
Abstract
Objective: Infection with hepatitis C virus is the leading indication for liver transplantation and most common cause of infectious disease-related mortality in the United States. BZF961 is a novel inhibitor of the hepatitis C virus NS3-4A protease.
Methods: This sequential, three part exploratory first-in-human study investigated the safety and pharmacokinetics of single and multiple ascending oral doses of BZF961 in healthy subjects. The first two parts were randomized, double-blind, placebo-controlled, time-lagged, single and multiple ascending oral dose segments. The third part analyzed the effect of ritonavir on BZF961 pharmacokinetics.
Results: BZF961 was generally safe and well-tolerated in single and multiple oral doses in healthy subjects. There were no deaths and no serious adverse events. The most common adverse events were nausea and other gastrointestinal symptoms. Co-administration of ritonavir with BZF961 was well tolerated and increased BZF961 exposure by up to 60-fold, as well as reduced the overall exposure variability.
Conclusions: BZF961 was generally safe and well-tolerated and its exposure was boosted by the co-administration of ritonavir.
Introduction
Hepatitis C virus (HCV) infection is a leading cause of chronic liver disease, cirrhosis, hepatocellular carcinoma, and liver transplantation [Citation1–4]. Most recent estimates suggest over 70 million people worldwide are infected with HCV [Citation5], with as few as 3% receiving treatment as recently as 2012 [Citation6]. HCV is classified into seven genotypes (GT), with GT 1 being the most common, followed by GT 3 [Citation7]; viral GT can be an important factor in response to the treatment [Citation8–10]. Until recently, all HCV treatment was interferon-based, with suboptimal response rates and significant side effects [Citation6,Citation11]. The first HCV direct acting antivirals were two NS3-4A protease inhibitors, telaprevir and boceprevir, which were introduced in 2011. These drugs resulted in improved outcomes, but had treatment complexity and tolerability challenges [Citation12]. Recently, additional NS3-4A inhibitors (simeprevir, paritaprevir, and grazoprevir) have become available for combination use with the NS5B inhibitor sofosbuvir or with NS5A inhibitors. Current combination regimens with or without new generation NS3-4A inhibitors have excellent efficacy and tolerability and do not require the co-administration of pegylated interferon. Expanding access to effective treatments is a strategic component of the WHO goal to eliminate the public health impact of viral hepatitis by 2030 [Citation13].
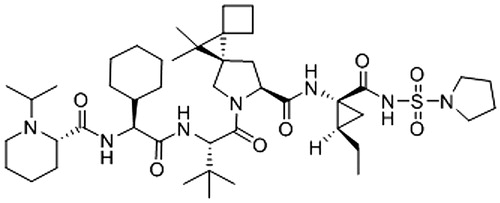
BZF961 () is a novel NS3-4A HCV inhibitor. Based on preclinical virology data, it is expected that BZF961 will be clinically active against HCV GT 1, 2, and 4, as well as against the majority of HCV strains with mutations that confer resistance to first generation NS3-4A inhibitors. In biochemical protease assays, BZF961 demonstrated IC50 values between 4 and 28 nM for GT 1, 2, and 4 and 290 nM for GT 3. BZF961 also showed potent activity in cellular replicon assays for GT 1a and 1 b, with a modest (up to 10-fold) decrease in activity in the presence of human serum. BZF961 is predominantly metabolized by CYP3A4 in vitro and in mice. It is expected that the co-administration of a potent CYP3A4 inhibitor would boost the exposure of BZF961.
Figure 1. Structure of BZF961.
Here we describe the results from the first study of BZF961 in humans, including safety and pharmacokinetics of single and multiple ascending doses, and the effect of ritonavir on BZF961 exposure.
Materials and methods
Study design
This was a single center, three part, placebo-controlled, randomized, double-blind, first-in-human study to determine the safety and tolerability of BZF961 in healthy subjects (). It was conducted from 11 April 2012 to 8 June 2013 at the Celerion Phase 1 unit in Lincoln, Nebraska.
Figure 2. Study design.
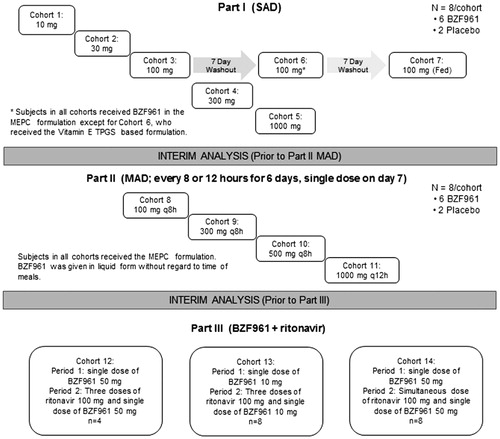
Part 1 was a single-ascending dose study (SAD) of escalating doses of BZF961 or placebo (vehicle only) administered to five cohorts of eight subjects each (randomized BZF961:placebo; 6:2) (Cohorts 1–5, 10 mg, 30 mg, 100 mg, 300 mg, and 1000 mg). The formulation for BZF961 in these cohorts was microemulsion pre-concentrate (MEPC). The 100 mg cohort of eight subjects (Cohort 3) received a second dose of BZF961 100 mg or placebo in an alternative formulation (Vitamin E TPGS-based, designated Cohort 6), and a third dose of BZF961 100 mg following a standard FDA breakfast [Citation14] (termed Cohort 7). The study consisted of a maximum 21-day screening period, a baseline period (Day 1), treatment period (Days 1 to 4), and a single study completion evaluation conducted between 8 and 10 days after the dose. The subjects were domiciled in the study center from the baseline visit through Day 4 (5 days total). An interim analysis for safety and pharmacokinetics was performed to inform the doses in Part 2 of the study. A total of 30 subjects received BZF961 and 11 subjects received placebo in Part 1 of the study.
Part 2 was a multiple-ascending dose study (MAD) of escalating doses of BZF961 or placebo administered to four cohorts of eight subjects each (randomized BZF961:placebo; 6:2) (Cohorts 8–11, 100 mg every 8 h, 300 mg every 8 h, 500 mg every 8 h, and 1000 mg every 12 h) administered for 6 days with a single dose on the morning of Day 7. The BZF961 formulation for all cohorts in Part 2 was MEPC. The study consisted of a maximum 21-day screening period, a baseline period (Day 1), treatment period (Days 1 to 10), and a single study completion evaluation conducted between Days 14 and 16. The subjects were domiciled in the study center from the baseline visit through Day 10 (11 days total). An interim analysis for safety and pharmacokinetics was performed to inform the doses in Part 3 of the study. A total of 24 subjects received BZF961 and 9 subjects received placebo in Part 2 of the study.
Part 3 was an open-label drug–drug interaction study of escalating doses of BZF961 (10 mg or 50 mg) co-administered with ritonavir (100 mg) to three cohorts (Cohort 12 n = 4, Cohort 13 n = 8, and Cohort 14 n = 8). All subjects received active drug in the MEPC formulation. Each of the three cohorts followed a fixed sequence design with variations in BZF961 and ritonavir doses. In all cohorts, subjects were dosed with BZF961 alone (50 mg, Cohorts 12 and 14; or 10 mg, Cohort 13) to obtain baseline BZF961 exposure data (Period 1). Following a washout period of approximately 5 days, subjects in Cohort 12 received a single evening dose of 100 mg ritonavir, two doses of 100 mg ritonavir 12 h apart on Day 2, and a single dose of 50 mg BZF961 on the morning of Day 2 concurrently with the morning dose of ritonavir (Period 2). An interim analysis was conducted on the data obtained from this cohort. Based on the results of the interim analysis and a review of the safety data, the doses in the next two cohorts were determined. Cohort 13 followed the same scheme as Cohort 12, but with 10 mg BZF961 administered. In Cohort 14, a single 100 mg dose of ritonavir was administered simultaneously with 50 mg BZF961; no evening dose of ritonavir was administered. Dosing periods were the same for all cohorts. A total of 20 subjects received BZF961 and ritonavir in Part 3 of the study.
Study treatment was administered with 240 mL of water. Subjects fasted for at least 10 h prior to BZF961 administration (no food or liquid except for water), except for those in the cohort receiving BZF961 after a standard FDA breakfast. No fluid intake apart from the fluid given at the time of drug intake was allowed from 2 h before until 2 h after dosing. Subjects did not consume any carbonated drinks; consumption of alcohol, caffeine, nicotine-containing products, and grapefruit or related citrus fruit was prohibited during the study. Subjects who were enrolled in the food effect cohort were given the standard FDA breakfast and dosed with BZF961 within 5 min after completion of the meal.
Prescription and non-prescription drugs and dietary supplements were not permitted and had to be discontinued at least 30 days or five half-lives (whichever was longer) prior to the enrollment. Co-administration of moderate to strong inhibitors of CYP3A4 (except ritonavir in Part 3) was prohibited. Additionally, co-administration of inducers of CYP3A4 was prohibited. Treatment was to be discontinued in the event of the development of one adverse event of moderate severity (common terminology criteria for adverse events [Citation15] Grade 3 or greater) believed to be related to the study drug. Additionally, study treatment would be terminated for an increase in total bilirubin ≥5-fold the upper limit of normal (ULN), ALT, AST, or alkaline phosphatase ≥3-fold the ULN, total bilirubin increase ≥2-fold ULN and ALT ≥3-fold ULN, or lipase ≥3-fold ULN. The subjects could discontinue from the study at their own request for safety, behavioral, or administrative reasons or for any other reason at the discretion of the investigator or sponsor.
This study was conducted in compliance with the Declaration of Helsinki and Good Clinical Practice Guidelines established by the International Conference on Harmonization. The final protocol, amendments, and informed consent documentation were reviewed and approved by the study center’s Institutional Review Board. All the subjects provided written, informed consent before participating in any study procedures.
Subjects
The study planned to enroll 92 subjects (40 subjects in Part 1, 32 in Part 2, and 20 in Part 3) including both healthy male and female subjects (of non-child-bearing potential), aged 19 to 55 years and at least 50 kg in weight (with body mass index between 18 and 30 kg/m2) who passed screening assessments, met inclusion/exclusion criteria, and provided written informed consent.
Health status was determined by medical history, physical examination, and laboratory tests at screening. Only women who were not of child-bearing potential were allowed to enroll, and all female subjects were required to have a negative pregnancy test result at screening and at the baseline visits. Male subjects were required to comply with barrier contraception methods during the study and up to 7 days after the last dose of study drug. Exclusion criteria included a history of clinically significant electrocardiogram abnormalities; history of organ system malignancy within the last 5 years (excluding skin basal cell carcinoma); use of tobacco products within the previous 3 months; use of prescription medications or herbal supplements within the previous 4 weeks or over-the-counter medication (except for incidental acetaminophen), dietary supplements, or vitamins within 2 weeks; blood or plasma donation or blood loss of ≥400 mL within the previous 8 weeks; and/or a hemoglobin level of below the lower limit of normal. Subjects were also excluded if found or reported to have had significant illness within the previous 2 weeks; history of food allergies or any surgical or medical condition that might significantly alter drug pharmacokinetics; history of liver disease or injury as indicated by abnormal transaminases or bilirubin; history or evidence of any inherited bilirubin disease or disorder, including but not limited to Dubin–Johnson, Gilbert’s, or Rotor syndrome; history of immunodeficiency disease; positive hepatitis B or C test result; and/or a history of drug or alcohol abuse in the previous 12 months or evidence thereof.
Safety assessment
Safety evaluations included monitoring for adverse events, monitoring for serious adverse events with the severity and relationship to the study drug, monitoring for pregnancies, and regular monitoring of hematology, blood chemistry, urinalysis, vital signs, physical condition, body weight, and electrocardiogram.
Pharmacokinetic parameters and assessment
In Part 1 of the study (single dosing), blood samples were collected pre-dosing and 0.5, 1, 2, 2.5, 3, 4, 6, 8, 12, 24, 36, 48, and 72 h post-dose. In Part 2 (daily dosing for 3 days), blood sampling on Day 1 occurred at pre-dose and 0.5, 1, 2, 2.5, 3, 4, 6, 8, and 12 h post-dose. Samples were also taken on Days 2–6 at pre-dosing, on Day 7 at pre-dosing and 0.5, 1, 2, 2.5, 3, 4, 6, 8, 12, 24, 36, 48, and 72 h post-dosing. In Part 3 of the study drug-drug interaction with ritonavir was measured. In Period 1, blood samples were collected for BZF961 concentration on Day 1 at pre-dosing and 0.5, 1, 2, 2.5, 3, 4, 6, 8, 12, 24, and 36 h post-dose. In Period 2, blood samples were collected for BZF961 concentration on Day 2 at pre-dosing and 0.5, 1, 2, 2.5, 3, 4, 6, 8, 12, 24, and 36 h post-dose. Additionally, blood samples were collected for ritonavir concentration on Day 1 at 2 h post-ritonavir evening dose and on Day 2 at 1, 2, 4, and 8 h post-ritonavir morning dose. The plasma samples from all the subjects were assayed for BZF961 concentrations using a validated liquid chromatography-tandem mass spectrometry assay (LC-MS/MS). Values below the lower limit of quantitation (LLOQ) of 1.00 ng/mL were reported as 0.00 ng/mL.
The pharmacokinetic parameters were calculated from the BZF961 plasma concentration–actual-time data for subjects administered BZF961 by standard non-compartmental methods using WinNonlin Phoenix software (version 5.2; Centara, Princeton, NJ). The pharmacokinetic parameters include area under the concentration–time curve from time zero to 24 h post-dose (AUC0–24h), AUC from time zero to the time of the last quantifiable concentration (mass × time/volume) (AUClast), AUC from time zero extrapolated to infinity (AUCinf), maximum observed concentration (Cmax), time of occurrence of Cmax (Tmax), elimination half-life (t1/2), apparent oral clearance (CL/F), apparent volume of distribution (Vz/F), and accumulation ratio (Racc), which was defined as the ratio of AUC0–24h (Day 7) to AUC0–24h (Day 1). Subjects for whom selected pharmacokinetic parameters could not be adequately characterized were excluded from the calculation of overall means and statistics for those parameters.
Statistical methods
Adverse events were summarized by counts. Laboratory tests, vital signs, and electrocardiogram parameters were summarized by descriptive statistics by part, cohort, and visit at each time point. Pharmacokinetic parameters were summarized using arithmetic means and standard deviations, except for Tmax, for which median values and ranges are reported. To test dose proportionality, the primary pharmacokinetic parameters were analyzed using a regression model on the log-transformed pharmacokinetic parameters (Cmax, AUC0–24h, and/or AUCinf) versus log-transformed dose, assessing dose proportionality with a lack-of-fit test. The analyses were done separately for each part of the study and separately on Days 1 and 7 for the multiple-dose cohorts. To assess food effect, data from the same subjects under fed and fasting conditions were pooled, and AUC, AUClast, and Cmax were analyzed by mixed-effect model analysis of variance with the fasting or fed condition as a fixed effect and a compound symmetric covariance within each subject.
Results
Subject demographics
A total of 94 subjects (41 subjects in Part 1 [30:11, BZF961:placebo], 33 subjects in Part 2 [24:9, BZF961:placebo], and 20 in Part 3 [20, BZF961]) were enrolled in the study, and 80 subjects completed the study. Demographic data are summarized in . All the subjects with evaluable data were included in safety and pharmacokinetics analyses.
Table 1. Subject demographics.
Safety and tolerability
All 94 subjects were included in the safety analysis. Subjects who returned to be included in the fed cohort and alternate formulation cohorts were counted twice or three times depending on their participation, resulting in a total of 56 subjects being counted in the safety analysis set of Part 1. There were no deaths or serious adverse events (SAE). The overall incidence of adverse events (AEs) was similar in treated subjects and placebo subjects in the single dose part of the study but was higher in treated subjects than placebo subjects in the multiple-dose part of the study ().
Table 2. Number and percentage of subjects with AEs by preferred term (safety analysis set).
In the single ascending dose cohorts, all subjects completed dosing as planned. The overall incidence of AEs is shown in . There were no SAEs. Among the 41 subjects (counted as a total of 56 subjects), there were a total of 13 AEs occurring in 8 subjects (6 [14.6%] BZF961-treated subjects and 2 [13.3%] placebo-treated subjects) (). Eight of the 13 AEs occurred in the 1000 mg dose group (66.7% of subjects dosed with 1000 mg); seven of the AEs were nausea or vomiting all of which were suspected by the investigator as being related to study drug. All of the AEs were considered Grade 1.
In the multiple ascending dose cohorts, 33 subjects were included in the safety analysis set (). Twenty-four subjects were dosed with BZF961 (six subjects each received 100 mg every 8 hours, 300 mg every 8 h, 500 mg every 8 h, or 1000 mg every 12 h) and nine subjects were dosed with placebo. Twenty three of the 33 subjects in Part 2 of the study experienced at least one AE; there were no SAEs. The majority of the AEs were nausea and/or vomiting. All the subjects in the BZF961 1000 mg cohort (six dosed with BZF961 and two dosed with placebo) developed Grade 2 nausea and/or Grade 1 or Grade 2 vomiting. This was suspected to be related to the MEPC-based formulation, which was dose limiting at the volume required for the 1000 mg BZF961 dosage and matching placebo. For seven of the eight subjects in this dose group, including two subjects that received placebo, the AEs resulted in discontinuation of the study drug. Study drug discontinuation resulted in cessation of nausea and vomiting. Two subjects (one who received BZF961 300 mg every 8 h and the other BZF961 1000 mg every 12 h) experienced elevations in total bilirubin, resulting in discontinuation of study drug. All elevations in bilirubin occurring during the study were resolved by the end of study visits. In the 500 mg every 8 h cohort, one subject withdrew from the study on Day 4 citing personal reasons. This subject is included in the safety analysis dataset and was replaced in the study. Another subject in this cohort withdrew on Day 2 due to Grade 2 nausea and vomiting. This subject was not replaced.
In Part 3 (co-administration with ritonavir) twenty subjects were randomized and were included in the safety analysis (). Seven subjects reported at least one of 12 AEs. Four of the AEs were reported after the administration of the single dose of BZF961 and prior to the administration of ritonavir, these were generalized body ache, upper respiratory tract infection and pre-syncope, and an insect bite. The other eight AEs were reported when BZF961 was co-administered with ritonavir 100 mg. Diarrhea was the most commonly reported AE. None of the subjects in Part 3 discontinued the study drug due to an AE. All AEs in this part of the study were Grade 1 in severity.
Although there were sporadic hematology, chemistry, or urinalysis parameters outside normal ranges among subjects receiving BZF961 and placebo, none were considered clinically significant or drug related. Mean values for most of the hematology and blood chemistry parameters remained within the normal range from baseline to the end of the study, and there were no significant differences between placebo and control mean values.
Overall, there were elevations in mean total bilirubin during the dosing period in subjects from the BZF961 300 mg every 8 h, 500 mg every 8 h, and 1000 mg every 12 h dosing cohorts. Mean bilirubin peaked on Day 2 and declined on Day 4 and later time points. For the 1000 mg every 12 h dosing cohort, which had the greatest change in bilirubin, the Day 2 mean bilirubin was 32.0 ± 26.3 µmol/L (1.87 ± 1.54 mg/dL) and the median was 22.0 µmol/L (1.29 mg/dL) (upper limit of normal is 27 µmol/L; 1.58 mg/dL). Upon fractionation, the elevation was due predominantly to an increase in unconjugated bilirubin. Four subjects in this study had elevated total bilirubin levels. A total of two subjects in the multiple ascending dose cohorts had elevations in total bilirubin levels that resulted in premature discontinuation of dosing. One subject who received BZF961 500 mg every 8 h for 7 days had a Grade 1 elevation of ALT, AST, and γ-GT associated with very high BZF961 exposure compared to the remainder of the subjects in the dose cohort. This subject was asymptomatic and the laboratory changes resolved during follow-up. This subject also had a baseline total bilirubin of 9 µmol/L (0.53 mg/dL) (normal range 3 to 27 µmol/L) that increased to 32 µmol/L (1.87 mg/dL) on Day 4 of dosing and peaked at 38 µmol/L (2.22 mg/dL) on Day 7 of dosing. Fractionation revealed that the bilirubin elevation consisted mainly of unconjugated bilirubin. On Day 7 of dosing, the subject had an ALT of 124 U/L from a baseline of 29 U/L (normal range 10–67 U/L). The ALT was within normal limits 10 days after completion of the study. The AST increased to 57 U/L on Day 7 of dosing (normal range 14–48 U/L) and decreased to 42 U/L the following day. The γ-GT increased to 84 U/L (normal range 7 to 62) on Day 7 of dosing and decreased to 65 U/L 10 days after completion of dosing. The elevated ALT and bilirubin were Grade 1 in severity. No other subject had Grade 1 or higher ALT, AST, or γ-GT elevations. There were no clinically significant changes in mean bilirubin in the single ascending dose cohorts or the ritonavir-boosted cohorts.
In all parts of the study, most vital sign measurements were within normal ranges. There were no recorded AEs related to vital signs and no trend in mean or median values indicating subclinical abnormality. Electrocardiogram measurement did not reveal any significant QTc prolongations nor any abnormalities determined to be clinically significant.
Overall, there were no deaths or SAEs in this study. None of the subjects enrolled in Part 1 (SAD) or Part 3 (co-administration of ritonavir) discontinued because of an AE. In Part 2 (MAD), 10 subjects in total discontinued because of an AE (Grade 2 nausea and vomiting or Grade 1 elevated bilirubin; eight subjects who received BZF961 and two subjects who received placebo).
Pharmacokinetics
The mean plasma concentration-time profiles of BZF961 are presented in . The pharmacokinetic parameters are summarized in .
Figure 3. Arithmetic mean plasma concentration–time profiles of BZF961 following oral administration of single ascending doses (Part 1). BZF961 10 mg (^), 30 mg (Δ), 100 mg ( □ ), 300 mg (⋄), and 1000 mg (*).
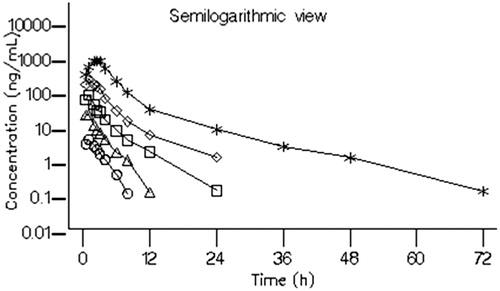
Table 3. Summary statistics of BZF961 pharmacokinetic parameters by treatment (pharmacokinetic analysis set).
Part 1: Single ascending dose
Following oral administration of BZF961 in the MEPC formulation in a fasted state, BZF961 was rapidly absorbed with the median Tmax in the range of 0.74 to 2.25 h post-dose (). After reaching peak concentrations, BZF961 was eliminated rapidly. The mean terminal elimination half-life, however, tended to increase with dose, from 1.54 h for the 10 mg single dose to 8.78 h for the 1000 mg single dose ().
BZF961 plasma mean exposure (Cmax and AUC) increased with the increase in BZF961 dose throughout the dose range studied (). The plasma exposure increased approximately linearly with doses up to 300 mg. A trend towards higher than dose proportionality was noted at the highest dose tested (1000 mg). Dose proportionality was assessed using a power model for Cmax, AUClast, and AUCinf over the dose range from 10 mg to 1000 mg.
Administration of BZF961 with food appeared to delay the time to peak plasma concentration from approximately 1 h to 2 h and to decrease the Cmax by approximately 44%; however, the overall AUC remained the same. Dosing of BZF961 100 mg using the Vitamin ETPGS formulation resulted in higher Cmax (39%) and exposures (approximately 17%) than with the MEPC formulation.
Part 2: Multiple ascending dose
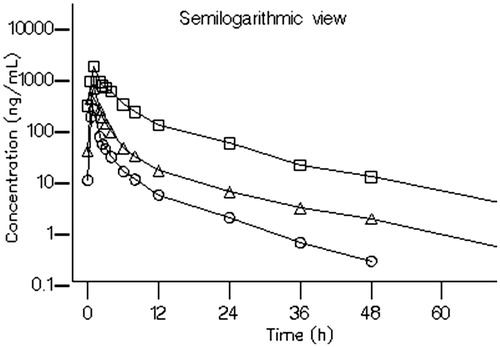
Following multiple dose administration, BZF961 exposure (Cmax and AUC) increased in an approximately dose proportional manner from 100 mg every 8 h to 500 mg every 8 h (). Slight accumulation of BZF961 was observed following multiple dosing for 7 days, ranging from 1.33-fold to 2.88 fold for AUC and 1.19-fold to 2.54-fold for Cmax. There was no change in time to peak concentration between Day 1 and Day 7, indicating that there were no changes in the potential absorption mechanisms of BZF961. The terminal half-life following multiple dose administration ranged between 10 and 13 h on Day 7.
Figure 4. Arithmetic mean plasma concentration-time profiles of BZF961 following oral administration of multiple ascending doses on Day 7 (Part 2). BZF961 100 mg q 8 h (^), 300 mg q 8 h (Δ), and 500 mg q 8 h ( □ ).
Part 3: Drug interaction with ritonavir
In Part 3 of the study, the impact of ritonavir on the pharmacokinetics of BZF961 was evaluated. A single dose of 10 or 50 mg BZF961 was administered either alone or with ritonavir 100 mg administered prior to, concomitantly with, and following the BZF961 dose for Cohorts 12 and 13, and only simultaneously with BZF961 for Cohort 14. Semilogarithmic plots of the arithmetic mean BZF961 plasma concentration at each sample collection time-point with and without concomitant administration of ritonavir 100 mg are shown in . Plasma concentrations of BZF961 are provided for each cohort in .
Figure 5. Arithmetic mean plasma concentration–time profiles of BZF961 following oral administration of BZF961 alone or in combination with ritonavir (Part 3). BZF961 50 mg alone (^), BZF961 50 mg + three doses of ritonavir (Δ), BZF961 10 mg alone ( □ ), BZF961 10 mg + three doses of ritonavir (⋄), BZF961 50 mg alone (*) and BZF961 50 mg + single dose of ritonavir (♣).
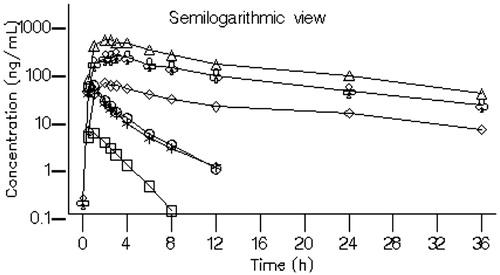
Ritonavir administered prior to, concomitantly with, and following a single 50 mg dose of BZF961 (Cohort 12) increased the mean plasma BZF961 Cmax approximately 10-fold from 47.8 (± 25.5) to 444 (± 105) ng/mL and the mean AUCinf approximately 43-fold from 128 (± 50.3) to 5440 (± 1590) h*ng/mL. A similar increase (approximately 50-fold) in the overall plasma BZF961 exposure was noted when a single dose of 10 mg BZF961 (Cohort 13) was administered with 100 mg ritonavir administered prior to, concomitantly with, and following the BZF961 dose, suggesting linearity in BZF961 exposure. Ritonavir administered concomitantly with a single 50 mg dose of BZF961 (Cohort 14) increased the mean plasma BZF961 Cmax from 41.7 (± 12.5) to 214 (± 67.7) ng/mL and the mean AUCinf from 107 (± 28.6) to 2770 (± 666) h*ng/mL. The terminal elimination half-life of BZF961 increased approximately 4-fold from ∼3 to ∼11.5 h when administered with ritonavir. The time to reach peak plasma BZF961 concentrations remained relatively unchanged with or without ritonavir. Ritonavir increased BZF961 Cmax, AUC, and T1/2 by approximately 5 to 10-fold, 26 to 60-fold and 5-fold, respectively. The overall variability in BZF961 Cmax and AUCinf was reduced by approximately 25% when dosed with ritonavir than without ritonavir.
Discussion
HCV infection has been the leading cause of liver-related deaths and indication for liver transplantation in Europe and the United States. Recently, treatment for chronic HCV infection has improved dramatically with decreased treatment duration and increased cure rates. Here we investigated a novel HCV NS3-4A protease inhibitor in a first-in-human study. BZF961 was safe and well tolerated when administered as single and multiple doses in healthy subjects; there were no serious AEs. Following administration of single doses of BZF961, all AEs were Grade 1 or 2, did not result in discontinuation, and were resolved. Following the administration of multiple doses of BZF961, the most frequent AEs were nausea, vomiting, and elevated bilirubin, which resulted in discontinuation for eight subjects. A significant proportion of the nausea and vomiting appeared to have been related to the MEPC formulation vehicle, as similar percentages of placebo-treated subjects and BZF961-treated subjects experienced this AE. Using lower doses of MEPC-formulated BZF961 in combination with ritonavir, or the development of a more palatable formulation may help mitigate this risk. The elevated bilirubin is thought to be secondary to the effect of BZF961 on bilirubin transporters, in particular OATP1B1 and OATP1B3 (data not shown). This effect has been seen with other protease inhibitors for HCV and HIV infection in the clinic and generally does not limit the ability to use the drugs for the treatment [Citation16,Citation17].
The pharmacokinetic results indicate that BZF961 systemic exposure increased with dose and the increase was slightly greater than dose proportional following single ascending doses from 10 mg to 1000 mg and multiple ascending doses from 100 mg to 500 mg on Days 1 and 7. The accumulation ratio ranged from 1.33-fold to 2.88 fold (based on AUC) after multiple doses compared to Day 1. Administration with food had minimal effect on BZF861 exposure.
BZF961 is predominantly metabolized by CYP3A4. Ritonavir is a potent CYP3A4 inhibitor and has been used to pharmacologically increase exposure of HIV and HCV protease inhibitors in the clinic. Co-administration of BZF961 with ritonavir was well tolerated and resulted in significant increases (up to 60-fold) in BZF961 exposure. Co-administration of BZF961 with ritonavir increased trough levels more than the AUC and Cmax levels, thereby resulting in a lower predicted efficacious dose and higher safety margins. Additionally, the inter-subject variability of BZF961 exposure was decreased with ritonavir co-administration. Based on the pharmacokinetic results and the preclinical virology data, it is expected that a 500 mg twice daily dose of BZF961 – or 50 mg twice daily dose with ritonavir – will be able to inhibit wild-type HCV GT 1, 2, 4, 5, and 6. Additionally, this dose may be adequate to cover most of the known GT 1a and 1 b resistant variants.
In conclusion, the strategy of co-administering BZF961 with the CYP3A4 inhibitor ritonavir considerably boosted exposure of the HCV NS3-4A protease inhibitor. This finding indicates the possibility of using lower doses, with improved safety margins, for efficacy studies.
Transparency
Declaration of funding
This study was funded by Novartis Institutes for BioMedical Research.
Declaration of financial/other relationships
AB, SM, CTJ, KD, CLJ, and RAC are, or were at the time of the study, employees of and/or shareholders in Novartis Pharmaceutical Corp. JDA peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
This manuscript is dedicated to Dr. Stephen Youngberg (1946–2013).
References
- Gower E, Estes C, Blach S, et al. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol. 2014;61:S45-S57.
- Casey LC, Lee WM. Hepatitis C therapy update. Curr Opin Gastroenterol. 2012;28:188-192.
- Ly KN, Xing J, Klevens RM, et al. The increasing burden of mortality from viral hepatitis in the United States between 1999 and 2007. Ann Intern Med. 2012;156:271-278.
- Muhlberger N, Schwarzer R, Lettmeier B, et al. HCV-related burden of disease in Europe: a systematic assessment of incidence, prevalence, morbidity, and mortality. BMC Public Health. 2009;9:34.
- WHO. Global Hepatitis Report. 2017. Available at: http://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/
- McGowan CE, Fried MW. Barriers to hepatitis C treatment. Liver Int. 2012;32:151-156.
- Messina JP, Humphreys I, Flaxman A, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology. 2015;61:77-87.
- Buti M, Esteban R. Hepatitis C virus genotype 3: a genotype that is not 'easy-to-treat'. Expert Rev Gastroenterol Hepatol. 2015;9:375-385.
- Smith DB, Bukh J, Kuiken C, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014;59:318-327.
- Simmonds P, Bukh J, Combet C, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42:962-973.
- McHutchison JG, Lawitz EJ, Shiffman ML, et al. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N Engl J Med.. 2009;361:580-593.
- Jacobson IM, Pawlotsky JM, Afdhal NH, et al. A practical guide for the use of boceprevir and telaprevir for the treatment of hepatitis C. J Viral Hepat. 2012;19 (Suppl 2):1-26.
- WHO. Global Health Sector Strategy on Viral Hepatitis 2016–2021: Towards ending viral hepatitis 2016. Available at: http://www.who.int/hepatitis/strategy2016-2021/ghss-hep/en/
- U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Guidance for Industry: Food-Effect Bioavailability and Fed Bioequivalence Studies. Available at: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070241.pdf
- Common Terminology Criteria for Adverse Events (CTCAE), Version 4.0. USDEPARTMENT OF HEALTH AND HUMAN SERVICES, National Institutes of Health, National Cancer Institute. Available at: https://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm#ctc_40
- Sanne I, Piliero P, Squires K, et al. Results of a phase 2 clinical trial at 48 weeks (AI424-007): a dose-ranging, safety, and efficacy comparative trial of atazanavir at three doses in combination with didanosine and stavudine in antiretroviral-naive subjects. J Acquir Immune Defic Syndr. 2003;32:18-29.
- Johnson M, Grinsztejn B, Rodriguez C, et al. Atazanavir plus ritonavir or saquinavir, and lopinavir/ritonavir in patients experiencing multiple virological failures. AIDS. 2005;19:685-694.