
ABSTRACT
The definition of epigenetics is still under intense debate; however, its concept has evolved since it was originally introduced in 1939 by Conrad Hal Waddington as a way to reconcile antagonistic views between the school of preformationism and the school of epigenesis. The characterization of a large number of phenomena that diverge from the dogmas of classical genetics, and the discovery of the molecular mechanisms through which these phenomena occur, has given rise to a new area of study with important implications for biological sciences. Interactions between the environment and the DNA through modifications on the chromatin are not only responsible for the expression of a normal phenotype, these may be involved in the development of various pathologies. The epigenome, as the bridge between the genome and the phenotype, is no doubt one of the most interesting current ideas in genetics and is so revolutionary that it may change our present notions about inheritance and evolution. In this review, we made a compilation of the most important events in the history of epigenetics, its implications and some perspectives to the future.
Abbreviations: DNA: deoxyribonucleic acid; RNA: ribonucleic acid; DNMT: DNA methyltransferase; MBP: methyl-CpG-binding proteins; HAT: histone acetyltransferase; HDAC: histone deacetylase; SAM: S-adenosyl methionine; ncRNA: non-coding RNA; rRNA: ribosomal RNA; miRNA: microRNA; siRNA: small interfering RNA; piRNA: Piwi-interacting RNA; XiRNA: X-inactivation RNA; lncRNA: long non-coding RNA; GR: glucocorticoid receptor; IGF2: insulin-like growth factor II; HPA: hypothalamic–pituitary–adrenal; TSA: trichostatin A; LINE: long interspersed nuclear elements; LOI: loss of genomic imprinting; MAS: McCune–Albright syndrome; AS: Angelman syndrome; PWS: Prader–Willi syndrome; FDA: Food and Drug Administration; AHEAD: International Human Epigenome Project; HEP: Human Epigenome Project; TMG: thiomethyl-β-D-galactoside
Introduction
The concept of epigenetics has significantly evolved since it was originally proposed more than 70 years ago (Waddington Citation1939). It has gone from being a term infrequently and inadequately used in the literature to being one of the most important areas of study in the post-genomics era and having important implications for the biological sciences. Science has traveled a long road toward a proper definition of epigenetics and of the phenomena that it encompasses. This road began with debates about the nature and localization of the components that drive organismal development and has passed through events such as the development of the chromosome theory of inheritance, the determination of the structural details of the genetic material and the mechanisms that mediate such inheritance and architecture, such as the modifications of the deoxyribonucleic acid (DNA) and histones, and the establishment of the biological activity of small non-coding ribonucleic acid (RNAs) (Jablonka & Lamb Citation2002; Allis et al. Citation2007). However, as will be evident along this review, the journey is not over yet.
The notion that the genome is programmed and regulated by the epigenome doubtless provides one of the most interesting current perspectives in genetics. The currently known molecular mechanisms of epigenetic regulation not only offer a comprehensive explanation for the functioning of biological systems, but they also again place the influence of the environment as a key determinant in the processes of cellular development and differentiation that give rise to a given phenotype (Richards et al. Citation2010; Faulk & Dolinoy Citation2011a). Epigenetics thus challenges the reductionist ideas of Darwinism, Neo-Darwinism and classical genetics by recognizing the possibility of a new system of rapidly emerging transgenerational phenotypic variation (as an alternative to mutation) and by recognizing the fact that under particular situations certain acquired traits could be heritable in organisms in line with Lamarck’s theory of evolution (Haig Citation2007; Rando & Verstrepen Citation2007; Jablonka Citation2013).
Epigenetics is a very extensive field and despite how much this area has evolved, there are not yet consensus about fundamental aspects, including a conceptual delimitation of what may or may not be considered as epigenetic; if it is valid that epigenetics be restricted only to phenomena of eukaryotic organisms; the inheritance of epigenetic changes and the demonstration of the real impact of this heritage in evolution of organisms. All these issues are controversial in such a heterogeneous area and the present work represents our position on several of them in the frame of a historical overview.
From Waddington’s concept and Nanney’s epigenetics to the present
The origin of the word ‘epigenetics’ dates back to 1939, when the Scotch embryologist and geneticist Conrad Hal Waddington established the term as a result of his analysis of the old debate between preformationism and epigenesis (Waddington Citation1939). The preformationist school of thought, which originated in classical Greece and was disseminated widely during the seventeenth century (M. Malpighi, J. Swammerdam, C. de Bonnet, A. von Haller and L. Spallanzani), claimed that the structures of organisms were preformed at a small scale in embryos from the beginning of development. The preformed specimen, known as the homunculus, which originally resided in the egg or in the sperm cell, simply grew after fertilization until it completed its development (Van Speybroeck et al. Citation2002; Wellner Citation2010). This simple and static idea disagreed with the observations made by Aristotle and presented in his book ‘On the generation of animals’, in which he logged the properties, fluids and means of reproduction of various animals, as well as the dynamics of avian embryological development. Aristotle claimed that in the embryo that resulted from a maternal and a paternal contribution, the different organs were formed from an amorphous material as a consequence of gradual changes. He also stated that differentiation and development were continuous processes of high biological complexity that involved various stages that were each dependent on the previous stage and that finally gave rise to the individual (Aristotle Citation2007). The fundamental concepts behind epigenesis were defended, broadened and shaped further by Galeno in his work ‘On the natural faculties’, by William Harvey in ‘Disputations touching the generation of animals’, by John Turberville Needham in ‘Observations on the generation, composition and decomposition of animal and vegetable substances’ and by other researchers between the eighteenth and ninteenth centuries (Van Speybroeck et al. Citation2002).
According to Waddington’s interpretation, preformationism and epigenesis were complementary theories. He tied the causal elements of development (genes) to embryology to devise the term ‘epigenetics’ (Van Speybroeck Citation2002). Waddington wrote in ‘An introduction to modern genetics’ (Citation1939):
… the fertilized egg contains constituents which have definite properties which allow only a certain limited number of reactions to occur; in so far as this is true, one may say that development proceeds on a basis of the ‘preformed’ qualities of the fertilized egg. However, equally it is clear that the interaction of these constituents gives rise to new types of tissue and organ which were not present originally, and in so far development must be considered as ‘epigenetic’ … . (Waddington Citation1939)
Later, he contemplated that genotype and phenotype were linked by a complex set of genetic and non-genetic developmental processes, which he named the ‘epigenotype’, and he proposed a shift from the scheme genotype + environment = phenotype to genotype + epigenotype + environment = specific phenotype (Van Speybroeck Citation2002; Waddington Citation2012). In this way, Waddington proposed the concept of the ‘epigenetic landscape’, in which a cell is ‘canalized’ to reach an expected phenotype by ‘rolling’ down an ‘undulating surface’ whose shape is determined by the interaction among diverse gene networks. In terms of development, once a cell is programmed to reach a specific stage of differentiation by crossing the ‘landscape’, the program cannot be changed to reach a different final stage (Choudhuri Citation2011).
Waddington’s concept of epigenetics essentially refers to cell differentiation, as ‘the process through which the genotype gives rise to the phenotype’. It was not until 1958 that the second important approach about this topic occurred, thus guiding the definition of epigenetics toward its current state. In his essay ‘Epigenetic control systems’, David L. Nanney claimed that the existence of two cell regulatory systems was evident. One system was related to the mechanisms of DNA template transcription (genetic), and a complementary system had different operational principles and was in charge of determining which information is expressed in a particular cell (epigenetic). Nanney was aware of the difficulties in distinguishing the properties of a cell due to changes in the genetic material from those due to other mechanisms and in explaining how the effects of these ‘auxiliary’ processes could persist in the phenotype across many generations (Citation1958).
The evidence that Nanney used to support his ideas was a set of phenomena deemed by the scientific community as ‘bizarre’ (Allis et al. Citation2007). For example, the observations by Muller (Citation1930) in his studies on translocations, inversions and deletions in irradiated Drosophila melanogaster chromosomes suggested that in the absence of any other alterations, the placement of genes within the genome could modify gene expression (eversporting, mottling, mosaic effects) (Muller Citation1930). Years later, Aloha Hannah (Citation1951) confirmed that the variegation effect observed by Muller was due to genes in euchromatic regions were moved to regions under the influence of heterochromatin (juxtaposition of euchromatin and heterochromatin). Around the same time, such phenomena were of interest to Barbara McClintock in maize plants (1945–1956). During her research, McClintock discovered that the movement of the loci Ds and Ac along the genome (transposition) was the cause of phenotypic alterations in maize as mosaic color, depending on the new location of the transposable element by the modification of the action of nearby genes (regulation of gene expression) (McClintock Citation1950; Felsenfeld Citation2014).
Similarly, the first questions had been asked regarding the phenotypic differences among cells that allegedly carried the same genetic material, such as variations in morphology and pigmentation among colonies arising from a pure culture of some bacteria. This phenomenon indicated that the expression of certain traits was not determined solely by the presence of certain genes within the cell. Phenomena such as serotype variation and bacterial phase variation were considered to be of epigenetic origin and induced by the environment. Phase variation is currently known as an inheritable but reversible gene regulatory mechanism that gives rise to phenotypic heterogeneity by inducing different levels of protein synthesis in individual cells of a clonal population (van der Woude Citation2011). Phenotypically, it manifests as the emergence of changes in microbial colony morphology and color and as the expression of pili, fimbriae and other proteins directly linked to pathogenicity (van der Woude Citation2008). Other examples described by Nanney include phenomena in which cell feedback regulation systems can give rise to epigenetic events. The discovery of the permease enzyme of the bacterial lac operon by Cohen, Cohn and Monod in 1957 allowed the demonstration that Escherichia coli was capable of maintaining alternative and inheritable states of induction of the lac operon (Riggs & Porter Citation1996). Cohen and his colleagues found that the concentration of lactose inside of the cell determined the rate of β-galactosidase synthesis. The concentration of lactose in the cytoplasm of E. coli was in turn linked to the activity of another lactose-inducible enzyme known as lactose permease, which is responsible for the uptake of this carbohydrate into the cell (Cohen & Monod Citation1957). Experimental evidence indicated that for the operon to be active, a high concentration of the inducer was needed. However, once induction was attained, the operon could remain functional at very low lactose concentrations or even in the presence of another carbon source, such as glucose. This effect occurred because the quantities of permease synthesized during pre-induction allowed the bacterium to reach sufficient internal inducer concentrations to overcome the inhibitory effect of glucose (Figure ) (Novick & Weiner Citation1957). Alternatively, while testing low inducer concentrations (thiomethyl-β-d-galactoside, TMG), Novick and Weiner (Citation1957) established that the activation of the lac operon occurred over time in a portion of the E. coli culture, as permease and β-galactosidase were produced, until they reached a maximal rate of synthesis. Although activated bacteria passed on the activated state to their progeny during cell division, it was not possible to achieve induction in the entire population of cells in the culture because of the limiting levels of inducer and because induced cells reproduced at a lower rate than uninduced cells (Figure ) (Novick & Weiner Citation1957). This result provided further evidence that genetically identical cells growing in the same environment could give rise to differences that were transmitted across many generations (bistability) (Morange Citation2002).
Figure 1. Scheme of the induction of the β-galactosidase synthesis in E. coli. (a) At high levels of TMG in the medium, cells rapidly reach the maximum rate of synthesis of β-galactosidase (full induction). In few minutes, the concentration of TMG inside the cells is notably higher than in the medium (about 100 times more). (b) At low levels of TMG (top panel) and in the presence of glucose (lower panel), preinduced bacteria maintain a high internal inducer concentration, and the synthesis of β-galactosidase and galactoside permease (in orange) remains in a high rate (there is no inhibition of the induction).
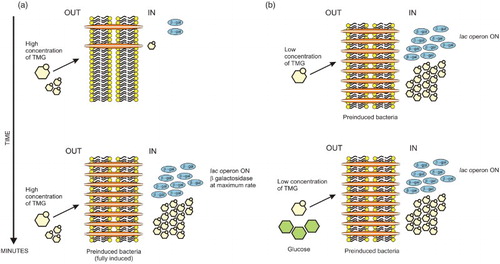
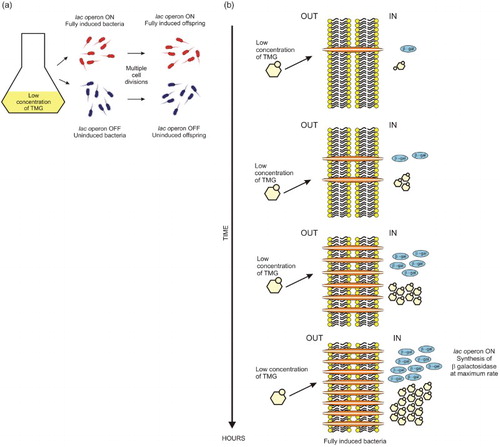
Figure 2. At low concentration of TMG, the induction of the lac operon is presented as an all-or-none phenomenon, a bacterium is fully induced and makes β-galactosidase or is uninduced and makes no β-galactosidase. (a) In the presence of a low concentration of TMG, all progeny of an induced bacterium are induced. Nevertheless, at low concentration of inducer, not all bacteria can be induced. Uninduced cells divide faster than those induced and their progeny remains uninduced, generating two populations of cells coexisting in the same culture. For Novick and Weiner (Citation1957), this phenomenon has a resemblance to mutation and today is considered an example of epigenetic inheritance through generations. (b) Scheme of the bacterial induction at low concentration of TMG. The formation of the first galactoside permease protein is a critical step in TMG accumulation and in the induction process.
Another similar and biologically balanced circuit that gives rise to two epiphenotypes is responsible for the shift from the lysogenic to the lytic phase of replication in the Lambda phage. The Lambda replication cycle is controlled by two mutually repressing proteins encoded by the viral genes cro and cl. During the lysogenic phase, the protein encoded by cl blocks the expression of cro by binding to two of the three operator sequences that regulate the system, thereby keeping the lytic state inactive and promoting its own expression (Satory et al. Citation2011). This state is strongly protected from environmental fluctuations, which allows it to be transmitted across many generations (Cao et al. Citation2010). Repression of cl and the subsequent activation of cro are driven by DNA damage in the host cell and by events that accelerate cl protein degradation to levels below the activation threshold of cro. After being synthesized, the Cro protein binds to the operator sequence responsible for cl silencing, and proteins involved in the phage lytic phase are then synthesized (Riggs & Porter Citation1996; Satory et al. Citation2011).
Work on the mechanisms of genetic regulation of protein synthesis in E. coli and the Lambda phage allowed Jacob and Monod to discuss for the first time the existence of a ‘developmental genetic program’ that encoded instructions for the generation of the molecular structures and phenotype of an organism. Although they did not recognize the epigenetic component of the phenomena described above, they did complement the concepts that had been presented by Waddington (Jacob & Monod Citation1961; Morange Citation2002; Lalucque et al. Citation2010).
Epigenetics in bacteria is still controversial given the genetic mechanisms differences between eukaryotes and prokaryotes; however, it has been shown that both α-Proteobacteria and γ-Proteobacteria have mechanisms that control transcription of specific genes through the formation of effectors (N6-methyl-adenine), which provides ‘additional information’ to the DNA without altering its sequence. That information can be inherited over multiple rounds of cell division and be considered as ‘epigenetic’. Furthermore, other bistable processes through circuits of positive feedback involving self-perpetuating states have been documented for example in the sporulation and motility of Bacillus subtilis (Veening et al. Citation2008; Piggot Citation2010).
The paramutation phenomenon, reported in corn in 1958 by Alexander Brink, was another piece of evidence pointing to the occurrence of epigenetics (Brink Citation1959). Paramutation is the transfer of information from one allele of a gene to another allele at the same locus to establish a state of gene expression that is inherited meiotically but without changes in the DNA sequences of the affected alleles. The exposure of a paramutable allele to a paramutagenic allele gives rise to a change in its phenotypic expression (for instance, a reduction in the intensity of a pigment in corn) by altering the synthesis of RNA; the paramutable allele then becomes paramutagenic itself (Hollick et al. Citation1997). The molecular mechanisms behind this phenomenon, which has been reported in some plants, Drosophila, and mice, remain unclear, although it has been linked to DNA methylation patterns and to the activity of small interfering RNA (siRNA) (Geoghegan & Spencer Citation2013).
By the time that Waddington coined the term ‘epigenetics’, the chromosome theory of inheritance had been demonstrated by the work of Thomas Morgan in D. melanogaster X-linked genes (Citation1911) and A.H. Sturtevant had generated the first genetic map based on recombination frequencies in this organism (Citation1913). Similarly, the change in the meaning of epigenetics that was proposed by Nanney emerged at a time when it was already known that DNA was the molecule that carries the genetic information of organisms (1952) (Avery et al. Citation1944; Hershey & Chase Citation1952) and when the structural details of nucleic acids were available (1953) (Watson & Crick Citation1953; Franklin & Gosling Citation1953a, Citation1953b). Several important discoveries in epigenetics were yet to be achieved, including X-chromosome inactivation in mammals (1961) (Lyon Citation1962), cytoplasmic inheritance in Paramecium (1965) (Beisson & Sonneborn Citation1965), characterization of the structure of nucleosomes (1974) (Kornberg & Thomas Citation1974) and the discovery of prions (self-replicating proteins capable of transmitting information, in analogy to nucleic acids) (1982) (Bolton et al. Citation1982; Prusiner et al. Citation1982; Halfmann & Lindquist Citation2010). Nonetheless, the studies that finally permitted the concepts of epigenetics presented today were performed primarily in eukaryotes and refer to the localization and organization of genetic material in cells and to the covalent modification of DNA and histones. The papers by Allfrey et al. (Citation1964) about histone acetylation and methylation and by Riggs (Citation1975) and Holliday and Pugh (Citation1975) about DNA methylation gave rise to a new stage in the study of epigenetics that has led to the elucidation of its main molecular mechanisms as well as the modern view of epigenetics (Figure ) (Felsenfeld Citation2014). However, the new advances have increased the difficulty of finding a definition covering all phenomena that alter the phenotype and are not dependent from the DNA sequence in organisms, especially when inheritance issue is involved.
Figure 3. Timeline of the principle events in the development of epigenetics. A long journey has been traveled since Waddington coined the concept of epigenetics (for details see the text).
Nowadays, epigenetics is commonly defined as the study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by alterations in the sequence of DNA (Riggs et al. Citation1996). Although for some time there was a relative consensus about this definition an important matter is still not clear, whether epigenetics need to encompass a heritability notion across cell divisions is under debate. As presented below, not all epigenetic modifications are inherited and some of them can only be transient. Additionally, despite the existence of evidence supporting the heritability of some epigenetic changes, mostly in plants and fungi, many researchers consider them poorly understood, uncertain and weak, principally in the case of mammals (Bird Citation2007).
For some scholars, the most practical solution would remove the requirement of heritability included in the definition previously submitted. Nevertheless, new efforts have been made in order to redefine the term epigenetics. For instance, Bird (Citation2007, p. 398) presented epigenetics as ‘the structural adaptation of chromosomal regions so as to register, signal or perpetuate altered activity states’. Deans and Maggert (Citation2015, p. 892) proposed that epigenetics is ‘the study of phenomena and mechanisms that cause chromosome-bound, heritable changes to gene expression that are not dependent on changes to DNA sequence’. Particularly, we do not feel these definitions sufficiently inclusive and by contrast, they are more exclusive than the definition proposed by Riggs et al. (Citation1996). These concepts try to solve the problem of the heritability but they ignore or underestimate phenomena such as feedback circuits, structural or tridimensional conformation inheritance (ciliates and prions), cytoplasmic memory, temporary modifications in chromatin and others, arguing lack of detailed knowledge or evidence. Epigenetics is not only the study of DNA methylation or post-translational histone modifications in eukaryotes. In this point, we feel closest to the proposal made by Mann (Citation2014) about to use an additional term when the epigenetic state is inherited (memigenetics), releasing the term epigenetics from the requirement of heritability to be used in a more general sense and according to their literal meaning (over DNA). In line with the idea of Mann, epigenetics could be considered as the study of both temporal and permanent (inheritable) changes in gene expression not caused by changes in the DNA sequence.
Additionally, we consider that the apparent discrepancies between the concepts of Waddington (Citation1939) and Riggs et al. (Citation1996) lie in the marked differences between the knowledge available in each period, which influenced their points of view; however, these concepts are part of the same phenomenon. Epigenetics actively participates in gene regulation of cells and provides to the genome diversity, using mechanisms that produce transient as permanent modifications in gene function, without alterations in the DNA sequence. These mechanisms are involved in cell differentiation, development and hence in the appearance of phenotype.
The epigenome and molecular mechanisms of epigenetic regulation
Epigenetic phenomena in eukaryotes are linked to the arrangement of genomic DNA into chromatin, a complex and dynamic structure primarily formed by the association between DNA, histones in the nucleosome and non-histone proteins. The nucleosome is the structural and functional unit of chromatin, and it contains 147 bp of DNA wrapped around a central histone octamer composed of two molecules of each of the four core histones H2A, H2B, H3 and H4. The chromatosome is formed by the addition of a fifth histone, histone H1, at the entry site of the DNA. Histones are small proteins that are basic and positively charged, which allows them to closely associate with DNA. Histones possess a globular domain, a fold domain, and tails at their amino and carboxyl ends whose residues undergo post-translational enzymatic modifications. Additional folding of nucleosomes at various levels of compaction allows for the packaging of DNA in the nucleus of the cell (Luger Citation2001; Battistini et al. Citation2010).
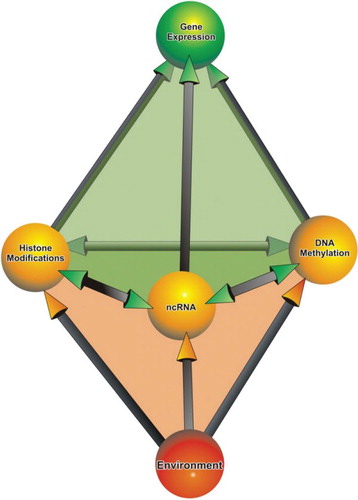
The arrangement of DNA into chromosomes has many purposes. Chromatin is a compact form of genetic material that protects it from external damage. Functionally and structurally, the chromatin is divided into two major types: euchromatin or heterochromatin. Euchromatin is gene rich, transcriptionally active chromatin whose microscopic appearance is uncondensed and is known to replicate early during the S phase. Conversely, heterochromatin is gene poor, mostly transcriptionally inactive and replicates late during the S phase. Microscopically, heterochromatin appears as compact and forms pyknotic aggregates in the nucleus. Only adequately packed DNA is segregated to each of the daughter cells during cell division. As a consequence of compaction and of the subsequent formation of heterochromatin, the accessibility of DNA to the components involved in replication, recombination and transcription is reduced. Therefore, a delicate balance between the mechanisms involved in establishing the heterochromatic and euchromatic states is required for the proper functioning of the cell (Watson et al. Citation2004). The DNA combined with the features of the chromatin, post-translational histone modifications, DNA methylation and nuclear organization, makes up the epigenome (Szyf Citation2009). Histone modifications, DNA methylation and the activity of non-coding RNA (ncRNA), represent some of the major mechanisms of epigenetic regulation in eukaryotes (Figure ) (Choudhuri Citation2011).
Figure 4. Major components of epigenetic regulation in eukaryotes. The interaction between the constituents of the epigenome and the environment controls gene expression in eukaryotic cells.
DNA methylation
DNA methylation was discovered in calf thymus cells by Hotchkiss (1948) (Jurkowska et al. Citation2011), yet it was years later that its link to the repression of gene expression was proposed by Riggs, Holliday and Pugh based on existing reports of adenine methylation in bacteria and the bacterial restriction modification system (Holliday & Pugh Citation1975). DNA methylation is an inheritable symmetrical epigenetic mark that, in high eukaryotes, is found almost exclusively on carbon 5 of cytosine residues (Jurkowska et al. Citation2011). The main targets of methylation are CpG dinucleotides. The heterochromatic regions of the genome (centromeres, transposons, telomeres and repetitive elements) are highly DNA methylated. In contrast, CpG islands, which are regions of at least 550 bp in length with a G + C content above 50% and an observed-to-expected CpG ratio >0.65, generally remain free of methylation. CpG islands are found in approximately 70% of the promoter regions of human genes (Chen & Riggs Citation2011; Jurkowska et al. Citation2011).
In mammals, DNA cytosine methylation is mediated through the action of DNA methyltransferase (DNMT) enzymes, which catalyze the transfer of a methyl group from an S-adenosyl-l-methionine (SAM) cofactor to a cytosine residue (Jurkowska et al. Citation2011). DNMT1, DNMT3a and DNMT3b are perhaps the most important proteins in the maintenance and the establishment of an epigenetic imprint. DNMT1 is associated with chromatin in the S phase of the cell cycle, and hence it is thought to be linked to DNA replication and the preservation of a methylated state. DNMT3a and DNMT3b are the enzymes responsible for establishing tissue-specific cytosine methylation de novo during embryonic development; such methylation is a decisive event in cell differentiation and development (Chen & Riggs Citation2011; Jurkowska et al. Citation2011; Auclair & Weber Citation2012). DNA cytosine methylation is involved in phenomena such as X-chromosome inactivation in female mammals and the establishment of the parental epigenetic imprint, in which one of the two inherited parental alleles of a gene is selectively inactivated (Choudhuri Citation2011).
There are three primary mechanisms through which methylation of the DNA is thought to lead to transcriptional repression: by inhibiting transcription factor binding to regulatory sequences, by recruiting methyl-CpG-binding proteins (MBPs) that block the response elements avoiding the binding of the transcriptional machinery and by forming complexes between MBPs and corepressors that condense chromatin (Attwood et al. Citation2002). Due to its significance, DNA cytosine methylation is considered by many as a fifth, ‘forgotten’ base of DNA.
Methylation can be actively reversed by enzymatic means (dioxygenases) or passively reversed when methylation mechanisms fail after DNA replication (Chen & Riggs Citation2011). The methylcytosine dioxygenase family of proteins TET (Ten-Eleven-Translocation) (TET1, TET2 and TET3) catalyzes the successive oxidation (processive) of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) in eukaryotes. Of these products, it has been determined that 5hmC is the most abundant and it is probable that 5hmC promotes passive DNA demethylation by replication-dependent loss of 5mC, when a cytosine in one DNA strand is oxidized and in the complementary strand is methylated (hemimethylated site). Additionally, new publications reveal that TET enzymes could be part of a pathway involved in the replacement of 5fC and 5caC by unmodified cytosine (TET/thymine-DNA-glycosylase/base excision repair), leading to DNA demethylation. There is not yet enough evidence to support the hypothesis that 5hmC, 5fC and 5caC could be new epigenetic marks (Rasmussen & Helin Citation2016).
Nonetheless, according to recent reports DNA cytosine methylation is not the only important DNA modification in eukaryotes. DNA adenine methylation could be considered soon a new important mark in both lower and higher eukaryotes with probable functions in regulation of gene and transposon expression, like an epigenetic modification. Studies in Caenorhabditis elegans and D. melanogaster have allowed the identification of potential enzymes responsible for DAMT-1 and demethylation (NMAD-1, Dmad), as well as the distribution and sequence motifs for this mark on the genome of these metazoans (Greer et al. Citation2015; Zhang et al. Citation2015). DAMT was previously reported in protists such as Chlamydomonas, Chlorella, Oxytricha, Paramecium, Tetrahymena, even in mosquitoes and plants; however, its role in these organisms need to be more studied (Ratel et al. Citation2006).
Post-translational histone modifications
In 1950, Stedman and Stedman proposed that histones could act as general repressors of gene expression and that each cell type contained within its nucleus a different type of histone; this idea was for them a possible explanation for the diversity of cellular archetypes observed in organisms (Stedman Citation1950). Work conducted by Allfrey et al. (Citation1964), Kornberg and Thomas (Citation1974) and Brownell and Allis (Citation1995), among many others, confirmed that histones played a fundamental role in controlling access to gene promoters by transcription factors and other components involved in RNA synthesis. The control of accessibility occurs primarily through covalent modifications at the amino termini of histones that can recruit ‘binders’ that facilitate transcription, or transcription factors themselves (Allis et al. Citation2007; Izzo & Schneider Citation2010).
According to the histone code hypothesis postulated by Turner (Citation2000) and Strahl and Allis (Citation2000), there is an underlying molecular language established by enzymes – the writers – that generate the different types of histone modifications – the signs –, the protein complexes in charge of recognizing these modifications – the readers –, the molecular effectors recruited to make changes in the conformation of chromatin, and the proteins responsible for reversing those changes – the erasers – (Strahl & Allis Citation2000; Turner Citation2000). Covalent histone modifications are interdependent and have the ability to crosstalk, which can culminate in specific histone landscapes. Thus, two modifications can be mutually exclusive, occur together or one modification can induce the emergence of another (Graff & Mansuy Citation2008). According to Jenuwein and Allis (Citation2001), the ‘histone code’ enables the establishment of different epigenetic states in chromatin and hence different readings of the information contained in DNA; these readings manifest as gene activation or silencing (Figure ) (Jenuwein & Allis Citation2001). Because ‘intercommunication’ between histone modifications and DNA methylation has been demonstrated, some authors have proposed the existence of an ‘epigenetic code’ that is responsible for controlling the expression of the genome (Richards & Elgin Citation2002; Turner Citation2007; Rothbart & Strahl Citation2014).
Figure 5. Chromatin and epigenetic modifications of DNA and histones. The components of epigenetic code and ncRNAs orchestrate the remodeling of chromatin. The delicate balance between heterochromatin and euchromatin is coordinated through the writers and erasers of each epigenetic modification. For instance, DNA methyltransferases and dioxygenases, and HATs and HDACs in the case of DNA and histones, respectively.
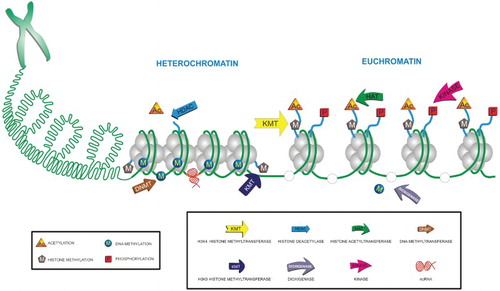
The major covalent histone modifications are acetylation, methylation, phosphorylation, ADP-ribosylation, ubiquitination, SUMOylation, citrullination, glycosylation, hydroxylation and isomerization, although many other modifications have been recently discovered. Although the majority of these post-translational modifications occur at the amino- or carboxy-terminal domains, some can occur at the histone fold or on the globular domain. Acetylation, methylation and phosphorylation are the best-studied modifications in the context of regulation of gene expression, establishment of chromatin structure, replication and DNA repair (Rothbart & Strahl Citation2014). Typically, there are three mechanisms through which epigenetic marks operate over chromatin to control gene expression: a loss of contact between adjacent nucleosomes or between nucleosomes and DNA by inducing a change in charge (cis effect), interference with transcription factor binding due to the coupling of proteins that recognize chromatin marks, and the recruitment of effector protein complexes specific to each type of modification (trans effects) (Gibney & Nolan Citation2010; Izzo & Schneider Citation2010).
Histone acetylation was directly linked to gene transcription after the discovery by Brownell and Allis (Citation1995) of a histone acetyltransferase (HAT) in Tetrahymena thermophila that was homologous to the yeast transcription factor Gcn5. This result, in concert with findings from histone deacetylase (HDAC) inhibitory assays in yeast, the discovery of transcriptional regulatory proteins with intrinsic acetyltransferase activity and the fact that the promoters of active genes are highly acetylated, allowed the role of acetylation as a transcriptional activator to be confirmed (Struhl Citation1998; Gregory et al. Citation2001). Acetylation occurs specifically at the N-epsilon position of lysine residues of the histones through the action of HAT enzymes, which transfer an acetyl group from a donor acetyl-CoA molecule (Choudhuri et al. Citation2010). HATs function as catalytic subunits in protein complexes. In mammals, HATs are classified into two groups, type A and type B, according to their localization within the cell. Type A HAT enzymes are found in the nucleus, where they perform their catalytic activity after nucleosome assembly. Type A HATs are further divided into three subclasses, namely the Gcn5-related N-acetyltransferase (GNAT) and MOZ, Ybf2/Sas3, Sas2, Tip60 (MYST) families and nuclear receptor coactivators, based on their homology to yeast proteins. Type B HAT enzymes are localized to the cytoplasm, and they act on free histones before their entry into the nucleus (Peserico & Simone Citation2011; Zhou et al. Citation2014). Acetylated lysine residues are recognized by proteins that possess evolutionarily conserved regions known as bromodomains. The initial recognition step then permits the recruitment of effector proteins responsible for chromatin decompaction or of proteins from the basal transcriptional machinery. Moreover, acetylation neutralizes the positive charge of histones, lowering their affinity for DNA and thereby altering the configuration of chromatin and generally favoring gene accessibility for transcription (Cruickshank et al. Citation2010).
HDACs are the enzymes responsible for deacetylating lysine residues. They are therefore involved in gene repression in euchromatic regions, but also in promoting heterochromatin formation. These proteins, along with HATs, are responsible for preserving the balance of chromatin acetylation. In mammals, HDACs are classified into two groups, namely classical HDACs and sirtuins (SIR 1–7). Classical HDACs are further divided into four families (classes I, IIa, IIb and IV) based on their homology to yeast proteins. Like HATs, HDACs do not act individually but instead act as a part of large multiprotein complexes that participate in transcriptional regulation pathways (Haberland et al. Citation2009; Chang & Guarente Citation2014).
Histone methylation is a post-translational modification that involves the transfer of a methyl group from an S-adenosyl methionine (SAM) donor to arginine and lysine residues (An Citation2007). Unlike acetylation, methylation does not directly alter the histone structure but instead changes its hydrophobic and steric properties. Histone methylation activates or represses transcription depending on the position of the methylated amino acid (Cruickshank et al. Citation2010; Upadhyay & Cheng Citation2011). In mammals, the enzymes responsible for catalyzing this reaction are known as lysine methyltransferases and protein arginine methyltransferases. In the case of lysine, three possible states of methylation have been described: mono-, di- and trimethylation. Meanwhile, only mono- and dimethylation have been described for arginine, and the latter can be symmetric or asymmetric (An Citation2007; Di Lorenzo & Bedford Citation2011). Enzymes reversing these changes are known as lysine demethylases which belong to two groups, the LSD family and JmjC domain-containing protein family (Liu et al. Citation2015). To date, at least 24 different histone sites that are prone to methylation have been identified, and they are recognized by proteins that possess regions known as chromodomains or Tudor domains. Such proteins include HP1 (heterochromatin protein 1), which is involved in the formation of heterochromatin, and Chd1, which is linked to transcriptional activation (An Citation2007).
Non-coding RNA
ncRNAs are biologically active RNA molecules that are not translated into protein but that participate in the regulation of gene expression (up-regulation and down-regulation), translation, splicing and catalysis in cells. ncRNAs encompass transcripts corresponding to transfer RNA, ribosomal RNA (rRNA), microRNA (miRNA), interfering RNA, siRNA, small nucleolar RNA, Piwi-interacting RNA (piRNA), X-inactivation RNA (XiRNA), long non-coding RNA (lncRNA) and enhancer RNA. However, only miRNA, piRNA, siRNA, lncRNA and XiRNA have been linked to epigenetic phenomena (Table ) (Collins et al. Citation2011). ncRNAs have been reported in viruses, bacteria, unicellular and multicellular eukaryotic organisms (Repoila & Darfeuille Citation2009; Chacko & Lin Citation2013; Tycowski et al. Citation2015). The mechanisms through which ncRNAs participate in the epigenetic regulation of the genome remain unclear, as ncRNAs represent a relatively recent field of study. Although it is known that ncRNA-mediated gene regulation does not alter the primary sequence of DNA, the process through which induced changes are inheritable has not been described. A new study in C. elegans proposes a ‘transgenerational timer’ based in a feedback loop that control the duration of the transgenerational RNAi responses in this organism (Houri-Ze'evi et al. Citation2016). Mutations in the enzymes responsible for ncRNA synthesis, such as Argonaute or Rdp1, have been shown to cause alterations in the formation of heterochromatin (Djupedal & Ekwall Citation2009; Huisinga & Elgin Citation2009). In the case of lncRNA, there is evidence that suggests that these molecules can bind to chromatin-remodeling enzymes and guide them to specific molecular targets (Yamada & Ogawa Citation2015).
Table 1. Characteristics of ncRNA associated to epigenetic phenomena in eukaryotic organisms.
miRNAs are 18–25 nucleotides long, they are present in almost all eukaryotes with some exceptions like Saccharomyces cerevisiae. miRNAs regulate gene expression post-transcriptionally, although their mechanisms of action are under debate. Three no mutually exclusive modes have been proposed: destabilization of mRNA, inhibition of translation initiation and blocking translation after translation initiation (Fabian et al. Citation2010). The genes that encode miRNAs represent approximately 1% of the genome in different species, and it is estimated that approximately 30% of genes are regulated by at least one miRNA. Thus, it follows that miRNAs are involved in important cellular processes, such as differentiation, development, metabolism, cell cycle regulation and aging. Nonetheless, the expression of genes that encode miRNAs is regulated by the aforementioned epigenetic mechanisms, providing evidence that such regulation can be tissue specific. In addition, some miRNAs (epi-miRNAs) are capable of regulating the expression of components of the epigenetic machinery (eg DNMTs and HDACs), giving rise to a tightly controlled feedback loop. When this regulatory circuit is altered, normal biological functions are disrupted (Iorio et al. Citation2010; Sato et al. Citation2011).
piRNAs are 26–32 nucleotides long and have been linked to the transcriptional regulation of de novo DNA methylation in germ cells. piRNAs also control the transcriptional silencing of mobile DNA components and maintain genomic integrity in germ cells. piRNAs were recently found in somatic cells such as neurons, where they may have a key role in memory storage (Landry et al. Citation2013; Weick & Miska Citation2014; Iwasaki et al. Citation2015). siRNA are ncRNAs composed of two RNA complementary molecules with a length of 21–25 nucleotides, homologous to a target gene. siRNAs have been considered defenders of the genome integrity (foreign nucleic acids, transposons) and participate in the organization of chromatin (Fire et al. Citation1998; Carthew & Sontheimer Citation2009). siRNA are involved in the formation of heterochromatin in the centromeres of Schizosaccharomyces pombe. In plants, they are involved in RNA-directed DNA methylation (RdDM) (Lejeune & Allshire Citation2011; Zhang et al. Citation2014).
lncRNA are RNA molecules of more than 200 nucleotides of length with functions in important biological processes as X-inactivation and imprinting in mammals. XiRNA (17 kb) is a lncRNA that is expressed specifically from the inactive X chromosome in females as part of the normal genetic dosage compensation. The function of XiRNA is to recruit chromatin-remodeling complexes (Polycomb) and repressive marks such as H3K9 and H3K27 methylations to the Xi chromosome for its silencing. The activity of XiRNA is necessary and sufficient for the inactivation of the entire chromosome (Ahn & Lee Citation2008). XiRNA is encoded by Xist, this gene is located within the X-inactivation center (Xic), a chromosomal region required for X-inactivation. Similarly, other genes for lncRNAs are part of the Xic: Tsix, Jpx, Ftx, Xite, RepA and Tsx; they regulate Xist expression (Yamada & Ogawa Citation2015). In a recent study, Yildirim et al. (Citation2013) showed that deletion of Xist induces an aggressive and lethal blood cancer in female mice. This result indicates that XiRNA also has an important role in cancer suppression (Yildirim et al. Citation2013).
Still, ncRNAs are not the only factors involved in epigenetic phenomena. The discovery that the FTO (human fat mass and obesity-associated protein) enzyme targets mRNA and lncRNA for m6A modification (adenine methylation) raises the possibility that FTO acts as an ‘eraser’ (demethylase) of the epigenetic code in RNA. m6A is the most prevalent modification in mRNAs and lncRNA in higher eukaryotes. Such modifications also have ‘writers’ (methyltransferases METTL3, METTL14) and ‘readers’ (YTHDF2, hnRNPs), and they are thought to be important in many aspects of human biology (Liu & Pan Citation2015). ALKBH5 is another recently reported demethylase of m6A which participates in mRNA export and metabolism (Zheng et al. Citation2013; Fu et al. Citation2014). Although there are several RNA modifications that have been known for many years (more than 100), few studies have examined their biological functions or the possibility that they are part of an ‘RNA epigenetics’ (Liu & Jia Citation2014).
Implications and perspectives: epigenome, environment and disease
The epigenome can be thought of as the link between the genome and the environment. The cells of a given organism have the same genome; yet, starting from the embryonic stage, different types of cells arise that express different groups of genes, thereby granting them an individual identity (cell differentiation) (Sarkies & Sale Citation2012). The identity of a cell – its phenotype – represents its pattern of gene activation and silencing (Turner Citation2002). The different transcriptional profiles of different cells are not determined by changes in the DNA sequence but instead by the concerted activity of the components of the epigenome. Epigenetics inheritance is defined by Feinberg and Tycko (Citation2004) as cellular information, other than the DNA sequence itself, that is heritable during cell division. In the case of DNA methylation, these changes are preserved over successive cell divisions by the enzyme DNMT1. However, the mechanisms behind the inheritance of changes induced by histone and ncRNA modification remain unclear. It is known that histones carry some covalent modifications after their synthesis and at the point of DNA incorporation that are different from those in pre-existing histones. It has been proposed that the transmission of histone modifications may occur by several mechanisms: by DNA-binding factors and genetic elements (eg cis-acting Polycomb response elements); by dependent processes, such as the transmission of transcription-coupled marks; by interaction with other marks (DNA methylation and histone modifications); by spatiotemporal regulation during replication; by the use of marks on parental histones to facilitate copying of modifications and by enzymatic interactions during DNA replication between nucleosomes that possess old histone dimers or tetramers and nucleosomes that contain new histone dimers or tetramers (Jasencakova & Groth Citation2010; Sarkies & Sale Citation2012).
Transgenerational inheritance
According to the classical theory of evolution, phenotypic variations originate from random mutations that are subsequently conserved or eliminated through the process of natural selection (Rando & Verstrepen Citation2007; Ho Citation2014). Nonetheless, the importance of the environment on development, as presented by Jean-Baptiste Lamarck (1744–1829), who claimed that descendants can inherit traits acquired by the habits of their parents, is more important than originally thought (Faulk & Dolinoy Citation2011a). Lamarck believed that the interactions between individuals and their environments were a key factor in the evolution of species, but his theory was strongly rejected (Ho Citation2014). Currently, it is suggested that environmental events activate specific signaling pathways in cells that, through the mechanisms of epigenetic regulation, favor the stable remodeling of regions of the genome and thus generate heritable transcription profiles. This effect could represent a source of phenotypic variation and evolutionary diversity. The routes of epigenetic transmission include germline transmission, transmission from soma to germline and transmission from soma to soma. The study of non-genomic trait transmission from parents to progeny is known today as transgenerational epigenetics (Zhang et al. Citation2013; Ho Citation2014; Uller Citation2014). One piece of evidence for transgenerational epigenetics in humans was the link between maternal weight and infant weight at birth that was observed in a British cohort of three generations (Hypponen et al. Citation2004; Roseboom & Painter Citation2014). Nevertheless, for some authors an example of true transgenerational epigenetic inheritance has to demonstrate effects even in the fourth generation (F3) (Mitchell et al. Citation2016).
Several studies indicate that during the initial stages of embryo development and after birth, the environment influences the program of the genome, giving rise to changes in gene expression. These expression changes affect behavior, metabolism, immunity and disease development (Szyf Citation2009; Portha et al. Citation2014). According to Faulk and Dolinoy (Citation2011b) , environmental influences can be dietary, physical (maternal care, mistreatment or abuse), chemical (exposure to toxic, xenobiotic, hallucinogenic and pharmacological compounds) or of unknown origin (stochastic behavior). For instance, Lillycrop et al. (Citation2005) evaluated the methylation status and gene expression of the glucocorticoid receptor (GR) and the peroxisomal receptor PPAR-α in the livers of the progeny of rats exposed to protein-restricted diets during pregnancy. The results showed that restricted diets caused a specific decrease in the methylation of certain examined genes and thus an increase in their transcription; this result was deemed as evidence of stable epigenetic modification (Lillycrop et al. Citation2005). Later, Burdge et al. (Citation2007) found that this type of epigenetic alteration, induced in the first filial generation (F1), was transmitted to the second filial generation (F2), even though the diet of the F1 animals during pregnancy was normal.
A similar study was performed by Aagaard-Tillery et al. (Citation2008) in primate fetuses whose mothers were exposed during pregnancy to a control diet or a diet rich in fat. High fat intake led to an increase in triglycerides and the development of a fatty liver in the fetuses. These changes were accompanied by significant hyperacetylation of histone H3 on its lysine 14 residue (H3K14) and by a tendency toward increased H3K9 acetylation and reduced synthesis of the HDAC1 enzyme. Thus, a direct link was made between diet and the establishment of histone modifications. The availability of SAM or acetyl-CoA, which are the donors of the functional groups needed for DNA and histone modification, is dependent on diet. The synthesis of SAM in cells requires folates, vitamin B12, methionine, choline and betaine (Zeisel Citation2009). It has been shown in mice that a diet high in methyl supplements can cause a permanent alteration (hypermethylation) in the expression of the Avy Agouti allele, favoring the emergence of a blonde phenotype and also causing obesity. This alteration, observed in the F1 generation, can be inherited by the F2 generation, demonstrating the epigenetic modification of germline (Wolff et al. Citation1998).
Heijmans et al. (Citation2008) found a reduction in DNA methylation of the IGF2 (insulin-like growth factor II) gene in individuals exposed periconceptionally to the Dutch famine during World War II. The activity of IGF2 is important for development and growth, and this gene is normally regulated by the maternal genomic imprint. This was the first evidence in humans that environmental conditions during early stages of gestation can result in persistent changes in the epigenetic information of individuals (Heijmans et al. Citation2008). A similar study evaluated the DNA methylation levels at cardiovascular and metabolic disease candidate gene loci. In individuals exposed to early prenatal famine, there was an increase in the degree of DNA methylation in the GNASAS and MEG3 genes and a decrease in the methylation of the INSIGF gene promoter, all of which are subject to genomic imprinting. There was also an increase in the methylation of the proximal promoters of the IL10, ABCA1 and LEP genes, which are not linked to the genomic imprint. The link that was uncovered was gender dependent for the INSIGF, GNASAS and LEP loci. Only the methylation of GNASAS and LEP (the latter in males only) was altered in individuals exposed to prenatal famine during late stages of gestation (Tobi et al. Citation2009).
Abuse, neglect, stress and mistreatment during early childhood also lead to epigenetic modifications that can be transmitted to progeny in mammalian species. Francis et al. (Citation1999) showed evidence that differences in maternal care (high or low licking-grooming) are linked to differences in behavior and in the endocrine response to stress in rat progeny. Female progeny of mothers that seldom groomed their pups also recapitulated this behavioral pattern when caring for their own progeny. A corresponding response occurred with the progeny of mothers that properly cared for their pups. These results show the transmission of individual differences in maternal behavior across generations. In addition, such differences gave rise to alterations in progeny of the behavior and responses of the hypothalamic–pituitary–adrenal (HPA) axis to stress. Upon reaching maturity, pups that receive generous care during infancy are less fearful and show a moderate HPA response to stress. This is not the case for pups that undergo precarious care, which show high stress response levels during adulthood (Francis et al. Citation1999). Weaver et al. (Citation2004) showed that maternal care gives rise to stable alterations in the structure of chromatin due to care-dependent differential methylation of specific sites within the GR promoter. When pups were exchanged to another type of mother, they acquired the methylation pattern of their caretaker and the corresponding behavior. The pups that received poor maternal care showed an increase in DNA methylation and a decrease in GR H3K4 acetylation, leading to a reduction in the synthesis of GR and an increase in corticosterone in plasma under stress (Weaver et al. Citation2004). In humans, environmentally induced changes in the components of the epigenome have been linked to late-onset pathologies, including mental illnesses (schizophrenia, depression, bipolar affective disorder, autism and some addictions), immune diseases, metabolic diseases (cardiovascular disease, obesity, diabetes) and cancer (Feinberg & Tycko Citation2004; Chen & Zhang Citation2011). In a study of suicide victims, it was found that those who had suffered from abuse during childhood showed greater total methylation of the promoters of the GR and rRNA genes in the hippocampus compared to the control group (McGowan & Kato Citation2008; Szyf Citation2009). In studies of schizophrenia, evidence has indicated that DNA methylation and chromatin remodeling participate in regulating the expression of reelin, a protein involved in neuronal migration, brain development and synaptic plasticity. The reln gene has a large number of CG dinucleotides in its promoter region. Hypermethylation of its promoter decreases the expression of the reln gene, yet drugs such as trichostatin A (TSA) and valproic acid can activate the gene by inhibiting HDAC activity and increasing histone H3 acetylation, although only TSA prevents the promoter hypermethylation (Graff & Mansuy Citation2008).
Environmental epigenetics
Environmental epigenetics can be considered as the area that studies the effects of environmental exposures on epigenetic states of organisms (Bollati & Baccarelli Citation2010). Environmental pollutants and some chemicals can induce important changes in the epigenome by altering the synthesis of SAM or other processes (Hala et al. Citation2014). Chemicals such as those found in cigarette smoke (acrolein, nicotine and benzopyrene), particulate matter, arsenic, cadmium, aflatoxin B1 and benzene give rise to changes in the methylome and have been linked to cancer and other diseases (Bollati & Baccarelli Citation2010; Pogribny et al. Citation2014).
From the emergence of the area of ‘epigenetic epidemiology’, defined by Waterland and Michels as the study of the associations between epigenetic variation and risk of disease (Waterland & Michels Citation2007); in the last few years it has increased the interest in hypothesis-free, epigenome-wide association studies (EWAS) principally evaluating differential DNA methylation across the genome in different types of exposures, lifestyle factors and pathologies, to understand the epigenetic bases for disease risk in humans (Flanagan Citation2015). For instance, the research of Joubert, Felix, et al. (Citation2016) in which was evaluated the association of maternal smoking during pregnancy, in newborn blood DNA methylation at over 450,000 CpG sites in 13 cohorts of children (6685 newborns). They found more than 6000 CpG sites differentially methylated harboring 2965 CpG sites corresponding to 2017 genes not previously related to this exposure in newborns or adults. Of these genes, 27 have been related to susceptibility to orofacial clefts including BMP4 and BHMT2. PRDM8 was the gene with the largest number of CpG sites associated with the exposition, it belongs to the SET domain family of histone methyltransferases conducting the H3K9 methylation of histones to repress transcriptional activity. The authors also found that the effects of the maternal smoking in newborns can persist into later childhood (Joubert, Felix, et al. Citation2016). In other related work, Rzehak et al. (Citation2016) investigated differential methylation (450 K) associated with smoke exposure beyond the 12th week of gestation in whole blood of 366 children at age 5.5 years. The authors reported association with the genes MYO1G, CNTNAP2 and FRMD4A and these results were consistent with previous studies in individuals in a range of 3–17 years old and newborns (Rzehak et al. Citation2016).
Joubert, den Dekker, et al. (Citation2016) examined the association between maternal plasma folate during pregnancy and epigenome-wide DNA methylation in cord blood of 1988 newborns (450 K). In total, 443 CpG sites were associated with maternal plasma folate and DNA methylation. Some important genes identified were APC2 and GMR8 with known functions in the nervous system, others such as SLC16A12, KLK4 and LHX1 have been implicated in developmental abnormalities different from neural tube defects. The authors also reported association with OPCML and PRPH, genes related to neurologic diseases and CSMD1, a gene previously linked to schizophrenia and autism. These results show a broader view of the role of maternal folate in the epigenome development and disease risk in the offspring (Joubert, den Dekker, et al. Citation2016). Rijlaarsdam et al. (Citation2016) evaluated the association between prenatal exposure to maternal stress and offspring genome-wide cord blood methylation (450 K) in more than 1700 neonates and their mothers. The maternal stress covered aspects as stress due to personal and family circumstances, financial difficulties, psychopathologies and substance abuse. However, this investigation did not find evidence supporting differential DNA methylation associated to the evaluated factors (Rijlaarsdam et al. Citation2016). In another important research, Gruzieva et al. (Citation2016) investigated the effect of maternal exposure during pregnancy to air pollution (NO2), in DNA methylation of cord blood. The authors found epigenome-wide significant associations with LONP1, HIBADH and SLC25A28, genes involved in mitochondria function. The result for SLC25A28 was permanent in older children. They also observed differential methylation in genes CAT and TPO, related to antioxidant defense pathways (Gruzieva et al. Citation2016).
Similar studies with remarkable findings in the investigation of a variety of diseases as type 2 diabetes mellitus (Soriano-Tarraga et al. Citation2016), psoriasis (Zhou et al. Citation2016), schizophrenia (Montano et al. Citation2016), metabolic syndrome/obesity (Ali et al. Citation2016) and many others have been recently published. It is expected that the new technologies that make possible this type of works and the technologies that will come allow epigenetic epidemiology to reach major objectives as the complete description of the epigenetic processes shaping the development of organisms, the interactions gene–epigene and epigene–environment, the establishment of the role of epigenetics in risk and emergence of diseases, as marker of disease or exposure, and the mechanisms of the inheritance of epigenetic changes over generations in humans (Bollati & Baccarelli Citation2010; Bakulski & Fallin Citation2014).
Epigenetics in disease
In cancer, a disease that has been long considered as of genetic origin, the epigenome acquires aberrant states that involves significant changes in the DNA methylation pattern (Sharma et al. Citation2010). As it was mentioned above, CpG islands, particularly those associated with promoters, are not methylated, allowing the access of transcription factors to the DNA (Kanwal & Gupta Citation2012). In some types of cancer, the genome is hypomethylated, but there are specific methylation of CpG islands located in the promoters of pro-apoptotic genes or in promoter genes related to tumor suppressor pathways, which are accompanied by the hypomethylation of oncogenes (Feinberg & Vogelstein Citation1983; Sharma et al. Citation2010; Taby & Issa Citation2010; Kanwal & Gupta Citation2012). In addition, these alterations join to changes in HATs, HMTs, HDMs and HDACs expression. There is an up-regulation of DNMT1 and DNMT3a in colorectal and ovarian cancer, of DNMT3 in breast, hepatocellular and colorectal cancer, and of several MBD proteins in prostate, stomach, colon and lung cancer. Similarly, HDACs remain up-regulated in colon, cervix, endometrium, prostate, breast, gastric and thyroid cancers (Kanwal & Gupta Citation2012). miRNAs have also been associated with the development of malignancies. Numerous miRNA act as oncogenes or tumor suppressor genes and are closely related to cellular processes involved in the formation of neoplasms, such as cell cycle control, DNA damage response, cell differentiation, epithelial to mesenchymal transition and metastasis. The expression of miRNA families, miR-15/16 and let-7, as well as of miRNAs miR-155, miR17-92 and miR-21, among many others, is deregulated in a variety of malignancies in animal models and humans. Approximately half of miRNA genes are associated with CpG islands (Malumbres Citation2013).
Deregulation of epigenetic mechanisms may be the cause of the initial genetic instability that induces the appearance of the numerous mutations in tumor suppressor genes and oncogenes. DNA hypomethylation in regions with abundant repetitive sequences promotes the appearance of rearrangements by genome destabilization. DNA demethylation leads to the activation of transposable elements and endogenous retroviruses, loss of the genomic imprinting and expression of genes in non-coding regions. All of them contribute to the establishment and the progression of the disease (Ting et al. Citation2006; Wilson et al. Citation2007; Sharma et al. Citation2010). In colorectal cancer occurs a reduction in 10–30% in the normal methylation level of the genome and this percentage can be as high as 50% for breast cancer. Hypomethylation can be considered an early even in the generation process of malignant cells in some types of cancer and it may be increased in parallel to the tumor progress (Wilson et al. Citation2007).
Demethylation of repetitive sequences Sat2, Satα and SatR1 is common in breast cancer. Likewise Sat2, Sat3, Satα in the Immunodeficiency syndrome, Centromere instability and Facial syndrome. The last one is an autosomal recessive disease that causes agammaglobulinemia or hypoglobulinemia and rearrangements of the heterochromatic region adjacent to the centromere of chromosome 1 and/or 16, and in some cases, chromosome 9 in mitogen-stimulated lymphocytes (Ehrlich et al. Citation2006; Wilson et al. Citation2007). Hypomethylation of L1 LINE (long interspersed nuclear elements) is found in hepatocellular, bladder, lung, prostate, stomach and esophagus cancers and chronic myeloid leukemia. Demethylation of LINE may be useful as a molecular marker in diagnostic purposes for early detection of cancer or as its prognosis indicator. Methylation status of several genes is related to many types of cancer. Hypomethylation of cytochrome P450 1B1, p-Cadherin, p53, BAGE and maspin genes is related to prostate cancer, invasive breast cancer, lung cancer, ovarian and breast cancer, and colorectal and thyroid cancer, respectively (Wilson et al. Citation2007).
Similarly, hypermethylation of tumor suppressor genes involved in the processes of cell division, DNA repair, adhesion, apoptosis and angiogenesis induces the accumulation of a large number or alterations at different functional levels that contribute to the development of neoplasm (Ting et al. Citation2006; Sharma et al. Citation2010). In genitourinary carcinomas, GSTP1 gene is hypermethylated in 90% of the prostate tumors, whereas in bladder and kidney tumors, it is present only in 10% of the cases. In bladder cancer, CCNA1, MINT1, CCND2, PGP9.5, CRBP and AIM1 genes are differentially methylated in cancer cells compared with healthy cells. These genes and many others, who have been similarly reported for various cancer types, have great potential as potential biomarkers (Brait & Sidransky Citation2011). It is estimated that over 300 genes and gene products are altered in cancer diseases (Kanwal & Gupta Citation2012).
The loss of genomic imprinting (LOI) is the alteration of the normal expression pattern of a gene with specific parental origin. This loss may occur by activation of the silenced copy as in the case of growth-promoting genes (ie IGF2), or by silencing of the active copy, in the case of the growth inhibitory genes (ie p57HIP2). LOI has been reported in hepatoblastoma, rhabdomyosarcoma, colorectal, lung, liver and bladder cancers and leukemias (Feinberg et al. Citation2002). The best known example is the LOI of IGF2-H19 locus in Wilms’ tumor (nephroblastoma), this alteration is also observed in Silver–Russell and Beckwith–Wiedemann syndromes; latter predisposes a variety of cancers. The McCune–Albright, Angelman and Prader–Willi syndromes (MAS, AS and PWS, respectively) exhibit imprinting defects. In AS and PWS, defects can occur due to deletions in the active gene-containing chromosome, the inheritance of both gene copies from the same parent or the presence of mutations in the components responsible for imprinting during gametogenesis (Feinberg et al. Citation2002; Uribe-Lewis et al. Citation2011).
Because DNA and chromatin alterations are reversible, the implementation of epigenetic therapies for the treatment of epigenetically based diseases can be useful. The U.S. Food and Drug Administration (FDA) has approved two such drugs for the treatment of cancer: DNA methylation inhibitors (azacitidine and decitabine) and HDAC inhibitors (vorinostat, sodium phenylbutyrate, etinostat and parabinostat). DNA methylation inhibitors are primarily cytosine analogs that are incorporated into DNA during replication and that capture and inactivate DNMT enzymes in cells undergoing division. Other types of DNMT inhibitors that are not nucleosides or that bind DNMTs directly have been developed. Although azanucleotides have been used in the treatment of various malignancies, they have shown the greatest success in the treatment of hematological neoplasms. HDAC inhibitors target the deacetylation catalytic domain by interfering with substrate recognition and by promoting the retention of acetyl groups at histone tails. Combined administration of a hypomethylating agent and an HDAC inhibitor more effectively inhibits breast cancer growth in cell culture and mouse models. During treatment of myelodysplastic syndrome and acute myelocytic leukemia, this combination promotes the re-expression of genes that had been epigenetically silenced and yields an improved clinical response (Gnyszka et al. Citation2013; Popovic et al. Citation2013; Byler et al. Citation2014).
Perspectives
Much remains to be known and understood about the epigenetic mechanisms by which the epigenome controls and confers diverse expression patterns to the genome. It remains surprising that a single DNA sequence can give rise to more than 400 different cell types in humans. In the search for a full characterization of the workings of the genome, several consortia of researchers, including AHEAD (the International Human Epigenome Project), HEP (the Human Epigenome Project) and the NIH Roadmap Epigenomics Project, are currently working to fully map the epigenetic marks characteristic of each cell type. Elucidation of the normal methylation pattern of gene promoters and of the entire genome (methylome) will allow the generation of a reference profile that will then be useful for identifying potential anomalous epigenomic states, detecting specific diagnosis and prognosis markers for diseases with an epigenetic component and developing new therapeutic strategies for treatment. The goal of deciphering the ‘human epigenetic code’ will only be achieved once the pattern of imprinted marks on histones in chromatin is determined. At present, there is already DNA methylation profiling of human chromosomes 6, 20 and 22, there are the reference epigenomes from 127 tissue and cell types and the integrative analysis of 111 of these epigenomes (Eckhardt et al. Citation2004; Eckhardt et al. Citation2006; Jones et al. Citation2008; Bernstein et al. Citation2010). Nowadays, the scientific community is making efforts for the development and improvement of tools and technologies that facilitate the storage, management and analysis of data obtained from study models. Consequently, the scientific activity, the creation of Biobanks and the development of new compounds for disease treatments with an epigenetic component is favored (Almouzni et al. Citation2014).
Another challenge in the near future will be to elucidate and understand how all of the epigenetic phenomena that have been reviewed in this paper participate in the process of organismal evolution in the contexts of non-genetic inheritance and the modern synthesis (Bonduriansky & Day Citation2009). Part of the picture includes the four epigenetic inheritance systems identified so far, namely inheritance through feedback circuits, structural or tridimensional conformation inheritance (ciliates and prions), DNA and chromatin marks, and inheritance through RNA (Jablonka & Lamb Citation2007; Jablonka & Raz Citation2009). The transmission of epigenetic information is indirect when environmental signals induce a behavioral or physiological change in an organism that is preserved in subsequent generations through a non-meiotic mechanism. Transmission is direct when an environmental influence directly affects the germline cells or when the modification is mediated through the interaction of somatic cells with germline cells and is preserved through meiosis (Duncan et al. Citation2014). Considering that heritable epigenetic variations could affect the processes of adaptation and divergence, Jablonka and Raz (Citation2009) proposed that it is necessary to consider within evolutionary models the changes caused by the selection of epigenetic variants and changes in which an epigenetic modification drives the selection of a genetic variant. The modifications that originate from mechanisms of epigenetic control and that arise from the possibilities and limitations that epigenetic inheritance imposes on development are considered to be equally important to the evolutionary variations that lead to new modes of epigenetic inheritance. Collectively, these considerations would redefine what has been known to date as evolution and inheritance (Jablonka & Raz Citation2009). Notwithstanding, Dickins and Rahman (Citation2012) in their strong and solid defense of the modern synthesis argue that epigenetic inheritance has place in the modern synthesis because epigenetic processes are proximate mechanisms designed to calibrate organisms to stochastic environments and these mechanisms are under genetic control. Epigenetic mechanisms cannot be considered as ultimate causes nor how DNA or genes work, for instance. Changes in the concepts of evolution require population demonstration of the effects of epigenetic inheritance and so far, there is no evidence presented about it. The linking and grooming behaviour in rodents could be an adaptation to a stressful situation in order to make a profit, in this case a faster maturation and reproduction of their puppies (calibration) (Dickins & Rahman Citation2012).
As mentioned above, it has been a long road since Conrad Hal Waddington coined the word ‘epigenetics’, yet the trajectory toward a full understanding of the significance of epigenetics in biology, genetics and organismal evolution remains long. It is clear that much remains to be discovered, described, and understood in a field of study that is relatively new and, one might say, exciting.
Conclusions
Epigenetics is an increasingly important field because it provides a broader outlook of biological systems. Nowadays, many biological phenomena are approached from the epigenetics perspective; however, this area has not been so well known. Despite that the concept of epigenetics firstly appeared in 1939, only in recent years the term has been popularly used in the scientific literature. The idea of epigenetics of C. Waddington related the causal elements of the development and the embryology for explaining that in the development of organisms, both preformed and no preformed elements are involved. After the report of genetically identical cells with different phenotypes, the detailed studies on gene regulation of proteins, the existence of a genetic program of development in organisms, the evidence of no genetic transmission of traits and the molecular mechanisms of DNA organization in the nucleus; the current concept of epigenetics began, but even today, it is still under intense debate.
Currently, we know that in eukaryotes the occurrence of epigenetic phenomena is related to the structure of chromatin and its configurations. DNA is organized into chromatin within the nucleus. Chromatin is a complex with DNA, histone proteins and no histone proteins, which have two functional states, heterochromatin and euchromatin. The nucleosome is the first level of chromatin organization that enables DNA packaging, regulating gene expression and making possible epigenetic phenomena. Chromatin allows different configurations of the same genome, causing various epigenomes and then different phenotypes. Histone modifications, DNA methylation and the activity of nonncRNA, are the major mechanism of epigenetic regulation in eukaryotes.
Epigenome represents the connection between environment and genome. Environmental influences can affect the programming of the genome, activating specific pathways in cells that, through the mechanisms of epigenetic regulation, promote the stable remodeling of chromatin, changing gene expression and phenotype. There is evidence that some of these changes can be inherited to the progeny even when the environmental trigger is over. In humans, environmentally induced modification in the epigenome has also been linked to a variety of pathologies, nevertheless, DNA and chromatin alterations are reversible and the implementation of epigenetic therapies for the treatment of epigenetically based diseases is an area of ongoing research and shows amazing perspectives.
Undoubtedly, a major breakthrough could be expected. Parallel to the report of more reference epigenomes, the development of new technologies, laboratory techniques and tools for management and integration of information obtained, it will be possible to detail the interactions among environment, epigenome and genome and understand the role of these interactions in the inheritance of traits, adaptation and evolution of organisms.
Acknowledgements
The authors thank Maria Elena Torres Padilla for the critical revision of this manuscript and Jorge Eugenio Reynoso Jiménez for his help in figures preparation. NAVS acknowledges support by CONACYT and PIFI-BEIFI-IPN for the scholarships provided.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Aagaard-Tillery KM, Grove K, Bishop J, Ke X, Fu Q, McKnight R, Lane RH. 2008. Developmental origins of disease and determinants of chromatin structure: maternal diet modifies the primate fetal epigenome. J Mol Endocrinol. Aug;41:91–102.
- Ahn JY, Lee JT. 2008. X chromosome: X inactivation. 1. Available from: http://www.nature.com/scitable/topicpage/x-chromosome-x-inactivation-323
- Ali O, Cerjak D, Kent JWJr., James R, Blangero J, Carless MA, Zhang Y. 2016. Methylation of SOCS3 is inversely associated with metabolic syndrome in an epigenome-wide association study of obesity. Epigenetics. Aug;26:1–9.
- Allfrey VG, Faulkner R, Mirsky AE. 1964. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. May;51:786–794. Epub 1964 May 1.
- Allis CD, Jenuwein T, Reinberg D. 2007. Epigenetics. New York: Cold Spring Harbor Laboratory Press.
- Almouzni G, Altucci L, Amati B, Ashley N, Baulcombe D, Beaujean N, Bock C, Bongcam-Rudloff E, Bousquet J, Braun S, et al. 2014. Relationship between genome and epigenome – challenges and requirements for future research. BMC Genomics. 15:487. doi: 10.1186/1471-2164-15-487
- An W. 2007. Histone acetylation and methylation: combinatorial players for transcriptional regulation. Subcell Biochem. 41:351–369.
- Aristotle. 2007. On the generation of animals. Arthur Platt, Translator; Australia: University of Adelaide. Available from: http://ebooks.adelaide.edu.au/a/aristotle/generation/complete.html
- Attwood JT, Yung RL, Richardson BC. 2002. DNA methylation and the regulation of gene transcription. Cell Mol Life Sci. Feb;59:241–257. Epub 2002 March 28.
- Auclair G, Weber M. 2012. Mechanisms of DNA methylation and demethylation in mammals. Biochimie. Nov;94:2202–2211.
- Avery OT, Macleod CM, McCarty M. 1944. Studies on the chemical nature of the substance inducing transformation of pneumococcal types: induction of transformation by a deoxyribonucleic acid fraction isolated from pneumococcus type Iii. J Exper Med. Feb;79:137–158. Epub 1944 Feb 1.
- Bakulski KM, Fallin MD. 2014. Epigenetic epidemiology: promises for public health research. Environ Mol Mutagen. Apr;55:171–183.
- Battistini F, Hunter CA, Gardiner EJ, Packer MJ. 2010. Structural mechanics of DNA wrapping in the nucleosome. J Mol Biol. 396:264–279.
- Beisson J, Sonneborn TM. 1965. Cytoplasmic inheritance of the organization of the cell cortex in Paramecium Aurelia. Proc Natl Acad Sci USA. Feb;53:275–282. Epub 1965 Feb 1.
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. 2010. The NIH roadmap epigenomics mapping consortium. Nat Biotechnol. Oct;28:1045–1048.
- Bird A. 2007. Perceptions of epigenetics. Nature. May;447:396–398.
- Bollati V, Baccarelli A. 2010. Environmental epigenetics. Heredity. Jul;105:105–112.
- Bolton DC, McKinley MP, Prusiner SB. 1982. Identification of a protein that purifies with the scrapie prion. Science. Dec;218:1309–1311.
- Bonduriansky R, Day T. 2009. Nongenetic inheritance and its evolutionary implications. Annu Rev Ecol Evol Syst. 40:103–125.
- Brait M, Sidransky D. 2011. Cancer epigenetics: above and beyond. Toxicol Mech Method. May;21:275–288. Epub 2011/04/19.
- Brink RA. 1959. Paramutation at the R locus in maize plants trisomic for chromosome 10. Proc Natl Acad Sci USA. Jun;45:819–827. Epub 1959 Jun 1.
- Brownell JE, Allis CD. 1995. An activity gel assay detects a single, catalytically active histone acetyltransferase subunit in Tetrahymena macronuclei. Proc Natl Acad Sci USA. Jul;92:6364–6368. Epub 1995/07/03.
- Burdge GC, Slater-Jefferies J, Torrens C, Phillips ES, Hanson MA, Lillycrop KA. 2007. Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. Brit J Nutr. Mar;97:435–439. Epub 2007 Feb 23.
- Byler S, Goldgar S, Heerboth S, Leary M, Housman G, Moulton K, Sarkar S. 2014. Genetic and epigenetic aspects of breast cancer progression and therapy. Anticancer Res. Mar;34:1071–1077.
- Cao Y, Lu HM, Liang J. 2010. Probability landscape of heritable and robust epigenetic state of lysogeny in phage lambda. Proc Natl Acad Sci USA. Oct;107:18445–18450. Epub 2010 Oct 13.
- Carthew RW, Sontheimer EJ. 2009. Origins and mechanisms of miRNAs and siRNAs. Cell. Feb;136:642–655.
- Chacko N, Lin X. 2013. Non-coding RNAs in the development and pathogenesis of eukaryotic microbes. Appl Microbiol Biot. Sep;97:7989–7997.
- Chang HC, Guarente L. 2014. SIRT1 and other sirtuins in metabolism. Trends Endocrin Met. Mar;25:138–145.
- Chen M, Zhang L. 2011. Epigenetic mechanisms in developmental programming of adult disease. Drug Discov Today. Dec;16:1007–1018.
- Chen ZX, Riggs AD. 2011. DNA methylation and demethylation in mammals. J Biol Chem. May;286:18347–18353.
- Choudhuri S. 2011. From Waddington’s epigenetic landscape to small noncoding RNA: some important milestones in the history of epigenetics research. Toxicol Mech Method. May;21:252–274. Epub 2011 Apr 19.
- Choudhuri S, Cui Y, Klaassen CD. 2010. Molecular targets of epigenetic regulation and effectors of environmental influences. Toxicol Appl Pharm. Jun;245:378–393. Epub 2010 Apr 13.
- Cohen GN, Monod J. 1957. Bacterial permeases. Bacteriol Rev. Sep;21:169–194.
- Collins LJ, Schönfeld B, Chen XS. 2011. The epigenetics of non-coding RNA. In: Handbook of epigenetics: the new molecular and medical genetics. San Diego: Elsevier; p. 49–61.
- Cruickshank MN, Besant P, Ulgiati D. 2010. The impact of histone post-translational modifications on developmental gene regulation. Amino Acids. Nov;39:1087–1105.
- Deans C, Maggert KA. 2015. What do you mean, ‘epigenetic’? Genetics. Apr;199:887–896.
- Di Lorenzo A, Bedford MT. 2011. Histone arginine methylation. FEBS Lett. Jul;585:2024–2031. Epub 2010 Nov 16.
- Dickins TE, Rahman Q. 2012. The extended evolutionary synthesis and the role of soft inheritance in evolution. Proc R Soc Biol Sci. Aug;279:2913–2921.
- Djupedal I, Ekwall K. 2009. Epigenetics: heterochromatin meets RNAi. Cell Res. Mar;19:282–295. Epub 2009 Feb 04.
- Duncan EJ, Gluckman PD, Dearden PK. 2014. Epigenetics, plasticity, and evolution: how do we link epigenetic change to phenotype? J Exp Zool Part B. Jun;322:208–220.
- Eckhardt F, Beck S, Gut IG, Berlin K. 2004. Future potential of the human epigenome project. Expert Rev Mol Diagn. 4:609–618.
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, et al. 2006. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. Dec;38:1378–1385.
- Ehrlich M, Jackson K, Weemaes C. 2006. Immunodeficiency, centromeric region instability, facial anomalies syndrome (ICF). Orphanet J Rare Dis. 1:2. Epub 2006 May 26.
- Fabian MR, Sundermeier TR, Sonenberg N. 2010. Understanding how miRNAs post-transcriptionally regulate gene expression. In: Rhoads RE, editor. miRNA regulation of the translational machinery. Berlin: Springer; p. 1–20.
- Faulk C, Dolinoy DC. 2011a. Timing is everything. The when and how of environmentally induced changes in the epigenome of animals. Epigenetics. 6:791–796.
- Faulk C, Dolinoy DC. 2011b. Timing is everything: the when and how of environmentally induced changes in the epigenome of animals. Epigenetics. Jul;6:791–797.
- Feinberg AP, Cui H, Ohlsson R. 2002. DNA methylation and genomic imprinting: insights from cancer into epigenetic mechanisms. Semin Cancer Biol. Oct;12:389–398. Epub 2002 Aug 23.
- Feinberg AP, Tycko B. 2004. The history of cancer epigenetics. Nat Rev Cancer. Feb;4:143–153.
- Feinberg AP, Vogelstein B. 1983. Hypomethylation of ras oncogenes in primary human cancers. Biochem Bioph Res Co. Feb;111:47–54.
- Felsenfeld G. 2014. A brief history of epigenetics. Cold Spring Harb Perspect Biol. Jan;6. doi: 10.1101/cshperspect.a018200
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. 1998. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. Feb;391:806–811.
- Flanagan JM. 2015. Epigenome-wide association studies (EWAS): past, present, and future. Meth Mol Biol. 1238:51–63.
- Francis D, Diorio J, Liu D, Meaney MJ. 1999. Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science. Nov;286:1155–1158.
- Franklin RE, Gosling RG. 1953a. Molecular configuration in sodium thymonucleate. Nature. Apr;171:740–741.
- Franklin RE, Gosling RG. 1953b. The structure of sodiumthymonucleate fibres. I. The influence of water content. Acta Crystallogr 6:673–677.
- Fu Y, Dominissini D, Rechavi G, He C. 2014. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet. May;15:293–306.
- Geoghegan JL, Spencer HG. 2013. The evolutionary potential of paramutation: a population-epigenetic model. Theor Popul Biol. Sep;88:9–19.
- Gibney ER, Nolan CM. 2010. Epigenetics and gene expression. Heredity. Jul;105:4–13. Epub 2010 May 13.
- Gnyszka A, Jastrzebski Z, Flis S. 2013. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. Aug;33:2989–2996.
- Graff J, Mansuy IM. 2008. Epigenetic codes in cognition and behaviour. Behav Brain Res. Sep;192:70–87. Epub 2008/03/21.
- Greer EL, Blanco MA, Gu L, Sendinc E, Liu J, Aristizabal-Corrales D, Hsu CH, Aravind L, He C, Shi Y. 2015. DNA Methylation on N6-Adenine in C. elegans. Cell. May;161:868–878.
- Gregory PD, Wagner K, Horz W. 2001. Histone acetylation and chromatin remodeling. Exp Cell Res. May;265:195–202. Epub 2001 Apr 17.
- Gruzieva O, Xu CJ, Breton CV, Annesi-Maesano I, Anto JM, Auffray C, Ballereau S, Bellander T, Bousquet J, Bustamante M, et al. 2016. Epigenome-Wide meta-analysis of methylation in children related to prenatal NO2 air pollution exposure. Environ Health Persp. Jul. doi: 10.1289/EHP36
- Haberland M, Montgomery RL, Olson EN. 2009. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. Jan;10:32–42.
- Haig D. 2007. Weismann rules! OK? Epigenetics and the Lamarckian temptation. Biol Philos. 22:415–428.
- Hala D, Huggett DB, Burggren WW. 2014. Environmental stressors and the epigenome. Drug Discov Today. 12:3–8.
- Halfmann R, Lindquist S. 2010. Epigenetics in the extreme: prions and the inheritance of environmentally acquired traits. Science. Oct;330:629–632.
- Hannah A. 1951. Localization and function of heterochromatin in Drosophila melanogaster. Adv Genet. 4:87–125.
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. 2008. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA. Nov;105:17046–17049.
- Hershey AD, Chase M. 1952. Independent functions of viral protein and nucleic acid in growth of bacteriophage. J Gen Physiol. May;36:39–56. Epub 1952 May 01.
- Ho DH. 2014. Historical perspective of transgenerational epigenetics. In: Tollefsbol T, editor. Transgenerational epigenetics. Burlington: Elsevier; p. 17–23.
- Hollick JB, Dorweiler JE, Chandler VL. 1997. Paramutation and related allelic interactions. Trends Genet. Aug;13:302–308. Epub 1997 Aug 1.
- Holliday R, Pugh JE. 1975. DNA modification mechanisms and gene activity during development. Science. Jan;187:226–232. Epub 1975 Jan 24.
- Houri-Ze'evi L, Korem Y, Sheftel H, Faigenbloom L, Toker IA, Dagan Y, Awad L, Degani L, Alon U, Rechavi O. 2016. A tunable mechanism determines the duration of the transgenerational small RNA inheritance in C. elegans. Cell. Mar;165:88–99.
- Huisinga KL, Elgin SC. 2009. Small RNA-directed heterochromatin formation in the context of development: what flies might learn from fission yeast. BBA. Jan;1789:3–16. Epub 2008 Sep 16.
- Hypponen E, Power C, Smith GD. 2004. Parental growth at different life stages and offspring birthweight: an intergenerational cohort study. Paediatr Perinat Epidemiol. May;18:168–177.
- Iorio MV, Piovan C, Croce CM. 2010. Interplay between microRNAs and the epigenetic machinery: an intricate network. BBA. Oct–Dec;1799:694–701. Epub 2010 May 25.
- Iwasaki YW, Siomi MC, Siomi H. 2015. PIWI-interacting RNA: its biogenesis and functions. Annu Rev Biochem. 84:405–433.
- Izzo A, Schneider R. 2010. Chatting histone modifications in mammals. Brief Funct Genomics. Dec;9:429–443.
- Jablonka E. 2013. Epigenetic inheritance and plasticity: the responsive germline. Prog Biophys Mol Biol. Apr;111:99–107.
- Jablonka E, Lamb MJ. 2002. The changing concept of epigenetics. Ann NY Acad Sci. Dec;981:82–96.
- Jablonka E, Lamb MJ. 2007. Precis of evolution in four dimensions. Behav Brain Sci. Aug;30:353–365; discussion 365–389. Epub 2007 Dec 18.
- Jablonka E, Raz G. 2009. Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Q Rev Biol. Jun;84:131–176. Epub 2009 Jul 18.
- Jacob F, Monod J. 1961. Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol. Jun;3:318–356.
- Jasencakova Z, Groth A. 2010. Restoring chromatin after replication: how new and old histone marks come together. Semin Cell Dev Biol. Apr;21:231–237.
- Jenuwein T, Allis CD. 2001. Translating the histone code. Science. Aug;293:1074–1080. Epub 2001 Aug 11.
- Jones PA, Archer TK, Baylin SB, Beck S, Berger S, Bernstein BE, Carpten JD, Clark SJ, Costello JF, Doerge RW, et al. 2008. Moving AHEAD with an International Human Epigenome Project. Nature. 454:711–715.
- Joubert BR, den Dekker HT, Felix JF, Bohlin J, Ligthart S, Beckett E, Tiemeier H, van Meurs JB, Uitterlinden AG, Hofman A, et al. 2016. Maternal plasma folate impacts differential DNA methylation in an epigenome-wide meta-analysis of newborns. Nat Commun. 7:10577. doi: 10.1038/ncomms10577
- Joubert BR, Felix JF, Yousefi P, Bakulski KM, Just AC, Breton C, Reese SE, Markunas CA, Richmond RC, Xu CJ, et al. 2016. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am J Hum Genet. Apr;98:680–696.
- Jurkowska RZ, Jurkowski TP, Jeltsch A. 2011. Structure and function of mammalian DNA methyltransferases. Chembiochem. Jan;12:206–222.
- Kanwal R, Gupta S. 2012. Epigenetic modifications in cancer. Clin Genet. Apr;81:303–311. Epub 2011 Nov 16.
- Kornberg RD, Thomas JO. 1974. Chromatin structure; oligomers of the histones. Science. May;184:865–868.
- Lalucque H, Malagnac F, Silar P. 2010. Prions and prion-like phenomena in epigenetic inheritance. In: Tollefsbol T, editor. Handbook of epigenetics: The new molecular and medical genetics. San Diego: Elsevier; p. 63–76.
- Landry CD, Kandel ER, Rajasethupathy P. 2013. New mechanisms in memory storage: piRNAs and epigenetics. Trends Neurosci. Sep;36:535–542.
- Lejeune E, Allshire RC. 2011. Common ground: small RNA programming and chromatin modifications. Curr Opin Cell Biol. Jun;23:258–265.
- Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. 2005. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J Nutr. Jun;135:1382–1386. Epub 2005 Jun 3.
- Liu J, Jia G. 2014. Methylation modifications in eukaryotic messenger RNA. J Genet Genomics. Jan;41:21–33.
- Liu K, Liu Y, Lau JL, Min J. 2015. Epigenetic targets and drug discovery Part 2: Histone demethylation and DNA methylation. Pharmacol Ther. Jul;151:121–140.
- Liu N, Pan T. 2015. RNA epigenetics. Transl Res. Jan;165:28–35.
- Luger K. 2001. Nucleosomes: structure and function. In: Finazzi-Agró A, editor. Encyclopedia of life sciences. Wiley-Blackwell; p. 1–8.
- Lyon MF. 1962. Sex chromatin and gene action in the mammalian X-chromosome. Am J Hum Genet. Jun;14:135–148. Epub 1962 Jun 1.
- Malumbres M. 2013. miRNAs and cancer: an epigenetics view. Mol Aspects Med. Jul–Aug;34:863–874.
- Mann JR. 2014. Epigenetics and memigenetics. Cell Mol Life Sci. Apr;71:1117–1122.
- McClintock B. 1950. The origin and behavior of mutable loci in maize. Proc Natl Acad Sci USA. Jun;36:344–355.
- McGowan PO, Kato T. 2008. Epigenetics in mood disorders. Environ Health Prev Med. Jan;13:16–24. Epub 2008 Jan 01.
- Mitchell C, Schneper LM, Notterman DA. 2016. DNA methylation, early life environment, and health outcomes. Pediatr Res. Jan;79:212–219.
- Montano C, Taub MA, Jaffe A, Briem E, Feinberg JI, Trygvadottir R, Idrizi A, Runarsson A, Berndsen B, Gur RC, et al. 2016. Association of DNA methylation differences with schizophrenia in an epigenome-wide association study. JAMA. May;73:506–514.
- Morange M. 2002. The relations between genetics and epigenetics: a historical point of view. Ann NY Acad Sci. Dec;981:50–60. Epub 2003 Jan 28.
- Morgan TH. 1911. An attempt to analyze the constitution of the chromosomes on the basis of sex-limited inheritance in Drosophila. J Exp Zool. 11:365–413.
- Muller HG. 1930. Types of visible variations induced by X-rays in Drosophila. J Gen. XXII:299–337.
- Nanney DL. 1958. Epigenetic control systems. Proc Natl Acad Sci USA. Jul;44:712–717. Epub 1958 Jul 15.
- Novick A, Weiner M. 1957. Enzyme induction as an all-or-none phenomenon. Proc Natl Acad Sci USA. Jul 15;43:553–566. Epub 1957 Jul 15.
- Peserico A, Simone C. 2011. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J Biomed Biotechnol. 2011:1–10, Article no. 371832. doi:10.1155/2011/371832
- Piggot P. 2010. Epigenetic switching: bacteria hedge bets about staying or moving. Curr Biol. Jun;20:R480–482.
- Pogribny IP, Kobets T, Beland FA. 2014. Alterations in DNA methylation resulting from exposure to chemical carcinogens. In: Cooper DN, editor. Encyclopedia of life sciences. Wiley; p. 1–8. doi: 10.1002/9780470015902.a0025456
- Popovic R, Shah MY, Licht JD. 2013. Epigenetic therapy of hematological malignancies: where are we now? Therap Adv Hematol. Apr;4:81–91.
- Portha B, Fournier A, Kioon MD, Mezger V, Movassat J. 2014. Early environmental factors, alteration of epigenetic marks and metabolic disease susceptibility. Biochimie. Feb;97:1–15.
- Prusiner SB, Bolton DC, Groth DF, Bowman KA, Cochran SP, McKinley MP. 1982. Further purification and characterization of scrapie prions. Biochemistry. Dec;21:6942–6950.
- Rando OJ, Verstrepen KJ. 2007. Timescales of genetic and epigenetic inheritance. Cell. Feb;128:655–668. Epub 2007 Feb /27.
- Rasmussen KD, Helin K. 2016. Role of TET enzymes in DNA methylation, development, and cancer. Gene Dev. Apr;30:733–750.
- Ratel D, Ravanat JL, Berger F, Wion D. 2006. N6-methyladenine: the other methylated base of DNA. BioEssays. Mar;28:309–315.
- Repoila F, Darfeuille F. 2009. Small regulatory non-coding RNAs in bacteria: physiology and mechanistic aspects. Biol cell. Feb;101:117–131.
- Richards CL, Bossdorf O, Pigliucci M. 2010. What role does heritable epigenetic variation play in phenotypic evolution? BioScience. 60:232–237.
- Richards EJ, Elgin SC. 2002. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell. Feb;108:489–500. Epub 2002 Mar 23.
- Riggs AD. 1975. X inactivation, differentiation, and DNA methylation. Cytogene Cell Gene. 14:9–25. Epub 1975 Jan 1.
- Riggs AD, Martienssen RA, Russo VEA. 1996. Introduction. In: Russo VEA, Martienssen R, Riggs AD, editors. Epigenetic mechanisms of gene regulation. New York: Cold Spring Harbor Laboratory Press; p. 1–4.
- Riggs AD, Porter TN. 1996. Overview of epigenetic mechanisms. In: Russo VEA, Martienssen R, Riggs AD, editors. Epigenetic mechanisms of gene regulation. New York: Cold Spring Harbor Laboratory Press; p. 29–45.
- Rijlaarsdam J, Pappa I, Walton E, Bakermans-Kranenburg MJ, Mileva-Seitz VR, Rippe RC, Roza SJ, Jaddoe VW, Verhulst FC, Felix JF, et al. 2016. An epigenome-wide association meta-analysis of prenatal maternal stress in neonates: a model approach for replication. Epigenetics. 11:140–149.
- Roseboom PH, Painter R. 2014. Transgenerational impact of nutrition on disease risk. In: Cooper DN, editor. Encyclopedia of life sciences. Wiley; p. 1–7.
- Rothbart SB, Strahl BD. 2014. Interpreting the language of histone and DNA modifications. BBA. Mar;12:627–643.
- Rzehak P, Saffery R, Reischl E, Covic M, Wahl S, Grote V, Xhonneux A, Langhendries JP, Ferre N, Closa-Monasterolo R, et al. 2016. Maternal smoking during pregnancy and DNA-methylation in children at age 5.5 years: epigenome-wide-analysis in the European childhood obesity project (CHOP)-study. PLoS one. 11:1–18, Article no. e0155554. doi: 10.1371/journal.pone.0155554
- Sarkies P, Sale JE. 2012. Cellular epigenetic stability and cancer. Trends Genet. Mar;28:118–127. Epub 2012 Jan 10.
- Sato F, Tsuchiya S, Meltzer SJ, Shimizu K. 2011. MicroRNAs and epigenetics. FEBS J. May;278:1598–1609. Epub 2011 Mar 15.
- Satory D, Gordon AJ, Halliday JA, Herman C. 2011. Epigenetic switches: can infidelity govern fate in microbes? Curr Opin Microbiol. Apr;14:212–217. Epub 2011 Apr 19.
- Sharma S, Kelly TK, Jones PA. 2010. Epigenetics in cancer. Carcinogenesis. Jan;31:27–36. Epub 2009 Sep 16.
- Soriano-Tarraga C, Jimenez-Conde J, Giralt-Steinhauer E, Mola-Caminal M, Vivanco-Hidalgo RM, Ois A, Rodriguez-Campello A, Cuadrado-Godia E, Sayols-Baixeras S, Elosua R, et al. 2016. Epigenome-wide association study identifies TXNIP gene associated with type 2 diabetes mellitus and sustained hyperglycemia. Hum Mol Genet. Feb;25:609–619.
- Stedman E. 1950. Cell specificity of histones. Nature. Nov;166:780–781.
- Strahl BD, Allis CD. 2000. The language of covalent histone modifications. Nature. Jan;403:41–45.
- Struhl K. 1998. Histone acetylation and transcriptional regulatory mechanisms. Gene Dev. Mar;12:599–606. Epub 1998 Apr 16.
- Sturtevant AH. 1913. A third group of linked genes in Drosophila ampelophila. Science. Jun;37:990–992.
- Szyf M. 2009. The early life environment and the epigenome. BBA. Sep;1790:878–885. Epub 2009 Apr 15.
- Taby R, Issa JP. 2010. Cancer epigenetics. CA-Cancer J Clin. Nov–Dec;60:376–392. Epub 2010 Oct 21.
- Ting AH, McGarvey KM, Baylin SB. 2006. The cancer epigenome--components and functional correlates. Gene Dev. Dec;20:3215–3231. Epub 2006 Dec 13.
- Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. 2009. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. Nov;18:4046–4053.
- Turner BM. 2000. Histone acetylation and an epigenetic code. BioEssays. Sep;22:836–845.
- Turner BM. 2002. Cellular memory and the histone code. Cell. Nov;111:285–291. Epub 2002 Nov 7.
- Turner BM. 2007. Defining an epigenetic code. Nat Cell Biol. Jan;9:2–6. Epub 2007 Jan 3.
- Tycowski KT, Guo YE, Lee N, Moss WN, Vallery TK, Xie M, Steitz JA. 2015. Viral noncoding RNAs: more surprises. Gene Dev. Mar;29:567–584.
- Uller T. 2014. Evolutionary perspectives on transgenerational epigenetics. In: Tollefsbol T, editor. Transgenerational epigenetics. Burlington: Elsevier; p. 175–185.
- Upadhyay AK, Cheng X. 2011. Dynamics of histone lysine methylation: structures of methyl writers and erasers. Prog Drug Res/Fortschritte der Arzneimittelforschung/Progres des recherches pharmaceutiques. 67:107–124. Epub 2010 Dec 15.
- Uribe-Lewis S, Woodfine K, Stojic L, Murrell A. 2011. Molecular mechanisms of genomic imprinting and clinical implications for cancer. Expert Rev Mol Med. 13:1–22, Article no. e2. doi: 10.1017/S1462399410001717
- van der Woude MW. 2008. Some types of bacterial phase variation are epigenetic. Microbe. 3:21–26.
- van der Woude MW. 2011. Phase variation: how to create and coordinate population diversity. Curr Opin Microbiol. Apr;14:205–211. Epub 2011 Feb 05.
- Van Speybroeck L. 2002. From epigenesis to epigenetics: the case of C. H. Waddington. Ann NY Acad Sci. Dec;981:61–81. Epub 2003 Jan 28.
- Van Speybroeck L, De Waele D, Van de Vijver G. 2002. Theories in early embryology: close connections between epigenesis, preformationism, and self-organization. Ann NY Acad Sci. Dec;981:7–49. Epub 2003 Jan 28.
- Veening JW, Smits WK, Kuipers OP. 2008. Bistability, epigenetics, and bet-hedging in bacteria. Annu Rev Microbiol. 62:193–210.
- Waddington CH. 1939. An introduction to modern genetics. New York: The MacMillan Company.
- Waddington CH. 2012. The epigenotype. Int J Epidemiol. Feb;41:10–13. Epub 2011 Dec 22.
- Waterland RA, Michels KB. 2007. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr. 27:363–388.
- Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick R. 2004. Molecular biology of the gene. 5th ed. San Francisco: Pearson/Benjamin Cummings.
- Watson JD, Crick FH. 1953. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. Apr;171:737–738. Epub 1953 Apr 25.
- Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. 2004. Epigenetic programming by maternal behavior. Nat Neurosci. Aug;7:847–854.
- Weick EM, Miska EA. 2014. piRNAs: from biogenesis to function. Development. Sep;141:3458–3471.
- Wellner K. 2010. “A History of Embryology (1959), by Joseph Needham”. Embryo Project Encyclopedia. http://embryo.asu.edu/handle/10776/2031
- Wilson AS, Power BE, Molloy PL. 2007. DNA hypomethylation and human diseases. BBA. Jan;1775:138–162. Epub 2006 Oct 19.
- Wolff GL, Kodell RL, Moore SR, Cooney CA. 1998. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. Aug;12:949–957. Epub 1998 Aug 26.
- Yamada N, Ogawa Y. 2015. Mechanisms of long noncoding Xist RNA-mediated chromosome-wide gene silencing in X-Chromosome inactivation. In: Kurokawa R, editor. Long noncoding RNAs: structures and functions. Tokyo: Springer; p. 151–171.
- Yildirim E, Kirby JE, Brown DE, Mercier FE, Sadreyev RI, Scadden DT, Lee JT. 2013. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell. Feb;152:727–742.
- Zeisel SH. 2009. Epigenetic mechanisms for nutrition determinants of later health outcomes. Am J Clin Nutr. May;89:1488S–1493S. Epub 2009 Mar 6.
- Zhang G, Huang H, Liu D, Cheng Y, Liu X, Zhang W, Yin R, Zhang D, Zhang P, Liu J, et al. 2015. N6-methyladenine DNA modification in Drosophila. Cell. May;161:893–906.
- Zhang M, Xie S, Dong X, Zhao X, Zeng B, Chen J, Li H, Yang W, Zhao H, Wang G, et al. 2014. Genome-wide high resolution parental-specific DNA and histone methylation maps uncover patterns of imprinting regulation in maize. Genome Res. Jan;24:167–176.
- Zhang TY, Caldji C, Diorio JC, Dhir S, Turecki G, Meaney MJ. 2013. The epigenetics of parental effects. In: Sweatt JD, Meaney MJ, Nestler EJ, Akbarian S, editors. Epigenetic regulation in the nervous system: basic mechanisms and clinical impact. London: Elsevier; p. 85–118.
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, et al. 2013. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. Jan;49:18–29.
- Zhou F, Wang W, Shen C, Li H, Zuo X, Zheng X, Yue M, Zhang C, Yu L, Chen M, et al. 2016. Epigenome-wide association analysis identified nine skin DNA methylation loci for psoriasis. J Invest Dermatol. Apr;136:779–787.
- Zhou Y, Peng J, Jiang S. 2014. Role of histone acetyltransferases and histone deacetylases in adipocyte differentiation and adipogenesis. Eur J Cell Biol. Mar;25:170–177.