
Abstract
In this study, metagenomics was applied to characterize microbial communities, specifically methanogens and methanotrophs, and to discover their functional activities under two different dietary treatments. To retrieve an overall rumen microbial community profile and to check the abundance of methanogenic and methanotrophic bacteria therein, semiconductor shotgun sequencing of DNA isolated from the rumen fluid of Mehsani buffalo treated with two different diets, i.e. 50% green roughage/50% concentrate (M50GL) and 100% green roughage (M100GL), was carried out. The study revealed that the M50GL group harboured more Proteobacteria than the M100GL group, which harboured more Bacteroidetes. The classes of Proteobacteria (methanotrophs) differed significantly in response to the change in diet. α-Proteobacteria and β-proteobacteria were found to be significantly (p < 0.05) higher in the M100GL group, whereas γ-proteobacteria were significantly more abundant in the M50GL group than in the M100GL group. Different species of methanogens were more abundant in the M100GL group than in the M50GL group. The enzymes involved in the serine pathway (glycine hydroxyl methyltransferase) carried out by type II methanotrophs, i.e. α-proteobacteria and β-proteobacteria, were found in higher abundance in the M100GL group, which correlates with the taxonomic abundance of the same classes in the M100GL group.
Introduction
The rumen can be simplistically described as an open system where discontinuous inputs of roughage and concentrate are fermented by a mixed microbial population, yielding volatile fatty acids, ammonium (NH4+), carbon dioxide (CO2) and hydrogen (H2) from proteins and polymeric carbohydrates (Howard Citation1959; Whitelaw et al. Citation1984). The H2 thus formed is utilized by methanogens, members of the domain Archaea (Moss et al. Citation2000), which comprise about 0.3–3.3% of the total microbial population in the rumen. Despite their low numbers, the methanogen group of archaea occupies an important niche in the rumen as it utilizes the H2 to reduce CO2 to methane (CH4) in a series of reactions that are coupled to ATP synthesis. Efficient H2 eradication may increase the fermentation rate by eliminating the inhibitory effect of H2 on microbial fermentation (Janssen & Kirs Citation2008).
There has been increasing interest in methane formation in the rumen, as 23% of global methane emission come from livestock, as recorded by the US Environmental Protection Agency (EPA), and 31% of New Zealand's greenhouse gases are released by livestock (Leahy et al. Citation2010). Looking at the Indian scenario, the percentage increase in enteric methane emission by Indian livestock was greater that for than world livestock (70.6% vs 54.3%) between the years 1961 and 2010, and the annual growth rate was highest for goats (1.91%), followed by buffalo (1.55%), swine (1.28%), sheep (1.25%) and cattle (0.70%) (Patra Citation2014). Estimates of livestock populations indicate that lactating dairy cattle and buffalo are expected to increase by 3.5 and 5.6 million, resulting in expected increases in methane emissions of approximately 36% and 17%, respectively, by 2021 (Chhabra et al. Citation2007).
The production of methane is influenced by the nature of the carbohydrate feed, particularly by higher roughage feed (McAllister et al. Citation1996). Although a great deal of information about methane production in the rumen is available, there is very little information about methane consumption. Methanotrophic bacteria, or methanotrophs, a subset of a physiological group of bacteria, have the ability to utilize methane as a sole carbon and energy source. Under aerobic conditions, they combine oxygen and methane to form formaldehyde, which is incorporated into organic compounds via the ribulose monophosphate pathway by type I methanotrophs (γ-proteobacteria) or the serine pathway by type II methanotrophs (α-proteobacteria) (Hanson & Hanson Citation1996).
In the present study, four buffalo of Mehsani breed were given a mixed ration of 50% green roughage and 50% concentrate for 6 weeks. After 6 weeks, the same four buffalo were shifted on to a 100% green roughage diet. Ruminal fluid collected after both treatments was fractionated to obtain the liquid portion. Shotgun semiconductor sequencing technology was used to generate the overall ruminal microbial community profile. The rumen microbial profile was analysed for its methanogen and methanotroph composition and their role in methane metabolism. This article describes how the change in diet contributes to ruminal methane metabolism with concomitant alterations in microbial colonization.
Material and methods
Ruminal sample collection
The experiment included four non-lactating Mehsani water buffalo (Bubalus bubalis) aged 4–5 years, which were assigned to two successive dietary treatments. The diet given to the animals was green roughage (sorghum bicolour) and concentrate (Amul pellets). In the first treatment, the buffalo were given 50% roughage and 50% concentrate as a mixed ration for a continuous 6 weeks. In the second treatment, the same animals were switched to a 100% roughage diet for another 6 weeks. On completion of each dietary treatment, the rumen fluid (approximately 500 ml) was collected using a flexible stomach tube approximately 2.0 h after the morning feed. The rumen content was filtered through a four-layered muslin cloth to separate the liquid portion from the solid fractions. The liquid fraction of the rumen fluid was stored at −80°C until DNA extraction.
DNA extraction
In total, eight ruminal fluid samples were used for DNA extraction using the QIAamp DNA Stool Mini Kit as per the manufacturer's protocol (Qiagen) (Wu et al. Citation2010). The DNA was eluted in TE buffer for better preservation and stored at −20°C until library preparation. The DNA concentration and purity were checked by ND1000 spectrophotometry.
Shotgun sequencing
The extracted DNA was used for shotgun library preparation with a start-up DNA concentration of approximately 300 ng for each of the eight samples. The samples were enzymatically fragmented and eight barcoded libraries were prepared as per 200 bp chemistry of the Ion Torrent Personal Genome Machine protocol. The libraries were then clonally amplified using the emulsion polymerase chain reaction technique as per the manufacturer's protocol. The positively amplified Ion Sphere particles were enriched and loaded on to an Ion 316 chip. Two sequencing runs for eight libraries were carried out, each run with four barcoded libraries of each treatment.
Data analysis
The sequencing data were obtained in FASTQ format and uploaded to MG-RAST for further analysis. To obtain the overall profile of the rumen microbial community, taxonomic classification against the M5nr database using 80% similarity and an e-value of 10−5 was carried out. Moreover, the taxon Archaea was focused on for its abundance as methanogens, whereas the Proteobacteria were focused on for their methanotroph subdivisions. Statistical Analysis of Metagenomic Profiles (STAMP) was used to derive significant differences in abundance between treatments. The functional classification was carried out using KEGG analysis, where the data were mapped with 80% identity against the KEGG pathway and the methane metabolism pathway was highlighted.
Results
The goal of the current study was to provide an in-depth description and characterization of how the ruminal archaeal diversity differs with the increasing proportion of green roughage. The study focuses on the phylogenetic and functional drift of methanogenic and methanotrophic organisms in response to changes in green roughage concentration in the feed of Mehsani buffalo.
Custom Perl script was used to convert the FASTQ files into a FASTA file, where the short reads (< 75 bp) and low-quality (Phred score < 20) sequence removal was carried out. The filtered reads were then uploaded to MG-RAST for further annotation. The data summary for four metagenomes each of M50GL and M100GL is shown in ; 96% of data for M50GL and 89% of M100GL passed the quality criteria of MG-RAST and were used for further taxonomic and functional annotation.
Table 1. Shotgun sequencing data summary for M50GL and M100GL.
Community structure of rumen metagenome
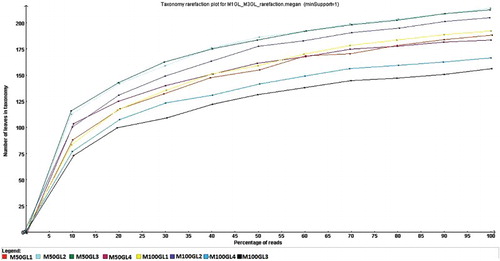
A rarefaction curve was drawn by MEGAN from the metagenomic dataset. Satisfactory coverage of the rumen microbiome in the rarefaction curve illustrates that the curve had already passed the steep region and levelled off to where fewer new species could be found ().
Figure 1. Rarefaction curve for M50GL and M100GL groups by MEGAN. x-Axis = percentage of reads; y-axis = number of leaves in taxonomy.
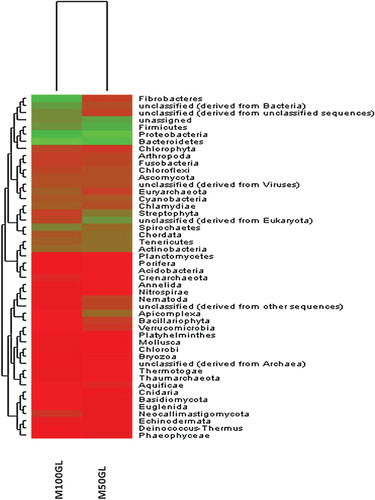
The M5nr database was used to determine community structure via MG-RAST at an e-value cut-off of 1E−5 and 80% identity. Annotation using the M5nr database discovered that Bacteroidetes was the major cellulose degrader, making up 78% of the M100GL population, whereas the major population of M50GL was comprised of Proteobacteria (56%) (). It is of note that the community structure of the M50GL and M100GL groups is made up of these phyla, since their colonization is beneficial to ruminal digestion and nutrition. For example, the phylum Fibrobacter had a relatively high proportion of annotated reads (5%) for M100GL compared to M50GL (0.05%) (). A change in the ruminal colonization by the phylum Firmicutes was also observed in response to the change in feed proportion, where it accounted for 6.17% in the M50GL group and 1.4% in the M100GL group.
Figure 2. Heatmap showing the taxonomic classification of the M50GL and M100GL groups.
Unwinding methanotroph diversity
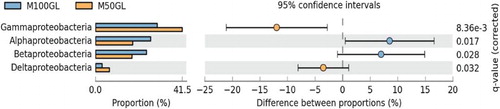
The taxonomic classification up to class level revealed that the abundance of different classes of the same phyla fluctuates in response to the change in feed proportion. For example, a greater abundance of Proteobacteria was observed in M50GL compared to M100GL. To check the difference in abundance of methanotrophs between treatments, a t-test using STAMP was performed. Upon unwinding at class level, α-proteobacteria accounted for significantly (p < 0.05) higher abundance in the M100GL group (67%) than in the M50GL group (43%) (). Similarly, as shown in , the population of β-proteobacteria was found to be significantly (p < 0.05) higher in the M100GL group (62%) than in the M50GL group (42%). The higher fraction of proteobacterial population was covered by γ-proteobacteria which, unlike α-proteobacteria and β -proteobacteria, accounted for significantly (p < 0.05) higher abundance in the M50GL group (100%) than in the M100GL group (74%) ().
Figure 3. Extended error bar chart showing comparative abundance between M50GL and M100GL for different groups of Proteobacteria.
Unwinding methanogen diversity
Of the total assigned domains, the archaeal domain was affiliated to 0.14% of the abundance for M50GL and 0.2% for M100GL. But, at phylum level, the Euryarcheota were found to be 91.6% abundant in the M100GL group compared to 81.8% in the M50GL group. Other phyla such as Thaumarcheota and unclassified sequences derived from archaea were found to be higher in the M50GL group (9.09% of each) than in the M100GL group (4.17%).
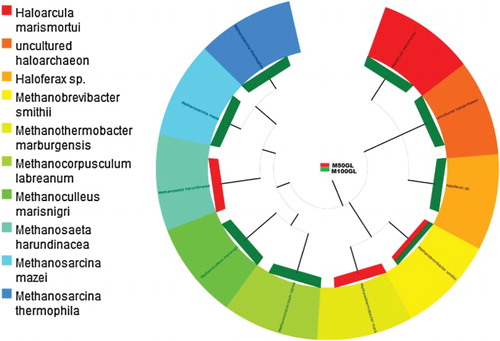
When the Euryarcheota were subdivided into different genera, Methanobrevibacter was found to be common in both groups, with a higher abundance in M100GL (). Methanocorpusculum labreanum was most abundant in M100GL but was lacking in M50GL (). Halobacteria were the least abundant in the M100GL group.
Figure 4. Tree showing species-level classification of phylum Euryarcheota for the M50GL and M100GL groups.
Methane metabolism in rumen of Mehsani buffalo
Carbohydrate metabolism is the major event carried out in all ruminants which reflects their ability to utilize fibrous feedstuffs not readily digested by monogastric animals. The polysaccharide degradation yields CO2 and H2 as the end-products, which are the major substrates of methanogens in the production of methane.
The methane metabolism pathway shown in reflects the different enzymes involved in methane utilization. Oxidation of methane to formaldehyde is carried out by methanotrophs, and further, under aerobic conditions, is incorporated into organic compounds via the serine pathway (Morgavi et al. Citation2010).
Figure 5. KEGG analysis showing enzymes involved in the methane metabolism pathway. The bar chart represents the abundance of enzymes for both M50GL and M100GL groups, where blue represents the M50GL group and red represents the M100GL group. x-Axis = abundance of enzymes; y-axis = E.C. number of enzymes involved in methane metabolism.
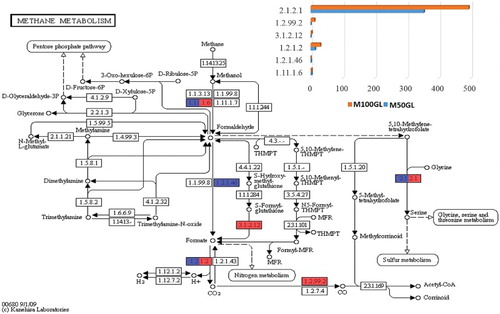
As shown in , under aerobic conditions, methane is oxidized to methanol in both groups, which is further oxidized to formaldehyde and ultimately to formate. Formate thus formed is then simplified to CO2 via the activity of the enzyme formate dehydrogenase (1.2.1.2), the activity of which was higher in the M100GL group compared to the M50GL group. Moreover, 5,10-methylene tetrahydrofolate generated from formaldehyde then enters the serine pathway, where two molecules of formaldehyde and one molecule of CO2 are utilized in each cycle, forming a three-carbon intermediate. In the present study, the glycine hydroxyl methyltransferase enzyme was found to be more abundant in the M100GL group (54%) than in the M50GL group (38%).
Discussion
The microbial community residing in the anaerobic environment of the rumen is extremely diverse. The majority of these microbes have not yet been cultivated. The next generation sequencing technologies have opened up new ways to explore these uncultured microbes for the assessment of the overall ruminal microbial community profile (Leahy et al. Citation2013).
Diet-dependent shifts in the microbial community of the rumen, where a change in the diet leads to an altered microbial community profile, are well established (Varel & Dehority Citation1989; Brulc et al. Citation2009; Fernando et al. Citation2010). The diet also has an effect on methane metabolism in the rumen of animals. Increases in the proportion of concentrate in the diet have been shown to reduce methane production in ruminants (Benchaar et al. Citation2001), whereas animals fed with a higher roughage diet produce large amounts of methane (Mathison et al. Citation1998). Methane emission from the rumen is a balance between the production of methane by methanogenic organisms (methanogenesis) and consumption by methanotrophic organisms. Although the rumen is considered an anaerobic environment, a significant amount of oxygen can enter with feed and water by diffusion across the rumen wall, in the presence of which methane oxidation takes place (Kajikawa et al. Citation2003).
The shotgun sequencing of the rumen metagenome revealed the overall profile of the microbial communities that were abundant at the time of the treatment, and which altered with the change in diet. To estimate the representation of the population, a rarefaction curve was plotted. The curve usually begins with a steep slope, which begins to flatten out as fewer species are discovered per sample (Wooley et al. Citation2010). In this study, all eight metagenomes formed a plateau, which clearly indicates that this much sampling is enough to cover and discover the rarest taxa. The taxonomic classification using the M5nr database revealed that in animals fed a mixed ration (M50GL), the Proteobacteria were abundant. The ruminants fed a diet with a high proportion of grain to hay (60:40, 80:20) were found to harbour more Proteobacteria compared to animals fed with a low grain to hay diet (20:80, 40:60) (Fernando et al. Citation2010). In the case of 100% roughage treatment, the animals harboured more Bacteroidetes, which comprised 78% of the total rumen population. Bacteroidetes are often found to fill an ecological niche in the rumen of animals, where they are increasingly regarded as specialists for high molecular weight organic compound degradation (Thomas et al. Citation2011). The higher proportion of Fibrobacter in animals fed 100% roughage compared to animals fed a mixed ration indicates their major role in fibre degradation in the rumen (Krause et al. Citation1999).
Methanogens, members of the domain Archaea, belong to the kingdom Euryarchaeota. In the rumen, they synthesize methane by contributing to the maintenance of ruminal efficiency, in that they prevent an increase in the partial pressure of H2 to a level that may inhibit normal functioning of the rumen (Wolin et al. Citation1997). A more detailed analysis of the behaviour of the archaeal community revealed that the diet may affect the composition of the methanogen community (Wright et al. Citation2004). Shin et al. (Citation2004) and Tajima et al. (Citation2001) reported the dominance of Methanomicrobium spp. among the total rumen archaea of cows (85.6% and 60.9%, respectively). In the present study, Methanocorpusculum labreanum, of the class Methanomicrobia, was found to comprise 36% of the total archaeal population in the M100GL group. Moreover, Methanobrevibacter spp. were more abundant in M100GL than in M50GL buffalo, and were found to be the dominant archaea (58.8%) in cattle (Whitford et al. Citation2001).
The role of methanotrophs in the rumen can be exploited to reduce methane emissions. Methane is the most stable carbon compound in anaerobic environments, and it escapes from the anaerobic environment to the atmosphere when it is not oxidized by methanotrophs (Hanson & Hanson Citation1996). Aerobic methanotrophic bacteria, a subset of a physiological group of bacteria, are a specialized group of microbes with the ability to utilize methane as a sole carbon and energy source (Matsen et al. Citation2013). Type I methanotrophs are γ –proteobacteria and use the ribulose monophosphate pathway to assimilate carbon, whereas type II methanotrophs are α-proteobacteria and use the serine pathway to assimilate carbon (Oremland & Culbertson Citation1992). In the present study, the higher abundance of Proteobacteria in the M50GL group, which was fed a mixed ration, strongly correlates with the results of a previous report, which found a higher abundance of Proteobacteria and lower abundance of Bacteroidetes following the consumption of a compositionally simple, sucrose-based diet that was low in fibre and devoid of isoflavones (Menon et al. Citation2013). The subdivisions of the phylum Proteobacteria were significantly altered in their abundance with the change in diet. At the phylum level, the Proteobacteria were abundant in the M50GL group, but unwinding the diversity at class level, α-proteobacteria and β -proteobacteria were significantly more abundant in the M100GL group, whereas γ-proteobacteria were significantly higher in the M50GL group compared to the M100GL group. This alteration in the abundance of subdivisions of Proteobacteria reflects their functional activity. In the present study, the methane metabolism pathway indicated that methane oxidation reactions take place through the activity of different enzymes. Methane is oxidized to methanol and formate, which is further oxidized to CO2 by the activity of the enzyme formate dehydrogenase (Ward et al. Citation2004). In the present study, the activity of formate dehydrogenase was found to be higher in the M100GL group. Moreover, the enzyme glycine hydroxyl methyltransferase, which is involved in the serine pathway, was found to be more abundant in the M100GL group than in the M50GL group. The higher abundance of enzymes involved in the serine pathway in M100GL correlates with the higher taxonomic abundance of α-proteobacteria and β-proteobacteria in this group.
Conclusion
Reducing enteric methane emissions has been identified as one way of lowering global methane emissions. Having an understanding of methanogenic and methanotrophic species among ruminant livestock by altering the animals' diet can help in the search for novel strategies to reduce greenhouse gases. In this study, the animals fed on 50% roughage and 50% concentrate harboured fewer methanogens than those fed on 100% roughage, which is the routine diet of Mehsani buffalo. The present study provides a basis for understanding the role of methanogens and methanotrophs with the alteration in diet.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Benchaar C, Pomar C, Chiquette J. 2001. Evaluation of dietary strategies to reduce methane production in ruminants: a modelling approach. Can J Anim Sci. 81:563–574. doi: 10.4141/A00-119
- Brulc JM, Antonopoulos DA, Miller MEB, Wilson MK, Yannarell AC, Dinsdale EA, Edwards RE, Frank ED, Emerson JB, Wacklin P. 2009. Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc Natl Acad Sci U S A. 106:1948–1953. doi: 10.1073/pnas.0806191105
- Chhabra A, Manjunath K, Panigrahy S. 2007. Assessing the role of Indian livestock in climate change. The International Archives of the Photogrammetry. Remote Sensing and Spatial Information Sciences. XXXVIII Part 8:W3.
- Fernando SC, Purvis HT2nd, Najar FZ, Sukharnikov LO, Krehbiel CR, Nagaraja TG, Roe BA, DeSilva U. 2010. Rumen microbial population dynamics during adaptation to a high-grain diet. Appl Environ Microbiol. 76:7482–7490. doi: 10.1128/AEM.00388-10
- Hanson RS, Hanson TE. 1996. Methanotrophic bacteria. Microbiol Rev. 60:439–471.
- Howard B. 1959. Metabolism of carbohydrates by rumen bacteria. Proc Nutr Soc. 18:103–108. doi: 10.1079/PNS19590028
- Janssen PH, Kirs M. 2008. Structure of the archaeal community of the rumen. Appl Environ Microbiol. 74:3619–3625. doi: 10.1128/AEM.02812-07
- Kajikawa H, Valdes C, Hillman K, Wallace RJ, Newbold CJ. 2003. Methane oxidation and its coupled electron-sink reactions in ruminal fluid. Lett Appl Microbiol. 36:354–357. doi: 10.1046/j.1472-765X.2003.01317.x
- Krause DO, McSweeney CS, Forster RJ. 1999. Molecular ecological methods to study fibrolytic ruminal bacteria: phylogeny, competition and persistence. International Symposium on Microbial Ecology. 15–19.
- Leahy SC, Kelly WJ, Altermann E, Ronimus RS, Yeoman CJ, Pacheco DM, Li D, Kong Z, McTavish S, Sang C et al. 2010. The genome sequence of the rumen methanogen methanobrevibacter ruminantium reveals new possibilities for controlling ruminant methane emissions. PLoS One. 5:e8926. doi: 10.1371/journal.pone.0008926
- Leahy SC, Kelly WJ, Ronimus RS, Wedlock N, Altermann E, Attwood GT. 2013. Genome sequencing of rumen bacteria and archaea and its application to methane mitigation strategies. Animal. 7; Suppl 2:235–243. doi: 10.1017/S1751731113000700
- Mathison GW, Okine EK, McAllister TA, Dong Y, Galbraith J, Dmytruk OIN. 1998. Reducing methane emissions from ruminant animals. J Appl Anim Res. 14:1–28. doi: 10.1080/09712119.1998.9706212
- Matsen JB, Yang S, Stein LY, Beck D, Kalyuzhnaya MG. 2013. Global molecular analyses of methane metabolism in methanotrophic alphaproteobacterium, Methylosinus trichosporium ob3b. Part I: transcriptomic study. Front Microbiol. 4:1–16. doi: 10.3389/fmicb.2013.00040
- McAllister T, Cheng K-J, Okine E, Mathison G. 1996. Dietary, environmental and microbiological aspects of methane production in ruminants. Can J Anim Sci. 76:231–243. doi: 10.4141/cjas96-035
- Menon R, Watson SE, Thomas LN, Allred CD, Dabney A, Azcarate-Peril MA, Sturino JM.2013. Diet complexity and estrogen receptor β status affect the composition of the murine intestinal microbiota. Appl Environ Microbiol. 79:5763–5773. doi: 10.1128/AEM.01182-13
- Morgavi D, Forano E, Martin C, Newbold C. 2010. Microbial ecosystem and methanogenesis in ruminants. Animal. 4:1024–1036. doi: 10.1017/S1751731110000546
- Moss AR, Jouany J-P, Newbold J. 2000. Methane production by ruminants: its contribution to global warming. Paris: Institut national de la recherche agronomique, 1960–2000. 231–254.
- Oremland RS, Culbertson CW. 1992. Importance of methane-oxidizing bacteria in the methane budget as revealed by the use of a specific inhibitor. Nature. 356:421–423. doi: 10.1038/356421a0
- Patra AK. 2014. Trends and projected estimates of GHG emissions from Indian livestock in comparisons with GHG emissions from world and developing countries. Asian Australas J Anim Sci. 27:592–599. doi: 10.5713/ajas.2013.13342
- Shin EC, Choi BR, Lim WJ, Hong SY, An CL, Cho KM, Kim YK, An JM, Kang JM, Lee SS . 2004. Phylogenetic analysis of archaea in three fractions of cow rumen based on the 16S rDNA sequence. Anaerobe. 10:313–319. doi: 10.1016/j.anaerobe.2004.08.002
- Tajima K, Nagamine T, Matsui H, Nakamura M, Aminov RI. 2001. Phylogenetic analysis of archaeal 16S rRNA libraries from the rumen suggests the existence of a novel group of archaea not associated with known methanogens. FEMS Microbiol Lett. 200:67–72. doi: 10.1111/j.1574-6968.2001.tb10694.x
- Thomas F, Hehemann JH, Rebuffet E, Czjzek M, Michel G. 2011. Environmental and gut bacteroidetes: the food connection. Front Microbiol. 2:1–16. doi: 10.3389/fmicb.2011.00093
- Varel VH, Dehority BA. 1989. Ruminal cellulolytic bacteria and protozoa from bison, cattle–bison hybrids, and cattle fed three alfalfa–corn diets. Appl Environ Microbiol. 55:148–153.
- Ward N, Larsen Ø, Sakwa J, Bruseth L, Khouri H, Durkin AS, Dimitrov G, Jiang L, Scanlan D, Kang KH. et al. 2004. Genomic insights into methanotrophy: the complete genome sequence of Methylococcus capsulatus (Bath). PLoS Biol. 2:1616–1628.
- Whitelaw FG, Eadie JM, Bruce LA, Shand WJ. 1984. Methane formation in faunated and ciliate-free cattle and its relationship with rumen volatile fatty acid proportions. Br J Nutr. 52:261–275. doi: 10.1079/BJN19840094
- Whitford MF, Teather RM, Forster RJ. 2001. Phylogenetic analysis of methanogens from the bovine rumen. BMC Microbiol. 1:5. doi: 10.1186/1471-2180-1-5
- Wolin M, Miller T, Stewart C. 1997. Microbe–microbe interactions. In: The Rumen Microbial Ecosystem. Editors: Hobson, P. N. Stewart, C. S. Springer. 467–491. doi: 10.1007/978-94-009-1453-7_11
- Wooley JC, Godzik A, Friedberg I. 2010. A primer on metagenomics. PLoS Comput Biol. 6:e1000667. doi: 10.1371/journal.pcbi.1000667
- Wright ADG, Williams AJ, Winder B, Christophersen CT, Rodgers SL, Smith KD. 2004. Molecular diversity of rumen methanogens from sheep in Western Australia. Appl Environ Microbiol. 70:1263–1270. doi: 10.1128/AEM.70.3.1263-1270.2004
- Wu GD, Lewis JD, Hoffmann C, Chen YY, Knight R, Bittinger K, Hwang J, Chen J, Berkowsky R, Nessel L et al. 2010. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol. 10:1–14. doi: 10.1186/1471-2180-10-1