microbial diversity in Daqu, a fermentation starter culture of Maotai liquor, using nested PCR-denaturing gradient gel electrophoresis. World J Microbiol Biotechnol. 28:2375–81.
- Qureshi T, Memon N, Memon SQ, Ashraf MA. 2015. Decontamination of ofloxacin: optimization of removal process onto sawdust using response surface methodology. Desalin Water Treat. doi:https://doi.org/10.1080/19443994.2015.1006825
- Shankar M, Priyadharshini R, Gunasekaran P. 2009. Quantitative digital image analysis of chromogenic assays for high throughput screening of a-amylase mutant libraries. Biotechnol Lett. 31:1197–201.
- Shi JH, Xiao YP, Li XR, Ma EB, Du XW, Quan ZX. 2009. Analyses of microbial consortia in the starter of Fen Liquor. Lett Appl Microbiol. 48:478–85.
- Surhio MA, Talpur FN, Nizamani SM, Amin FA, Bong CW, Lee CW, Ashraf MA, Shahd MR. 2014. Complete degradation of dimethyl phthalate by biochemical cooperation of the Bacillus thuringiensis strain isolated from cotton field soil. RSC Advances. 4:55960–55966.
- Tabassum N, Rafique U, Balkhair KS, Ashraf MA. 2014. Chemodynamics of Methyl Parathion and Ethyl Parathion: Adsorption Models for Sustainable Agriculture. Biomedical Research International. doi:https://doi.org/10.1155/2014/831989
- Wang HY, Gao YB, Fan QW, Xu Y. 2011. Characterization and comparison of microbial community of different typical Chinese liquor Daqus by PCR-DGGE. Lett Appl Microbiol. 53:134–40.
- Wang CL, Shi DJ, Gong GL. 2008. Microorganisms in Daqu: a starter culture of Chinese Maotai-flavor liquor. World J Microbiol Biotechnol. 24:2183–90.
- Zheng XW, Yan Z, Han BZ, Zwietering MH, Samson RA, Boekhout T, Nout MJR. 2012. Complex microbiota of a Chinese ‘Fen’ liquor fermentation starter (Fen-Daqu), revealed by culture-dependent and culture-independent methods. Food Microbiol. 31:293–300.