
Abstract
Objective: Long-term safety and efficacy of 10- and 20-mg rupatadine in Japanese patients with perennial allergic rhinitis (PAR) were investigated in a 52-week open-label study (JapicCTI-152952, clinicaltrials.jp).
Methods: The rupatadine dose was fixed to 10 mg once daily for the first 2 weeks. Thereafter, the study investigator was allowed to increase the dosage to 20 mg if the response was insufficient. Safety was evaluated on the basis of treatment-emergent adverse events, laboratory findings, and vital sign measurements. The primary efficacy endpoint was changed from baseline to Week 2 in the total 4 nasal symptom score. Secondary efficacy endpoints included changes over time in ocular symptoms, patient and physician clinical overall impression, and patient quality-of-life.
Results: Seventy-two immunoglobulin E positive patients (mean age = 32.1 years), consisting of 58 adults (age ≥ 18 years) and 14 adolescents (12–17 years), were enrolled. Ninety-four treatment-emergent adverse events were reported in 48 patients (66.7%), including nine adverse drug reactions in nine patients (12.5%). The most frequently reported adverse drug reaction was somnolence (9.7%). The primary and secondary efficacy endpoints demonstrated a statistically significant clinical benefit with rupatadine. The rupatadine dose was increased from 10 to 20 mg in 36 patients (50.0%), which resulted in better symptom management.
Conclusions: Rupatadine 10- and 20-mg once-daily doses were well tolerated in long-term use. Updosing to 20 mg is a reasonable option in PAR patients whose symptoms cannot be controlled effectively by the 10-mg dose.
Introduction
Allergic rhinitis (AR) is an immunoglobulin E (IgE)-mediated allergic response of the nasal mucous membrane following exposure to inhaled allergensCitation1. Depending on the type of allergens, AR is classified as perennial AR (PAR) or seasonal AR, which are characterized by persistent and intermittent responses, respectivelyCitation2,Citation3. The typical perennial allergens include house dust mites, cockroaches, cats, dogs, hamsters, and other domesticated pet animals with furCitation4. The common symptoms of AR include sneezing, rhinorrhea, nasal itching, and nasal congestionCitation1.
Oral and topical H1 antihistamines, particularly second-generation antihistamines, are the treatment of choice for patients with ARCitation1,Citation5–7. H1 antihistamines are effective for controlling sneezing, rhinorrhea, and nasal itching. Existing H1 antihistamines, such as desloratadine, fexofenadine, cetirizine, and levocetirizine, show modest effects for the treatment of nasal congestionCitation6.
Rupatadine is a new non-sedating H1 antihistamine that was first approved in 2001 in Spain for the treatment of AR. As of June 2018, this drug has been approved in more than 80 countries including Japan for the indication of AR and urticaria, and the indication includes skin pruritus in Japan. Its recommended dosing regimen is 10 mg once daily in adults and adolescents (age ≥12 years).
The pharmacokinetics of rupatadine suggest a favorable therapeutic effect. Rupatadine has a short maximum drug concentration time (tmax) of approximately 1 hour. Furthermore, rupatadine is metabolized to desloratadine, an H1 antihistamine with a longer tmax of 3.00 hCitation7, in the liver by cytochrome P450 3A4. Such features are expected to contribute to the fast onset and prolonged duration of action of rupatadine.
In addition to its H1 antihistamine action, rupatadine is an antagonist against the platelet-activating factor (PAF) receptorCitation8. This drug has a lutidinyl component, which has been shown in vitro and in vivo to prevent PAF from binding to its receptorCitation9. Histamine and PAF are major inflammatory mediators in ARCitation10. In a study by Shirasaki et al.Citation11, expression of the PAF receptor was evident in the nasal mucosa of patients with refractory nasal obstruction, insinuating the association between PAF and nasal airway resistance. The dual mechanism of action and results of such studies suggest that rupatadine is effective for the management of PAR.
To address the health problems related to PAR and other allergic diseases, the Japanese government launched a comprehensive program, including the development of new medications, particularly those for pediatric useCitation12. To investigate the safety and efficacy of long-term treatment with 10 and 20 mg rupatadine in Japanese adult and adolescent patients with PAR, we conducted a Phase III, single-center, open-label clinical trial from June 2015 to July 2016 (Japan Pharmaceutical Information Center Clinical Trial Information [JapicCTI] No. 152952). The study results are hereby reported.
Methods
Study procedure
This clinical trial was implemented according to the 1964 World Medical Association Declaration of Helsinki (as revised in October 2013 in Fortaleza, Brazil)Citation13 and relevant regulatory provisions and guidelines. The study investigator submitted the study protocol to the institutional review board of his medical center, and written informed consent was obtained from all patients before starting the study. Patient anonymity was maintained using methods approved by the institutional review board.
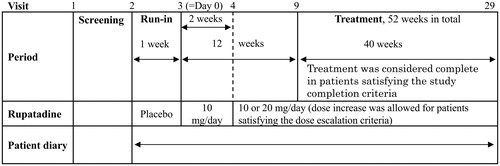
The design of this study is illustrated in . At Visit 1, patients were screened for study eligibility. At Visit 2, eligible patients underwent preliminary registration, were prescribed the run-in period medication (placebo), and began daily journaling using the patient diary. At Visit 3, patients who satisfied the eligibility criteria were enrolled into the study to start the study medication (rupatadine 10 mg once daily) on the following day. At Visit 4, the results of the 2-week rupatadine 10 mg treatment were evaluated. At Visit 4 or later, the dose was increased from 10 to 20 mg at the discretion of the study investigator if the 6-day mean total 4 nasal symptom score (T4NSS, defined as the sum of the four individual nasal symptom scores of the AR scoring system, TNSS, which is commonly used in assessments of AR symptomsCitation14) before the study visit did not show improvement for 1 or more points from the baseline score (3-day mean from Day −3 to Day –1). Dose reduction from 20 to 10 mg was left to the judgment of the study investigator. From Visit 9 onward (after 12 weeks of treatment), the study treatment was considered complete if the patient satisfied the study completion criteria and wished to terminate the study treatment.
Figure 1. Outline of the study procedure. Visit 1, screening, informed consent, and study ID assignment; Visit 2, assessment of study eligibility, preliminary registration, and start of placebo treatment; and Visit 3, reconfirmation of study eligibility, enrollment, and start of study medication.
Inclusion and exclusion criteria
Patients that satisfied the following inclusion criteria were enrolled: (i) 12–64 years of age at informed consent, (ii) history of typical PAR suggesting 12 or more weeks of continuing symptoms after study enrollment, (iii) positive for IgE specific to one or more PAR antigens (determined by CAP-RAST [capsulated hydrophilic carrier polymer radioallergosorbent test], AlaSTAT 3 g assay, or other immunoassays), (iv) presence of all four nasal symptoms (i.e. sneezing, rhinorrhea, nasal congestion, and nasal itching) for 3 days before the start of the study treatment, with a mean daily T4NSS score of 6 or greater, (v) capable of keeping the patient diary, and (vi) informed consent signed by the patient (age ≥ 20 years) or legal representatives (age < 20 years). Patients who were minors signed the informed assent document for participation.
Patients that met any of the following exclusion criteria were removed from the study: (i) nasal disorders (e.g. nasal polyps, nasal septum deviation, or hypertrophic rhinitis) or infectious diseases (e.g. upper respiratory tract inflammation, sinusitis, infectious rhinitis, or infectious eye disease) that could possibly interfere with the study evaluation, (ii) current or past vasomotor, infectious, or drug-induced rhinitis (non-allergic), (iii) patients positive for pollen grains that were expected to influence their condition within 12 weeks from study enrollment, (iv) intranasal laser coagulation or surgery performed within 1 year before signing informed consent, (v) severe or unmanaged mild or moderate bronchial asthma necessitating the use of injectable, oral, or inhaled corticosteroids, (vi) history of hypersensitivity to rupatadine, other antihistamines, or any ingredient of the study tablet, (vii) history of immunotherapy within 6 months before the start of the run-in period, or current immunotherapy ongoing for 7 months or more prior to the start of the run-in period but failing to achieve a stable condition, (viii) severe hepatobiliary, renourinary, or other disorders, (ix) women who were lactating, pregnant, possibly pregnant, or wishing to be pregnant, and men whose sexual partners were willing to be pregnant, and (x) current or history of participation in a separate clinical trial within 3 months before the screening. Furthermore, patients were excluded from the study if they could not abstain from the following: (i) from the start of the run-in period to the end of the study treatment: psychotropic drugs (tranquilizers, antipsychotics, anti-insomnia drugs, antidepressants), nebulizers, eye drops (excluding antibiotic and artificial lacrima formulations), eye irrigation, and nasal irrigation; (ii) from 1 week before the start of the run-in period to the end of the Week-2 treatment: anti-cold medicine and influenza vaccination; (iii) from 1 week before the start of the run-in period to the end of the study treatment: antihistamines (excluding H2 blockers, non-ocular and non-nasal applications permitted), leukotriene receptor antagonists, thromboxane A2 receptor antagonists, mast cell stabilizers, Th2 cytokine inhibitors, topical corticosteroids (non-ocular and non-nasal applications permitted), anticholinergics, biologics (excluding influenza vaccines, e.g. histamine-added human immunoglobulin formulations), immunosuppressants, and other agents that have similar pharmacological actions; and (iv) from 3 weeks before the start of the run-in period to the end of the study treatment: systemic corticosteroid preparations.
Endpoints
The primary efficacy endpoint was change in the T4NSS score from baseline to Week 2. Specifically, the daily T4NSS score was determined as the sum of sneezing, rhinorrhea, nasal congestion, and nasal itching scores, which were each evaluated on a 5-point scale: 0 = −, 1 = +, 2 = ++, 3 = +++, and 4 = ++++ (Supplementary Table S1). Therefore, the possible daily T4NSS score ranged from 0–16. The baseline T4NSS score was calculated as the mean of 3 consecutive days before the start of the treatment period, and the Week 2 T4NSS score was calculated as the mean of the last 7 days of treatment (in patients completing and prematurely discontinuing the 2-week treatment).
In the secondary efficacy analyses, the changes over time in the following variables were investigated: (i) T4NSS, (ii) Japan Rhinoconjunctivitis Quality of Life Questionnaire (JRQLQ) score, (iii) sneezing, rhinorrhea, nasal congestion, and nasal itching scores, and (iv) patient and physician clinical overall impression. The JRQLQ is a validated QoL instrument designed to meet the environment of AR patients in JapanCitation15,Citation16.
Patient safety was assessed post-treatment, on the basis of the new onset of adverse events or the worsening of pre-existing adverse events (hereinafter “treatment-emergent” adverse events), vital signs (systolic and diastolic blood pressure and pulse rate), and clinical laboratory findings (hematology, serum biochemistry, and urinalysis). Blood and urine samples were collected and sent to the contract laboratory (BML, Inc., Tokyo, Japan) for central measurement. Clinically abnormal laboratory findings were classified as adverse events. Adverse events were evaluated for causality, and those assessed as treatment-related were categorized as adverse drug reactions. Investigator-reported verbatim terms were coded using the Medical Dictionary for Regulatory Activities (MedDRA) Version 19.0Citation17. The severity of events were graded on the basis of the Common Terminology Criteria for Adverse Events Version 4.0Citation18. Adverse events with at least a reasonable possibility of causal association with the study were classified as adverse drug reactions.
Statistical analysis and study populations
The primary efficacy endpoint was statistically evaluated using the paired t-test at a 2-tailed significance of 0.05. The changes over time in the T4NSS score were also tested using the paired t-test in an exploratory manner. No multiplicity adjustments were made for exploratory analyses. Sub-group analyses were conducted on the safety, primary efficacy, and secondary efficacy variables by age (12–17 and 18–64 years), body weight (<50, 50–59, 60–69, and ≥70 kg), sex, and baseline T4NSS score (<9 and ≥9). Furthermore, post-hoc analyses were conducted to evaluate changes in the T4NSS score resulting from the dose increase from 10 to 20 mg. Analysis for the comparison between results of patients who received a fixed dose, updosed at Week 5, and updosed at Week 7 and later was performed by a two-tailed Student’s t-test at a Bonferroni-adjusted significance level of 0.0167.
The target sample size was set at 72 patients assuming a 12-week dropout rate of 20%. Approximately 60 patients were considered necessary in light of a long-term clinical trial of a similar product in patients with PAR. With this sample size, the probabilities of detecting at least one adverse drug reaction were 95.4% and 45.3% if the incidence rates were 5% and 1%, respectively. To investigate whether the safety and efficacy profile of rupatadine is similar between adults and adolescents, the minimum percentage of adolescents in the target patient population was set at 10% (n = 7).
The full analysis set (FAS) was defined as the group of patients who were administered at least one dose of rupatadine and provided at least one post-treatment efficacy measurement. The safety analysis set (SAS) included all patients that were administered one or more doses of the study medication. Efficacy and safety analyses were based on the FAS and SAS, respectively. In particular, safety was evaluated in a sub-set of the SAS consisting of patients who entered the treatment period (SAST).
Results
Patients
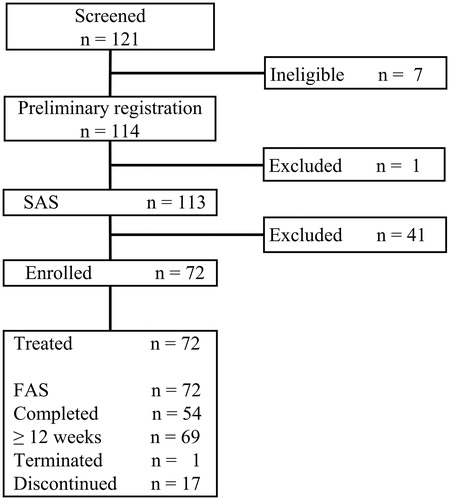
The disposition of study patients is illustrated in . Informed consent was obtained from 121 patients. After screening for study eligibility, 114 patients were preliminarily registered into the run-in period to receive placebo tablets. Among the patients who completed the run-in period, 42 were excluded according to the inclusion or exclusion criteria, and 72 were enrolled and started treatment with 10 mg rupatadine. Among the enrolled patients, 69 patients received rupatadine for 12 or more weeks, with 54 patients completing the 52-week treatment. One of the 69 patients left the study prematurely, and 17 patients prematurely discontinued the study during the treatment period. The reasons for discontinuation were: withdrawal of consent (n = 2), violation of eligibility criteria (i.e. use of prohibited concomitant medications, n = 5), and non-medical reasons (e.g. work-related reasons, n = 8).
Figure 2. Disposition of study patients. SAS indicates safety analysis set; and FAS, full analysis set.
The FAS, SAS, and SAST included 72, 113, and 72 patients, respectively. The SAST was identical to the FAS. The FAS consisted of 58 adults and 14 adolescents, with a mean age of 32.1 years (36.6 years for adults and 13.9 years for adolescents). The gender distribution was comparable between the overall population, adult sub-group, and adolescent sub-group of the FAS, with the proportions of males in each group being 56.9%, 56.9%, and 57.1%, respectively. The FAS had a mean body weight of 58.21 kg (61.00 kg for adults and 46.63 kg for adolescents). While 50.0% of the patients had a mean baseline T4NSS score ≥ 9, the proportion was 44.8% for adults and 71.4% for adolescents. These results indicated that the nasal symptoms were comparatively worse in the adolescent patients. All patients were positive (Class 2 or above) for house-dust allergen, and 59 patients (81.9%) were positive for cedar pollens. Further details on the demographic and other baseline characteristics of the FAS population are provided in Supplementary Table S2.
The mean (SD) drug adherence rate (i.e. the percentage of days with medication) during the treatment period was 99.48% (1.08%). All patients (n = 72) had a drug adherence rate ≥80%, with a mean (SD) rupatadine exposure of 4,554.4 (2,024.5) mg over a mean (SD) duration of 318.8 (89.7) days. The rupatadine dose was increased from 10 to 20 mg in 36 patients (50.0%). Only one patient (1.4%) underwent dose reduction, and this patient had the dose reduced to 10 mg after receiving 20 mg doses for 211 days. No dose reduction due to adverse events occurred. The mean duration of rupatadine 20 mg treatment in these patients was 275.0 days. The distribution of the patients with a dose increase to 20 mg is graphically depicted in Supplementary Figure S1. The graph shows a bimodal distribution, with the first peak from Day 18 to Day 103 (August 2015 to October 2015) and the second from Day 188 to Day 243 (January 2016 to March 2016).
Efficacy
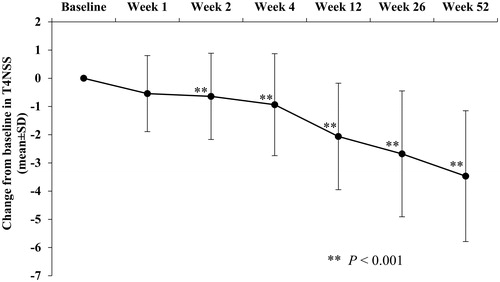
The primary efficacy analysis showed that the mean (95% CI) change from baseline in the T4NSS score to Week 2 was −0.642 (−1.002, −0.283), and this decrease was statistically significant (p < 0.001). The statistically significant decrease observed at Week 2 persisted to the end of the study treatment, exhibiting the long-term effect of rupatadine in alleviating AR nasal symptoms ( and Citation4). The mean T4NSS score showed a mild increase from Week 34 to Week 40 relative to Week 32 and Week 42. The changes over time in the individual scores (sneezing, rhinorrhea, nasal congestion, and nasal itching scores) showed a similar pattern (Supplementary Table S3).
Figure 3. Change of score over time in the total 4 nasal symptom score: full analysis set.
The results of the sub-group analyses of the mean T4NSS change from baseline to Week 2 by age showed that adolescents yielded a mean score increase of 0.075 at Week 2. However, the mean T4NSS changes from baseline were similar between the adolescent and adult sub-groups at Week 12 (−2.039 vs −2.068, respectively) and at Week 52 (−3.385 vs −3.489, respectively). The proportion of the adolescents that showed an increase in T4NSS (35.7%, 5/14) was lower than that of the adults with an increase in T4NSS (53.4%, 31/58). By body weight, the lowest weight sub-group (< 50 kg) had a smaller change (−0.115) than other weight sub-groups (50–59 kg, −0.691; 60–69 kg, −1.090; and ≥ 70 kg, −0.615). The analyses of the change from baseline to Week 2 in the T4NSS by gender and baseline T4NSS score showed no noteworthy differences between sub-groups (Supplementary Table S4).
The changes in the T4NSS score in patients who underwent rupatadine updosing from 10 to 20 mg (n = 36) and patients who did not undergo rupatadine updosing (n = 36) with similar mean baseline values were analyzed post-hoc (). Regarding the T4NSS score after 1 week of dose increase in patients updosed to 20 mg, the score was reduced to a comparable level to that of the T4NSS score achieved by patients administered with fixed dose at Week 4. The results indicated that rupatadine 20 mg was a viable option in PAR patients whose symptoms could not be effectively controlled with rupatadine 10 mg.
Table 1. Post-hoc analysis of change in the total 4 nasal symptom score (T4NSS) in patients whose rupatadine dose was increased from 10 to 20 mg (Updosed group) and patients whose rupatadine dose remained at 10 mg (10 mg group).
On the basis of the finding that the patients who underwent updosing to 20 mg showed an approximately 2-fold smaller mean reduction from baseline in the T4NSS score than the patients whose dose remained at 10 mg (−0.415 vs −0.869 at Week 2 and −0.665 vs −1.421 at Week 4), further post-hoc analyses were conducted by the timing of updosing (). Patients who underwent updosing at Week 5 (n = 12) showed an increase from baseline in the T4NSS score during the first 3 days of 10 mg treatment (0.222), during Week 1 (0.226), during Week 2 (0.071), and during Week 4 (0.095). In contrast, patients who underwent updosing at Week 7 or later (n = 24) and those who did not undergo updosing (n = 36) showed consistent decreases from baseline in the T4NSS score over time (): −0.319 and −0.565, respectively, during the first 3 days of treatment; −0.611 and −0.754, respectively, during Week 1; −0.659 and −0.869, respectively, during Week 2; and −1.046 and −1.421, respectively, during Week 4. However, the change in the T4NSS score from 1 week before updosing to the second week after updosing was comparable between the patients who underwent updosing at Week 5 (−1.679) and those who underwent updosing at Week 7 or later (−1.363) ().
The changes over time in the JRQLQ score are summarized in . Rupatadine treatment gradually improved the QoL of patients with PAR. In particular, the proportion of patients reporting “no” sleep disturbance increased from 12.5% (n = 9) at baseline to 33.3% (n = 18) at Week 52. Furthermore, the proportion of patients reporting “severe” or “very severe” sleep disturbance decreased from 29.2% (n = 21) to 3.7% (n = 2) within the same time frame. The sub-group analyses of the JRQLQ score changes showed no noteworthy trends by age, body weight, gender, or baseline T4NSS score (data not shown).
Table 2. Change over time in the JRQLQ score: full analysis set.
The patient and physician clinical overall impression rating results are graphically presented in Supplementary Figure S2. The proportions of patients and physicians who perceived the effect of rupatadine increased over time, and the majority of them assessed that rupatadine “extremely”, “very”, or “moderately” improved the clinical conditions at Week 12 and Week 52.
Safety
Treatment-emergent adverse events and adverse drug reactions reported in this clinical trial are summarized in . There were 94 treatment-emergent adverse events reported in 48 patients (66.7%), including nine adverse drug reactions reported in nine patients (12.5%). Only one treatment-emergent adverse event was classified as Grade 3 (increased blood creatine phosphokinase), and all other events were classified as Grade 1 or Grade 2. No treatment-emergent adverse events resulted in death or premature study discontinuation, and no serious or clinically significant cases were documented. The vital sign measurements indicated no symptomatic changes. The sub-group analyses of the occurrence of treatment-emergent adverse events and adverse drug reactions identified no specific trends by age, body weight, gender, or the presence or absence of complications.
Table 3. Treatment-emergent adverse events and adverse drug reactions reported in the treatment period.
The treatment-emergent adverse events reported at an incidence ≥2% were nasopharyngitis (40.3%), somnolence (12.5%), gastroenteritis (4.2%), increased blood creatine phosphokinase (4.2%), diarrhea (2.8%), back pain (2.8%), and eczema (2.8%). Among these adverse events, somnolence (9.7%) was the only adverse drug reaction to rupatadine.
The occurrences of treatment-emergent adverse events and adverse drug reactions are summarized by rupatadine dose and duration of treatment in . Regarding treatment-emergent adverse events with an incidence ≥2%, somnolence (4.2%, 3/72) and nasopharyngitis (2.8%, 2/72) were reported from Week 1 to Week 2; nasopharyngitis (12.5%, 9/72) and somnolence (6.9%, 5/72) were reported from Week 3 to Week 12; and nasopharyngitis (34.8%, 24/69), gastroenteritis (2.9%, 2/69), and increased blood creatine phosphokinase (2.9%, 2/69) were reported from Week 13 to Week 52. Only nasopharyngitis occurred three times or more frequently from Week 13 to Week 52 compared with Week 1 to Week 12. Regarding treatment-emergent adverse drug reactions with an incidence ≥2%, only somnolence was reported at 2.8% (2/72) from Week 1 to Week 2 and at 5.6% (4/72) from Week 3 to Week 12. No adverse drug reactions were documented at an incidence ≥2% from Week 13 to Week 52.
Table 4. Treatment-emergent adverse events and adverse drug reactions by duration of treatment and dose in 72 patients treated with rupatadine.
During the flexible-dose period (Week 3 to Week 52), nasopharyngitis (25.0%, 18/72), gastroenteritis (4.2%, 3/72), increased blood creatine phosphokinase (2.8%, 2/72), and eczema (2.8%, 2/72) were reported as treatment-emergent adverse events with rupatadine 10 mg, whereas nasopharyngitis (36.1%, 13/36), somnolence (13.9%, 5/36), diarrhea (5.6%, 2/36), gastritis (2.8%, 1/36), nasal herpes (2.8%, 1/36), back pain (2.8%, 1/36), and atopic dermatitis (2.8%, 1/36) were documented as treatment-emergent adverse events with rupatadine 20 mg. Among these adverse events, somnolence (11.1%, 4/36) and diarrhea (2.8%, 1/36) reported with rupatadine 20 mg were categorized as adverse drug reactions, but none of the adverse events reported with rupatadine 10 mg were classified as adverse drug reactions.
Discussion
This 52-week clinical trial evaluated the short- and long-term safety and efficacy of rupatadine in patients with PAR. After 2 weeks of 10 mg once-daily treatment of rupatadine, the dose was elevated to 20 mg at the discretion of the study investigator if the investigator considered it appropriate according to the clinical response of the patient. Patients were asked to remain on the study for a minimum of 12 weeks, and were allowed to remain on the study for 52 weeks. The excellent drug adherence rate of this clinical trial (mean: 99.48%) indicated that rupatadine was well tolerated and safe.
The efficacy results showed that rupatadine 10 mg effectively improves the AR nasal symptoms within 2 weeks, supporting the results of the previous placebo-controlled, 2-week clinical trial of rupatadine in Japanese seasonal AR patientsCitation19. The clinical benefit of rupatadine exhibited by Week 2 continued up to Week 52 ( and Supplementary Table S3). The patient and physician overall impression supported the long-term clinical benefit of rupatadine (Supplementary Figure S2). Moreover, the clinical benefit of rupatadine was supported by the JRQLQ results, which showed QoL improvements from baseline over time (). In particular, rupatadine markedly improved sleep disruption.
Sleep disorders can be a major health challenge in patients with ARCitation20,Citation21. In a French cross-sectional epidemiological study of 591 AR patients and 502 control subjects, AR was shown to impair all dimensions of sleepCitation22. The feeling of fatigue upon awakening was reported in 43.7% of patients. Daytime fatigue and sleepiness, as well as impaired memory, mood, and sexual health, were reported more frequently in patients than in the controls. The effect of rupatadine in improving sleep quality deserves further attention because sleep quality is important for physical and mental healthCitation23.
Rupatadine relieved nasal congestion as effectively as other AR nasal symptoms (Supplementary Table S3). Considering that the mechanism underlying nasal blockage involves histamine and PAF in addition to prostaglandin D2, kinin, leukotrienes, and other chemical mediators, the pharmacokinetic and pharmacodynamic features of rupatadine suggest its efficacy in treating nasal congestion. Our findings reflected the aforementioned characteristics of rupatadine, idiosyncratic to conventional second-generation oral antihistamines which are commonly described to have “little effect” on nasal congestionCitation24,Citation25. The characteristic of rupatadine as a PAF-antagonist possibly contributed to alleviating nasal blockage. Furthermore, desloratadine, a key metabolite with a longer tmax than its unmetabolized precursor, could have contributed in reducing nasal congestionCitation26.
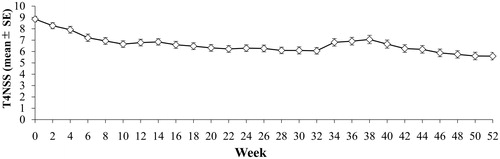
The mean T4NSS score showed a mild increase from Week 34 to Week 40 (February 11, 2016 to April 16, 2016) relative to Week 32 and Week 42, and this period coincided with the cedar pollen season in Japan (). This mild transient increase in the T4NSS score may be explained by the fact that more than 80% of the FAS patients were classified as cedar pollen-specific IgE antibody class 2 or higher (Supplementary Table S2).
Figure 4. Change over time in T4NSS: full analysis set. The upper and lower bars represent the SE. T4NSS indicates total 4 nasal symptom score; and SE, standard error.
This clinical trial provided additional evidence on the safety information of rupatadine 10 and 20 mg doses currently availableCitation8,Citation27,Citation28. All of the adverse drug reactions to rupatadine were Grade 1, with an incidence rate of 12.5%. Somnolence was the only adverse drug reaction reported at an incidence rate of 2% or higher (9.7%). The safety profile reported in this clinical trial was generally comparable with that reported in a placebo-controlled, Phase III clinical trial of rupatadine in Caucasian patients with PARCitation28. Somnolence was the only adverse event whose incidence was statistically higher with rupatadine than with placebo in Caucasians, with the incidence being 11% with rupatadine 10 mg and 20% with rupatadine 20 mg.
In this clinical trial, the effects of rupatadine dose increase from 10 to 20 mg were investigated. The results showed a significant reduction in the T4NSS score after a dose increase (). The benefit of rupatadine 20 mg observed in our clinical trial is in agreement with the results of the Phase III clinical trial of rupatadine in Caucasian patients with PARCitation28.
The number of patients with a dose increase to 20 mg showed a bimodal distribution, with the second peak appearing between Day 188 and Day 243 (Supplementary Figure S1). The first peak is logically attributable to insufficient response to 10 mg doses. Regarding the second peak (n = 4), the fact that it fell between January and March suggests the possibility that the dose increase was associated with cedar pollen exposure.
Reduction of T4NSS score during the first 4 weeks of 10 mg treatment in patients who underwent updosing at Week 5 was not observed, whereas the patients who underwent updosing at Week 7 or later exhibited a favorable initial treatment response, evident by the reduction in the mean T4NSS from baseline to Week 1, similar to that of the patients who did not undergo updosing (). These findings suggest that there were at least two types of patients who required updosing to 20 mg once daily in clinical settings; the patients who underwent updosing at Week 5 represented a group of patients who required a higher dose because of the lack of immediate improvement with the starting dose, and the patients who underwent updosing at Week 7 or later represented a group of patients who experienced disease aggravation, despite their favorable initial response to rupatadine 10 mg, possibly due to the exposure to cedar pollen and other seasonal allergens during the course of the treatment. Regardless of the presumed cause for updosing, patients who underwent updosing from both groups showed a similar drug response to 20 mg (). Furthermore, our post-hoc analyses showed that the baseline T4NSS score was comparable between the patients who did and did not require updosing to 20 mg. This suggested that the baseline disease severity (T4NSS) does not serve as a reliable predictor of the need for updosing.
Figure 5. Post-hoc analyses of change from baseline in total 4 nasal symptom score (T4NSS) by the timing of rupatadine updosing to 20 mg: updosing at week 5, updosing at week 7 or later, and no updosing. (a) The changes over the first 4 weeks of treatment. Patients who had no updosing and patients who underwent updosing at Week 7 or later showed the tendency of better improvement at Week 1 (p = 0.033 and p = 0.040, respectively) and Week 4 (p = 0.033 and p = 0.021, respectively) than patients who underwent updosing at Week 5. (b) The changes from 1 week before updosing to the second week after updosing.
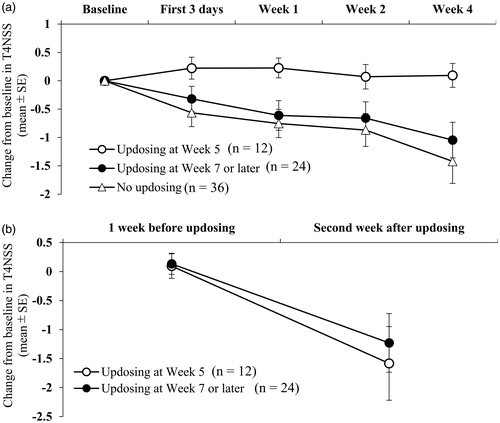
Altogether, our data suggested that the clinical starting dose of rupatadine should be 10 mg once daily as recommended in the package insert and that 20 mg is a reasonable option for patients whose AR nasal symptoms cannot be effectively controlled within 1 to 2 weeks of rupatadine 10 mg therapy. Given that the higher rupatadine dose is associated with an elevated frequency of somnolence and other adverse drug reactions (), clinicians should weigh the advantage and disadvantage for individual patients before prescribing the higher dose. In most cases, the side-effects are mild and manageable.
The sub-group analyses of the safety and efficacy results revealed no particular differences between adolescents and adults. Therefore, the results obtained in this clinical trial are generally applicable to both age sub-groups. However, the adolescent sub-group showed a minor increase in the mean T4NSS score from baseline at Week 2, although Week 12 and Week 52 results were comparable between the two age sub-groups. The apparent lack of short-term benefit in younger patients observed in this clinical trial may be attributable to the higher proportion of patients with T4NSS score ≥ 9 (worse conditions) at baseline in the adolescent than in the adult sub-group. Another explanation may be that adolescents require a longer treatment time than adults. The small sample size of the adolescent patients prevented reliable conclusions to be made on this point.
Study limitations included the open-label, uncontrolled, single-arm design of this clinical trial. The absence of a placebo control arm necessitates careful interpretation of the results, and the open-label nature of this clinical trial may have allowed the outcomes to be influenced by the power of expectancyCitation29. Second, the small sample size of adolescent patients in this clinical trial foreclosed meaningful statistical testing. A future study to investigate the effects of rupatadine in a larger cohort of adolescent patients will help overcome the limitations of this clinical trial. Third, most of the patients of this clinical trial were also allergic to cedar and other pollens, and this could have influenced the study results. For example, the incidence of nasopharyngitis reported from Week 13 to Week 52 was 3-times higher than the incidence of nasopharyngitis reported from Week 3 to Week 12. The fact that this period covered the Japanese cedar pollen season suggests that the 3-fold increase was caused, at least partly, by cedar pollen exposure. Despite these limitations, our clinical trial shows that rupatadine is an effective therapy for treating PAR in adult and adolescent patients.
Conclusions
This clinical trial showed the following: (i) rupatadine 10 and 20 mg are effective and safe for treating PAR in long-term use, (ii) a dose increase to 20 mg is a reasonable option in PAR patients whose symptoms are not effectively controlled by the 10 mg dose, and (iii) rupatadine improves the QoL of PAR patients.
Transparency
Declaration of funding
This study was funded by Teikoku Seiyaku Co., Ltd., the sponsor of this clinical trial.
Disclosure of financial/other interests
KO has received honoraria and/or consultant fees from Teikoku Seiyaku (the sponsor of this study), Taiho Pharmaceutical Co., Ltd, Mitsubishi Tanabe Pharma Corp., Torii Pharmaceutical Co., Ltd, Meiji Seika Pharma Co., Ltd, Kyorin Pharmaceutical Co. Ltd, and Hisamitsu Pharmaceutical. TS, AT, and HA are employees of Teikoku Seiyaku.
Supplemental Material
Download MS Word (249 KB)Acknowledgements
The sponsor of this study, Teikoku Seiyaku Co., Ltd., has borne the costs for the clinical trial and preparation and submission of this manuscript, including third-party writing assistance. The authors are indebted to the contribution of Dr Kazuhiro Hashiguchi of the Shinanozaka Clinic, Tokyo, Japan, who served as the study investigator of this clinical trial. The authors thank Dr Toshimitsu Hamasaki of the National Cerebral and Cardiovascular Center, Osaka, Japan, for his statistical advice and Dr Iñaki Izquierdo Pulido of J. Uriach y Cia S.A., Barcelona, Spain, for scientific review of this manuscript. The authors acknowledge the technical assistance of Intellim Corp., Tokyo, Japan with regard to study monitoring, data collection and management, and statistical analysis. The authors also acknowledge the editorial assistance of Mr Yasushi Sasaoka, Ms Shiori Mikami, and staff members at SunFlare Co., Ltd, Tokyo, Japan.
Related Research Data
References
- Seidman MD, Gurgel RK, Lin SY. Clinical practice guideline: Allergic rhinitis executive summary. Otolaryngol Head Neck Surg. 2015;152:197–206.
- Sibbald B, Rink E. Epidemiology of seasonal and perennial rhinitis: clinical presentation and medical history. Thorax. 1991;46:895–901.
- Bousquet J, Khaltaev N, Cruz AA, et al. Allergic Rhinitis and its Impact on Asthma (ARIA) 2008 update (in collaboration with the World Health Organization, GA2LEN and AllerGen). Allergy. 2008;63:8–160.
- Zahradnik E, Raulf M. Animal allergens and their presence in the environment. Front Immunol. 2014;5:76.
- Okubo K, Kurono Y, Ichimura K, et al. Japanese guidelines for allergic rhinitis 2017. Allergol Int. 2017;66:205–219.
- Scadding GK, Durham SR, Mirakian R, et al. BSACI guidelines for the management of allergic and non-allergic rhinitis. Clin Exp Allergy. 2007;38:19–42.
- Täubel J, Ferber G, Fernandes S, et al. Pharmacokinetics, safety and cognitive function profile of rupatadine 10, 20 and 40 mg in healthy Japanese subjects: a randomised placebo-controlled trial. PLoS One. 2016;11:e0163020.
- Shamizadeh S, Brockow K, Ring J. Rupatadine: efficacy and safety of a non-sedating antihistamine with PAF-antagonist effects. Allergo J Int. 2014;23:87–95.
- Carceller E, Merlos M, Giral M, et al. [(3-Pyridylalkyl)piperidylidene]benzocycloheptapyridine derivatives as dual antagonists of PAF and histamine. J Med Chem. 1994;37:2697–2703.
- Alfaro V. Role of histamine and platelet-activating factor in allergic rhinitis. J Physiol Biochem. 2004;60:101–111.
- Shirasaki H, Seki N, Kikuchi M, et al. Expression and localization of platelet-activating factor receptor in human nasal mucosa. Ann Allergy Asthma Immunol. 2005;95:190–96
- Ministry of Health, Labor and Welfare. Rheumatism and Allergy Countermeasure Committee Report. October 2005. Available at: http://www.mhlw.go.jp/shingi/2005/10/dl/s1031-6a.pdf. Accessed: June 14, 2015(in Japanese).
- World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. Jama. 2013;310:2191–2194.
- Ellis AK, Soliman M, Steacy L, et al. The Allergic Rhinitis – Clinical Investigator Collaborative (AR-CIC): nasal allergen challenge protocol optimization for studying AR pathophysiology and evaluating novel therapies. Allergy Asthma Clin Immunol. 2015;11(1):16.
- Okuda M, Ohkubo K, Goto M, et al. [Japanese Allergic Rhinitis Standard QOL Questionnaire (2002)]. Arerugi [Jpn J Allergol]. 2003;52:21–56 (in Japanese).
- Okuda M, Ohkubo K, Goto M, et al. Comparative study of two Japanese rhinoconjunctivitis quality-of-life questionnaires. Acta Otolaryngol. 2005;125:736–744.
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). MedDRA® the Medical Dictionary for Regulatory Activities. A registered trademark of the International Federation of Pharmaceutical Manufacturers and Associations (IFPMA); Chantilly, VA: Northrop Grumman MSSO (distributors). 2019[March 13]. http://www.meddra.org/
- National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) Version 4.03. 2019[March 13]. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm#ctc_40
- Okubo K, Suzuki T, Tanaka A, et al. Efficacy and safety of rupatadine in Japanese patients with seasonal allergic rhinitis: A double-blind, randomized, multicenter, placebo-controlled clinical trial. Allergol Int. 2018;pii:S1323–S8930. 18)30119-9.
- Koinis-Mitchell D, Craig T, Esteban CA, et al. Sleep and allergic disease: a summary of the literature and future directions for research. J Allergy Clin Immunol. 2012;130:1275–1281.
- Chiba S. [Nasal symptoms and sleep disorder in patients with allergic rhinitis]. Rinsho Meneki Arerugika [Clin Immunol Allergol] 2009;52:518–23 (in Japanese).
- Léger D, Annesi-Maesano I, Carat F, et al. Allergic rhinitis and its consequences on quality of sleep: An unexplored area. Arch Intern Med. 2006;166:1744–1748.
- Muliol J, Maurer M, Bousquet J. Sleep and allergic rhinitis. J Investig Allergol Clin Immunol. 2008;18:415–419.
- Sur DK, Scandale S. Treatment of allergic rhinitis. Am Fam Physician. 2010;81:1440–1446.
- Panigrahi R, Acharya SK. Recent trends in management of allergic rhinitis. Aijcr. 2016;9:130–136.
- Nayak AS, Schenkel E. Desloratadine reduces nasal congestion in patients with intermittent allergic rhinitis. Allergy. 2001; 56:1077–1080.
- Mullol J, Bousquet J, Bachert C, et al. Update on rupatadine in the management of allergic disorders. Allergy. 2015;70:1–24.
- Marmouz F, Giralt J, Izquierdo I. Morning and evening efficacy evaluation of rupatadine (10 and 20 mg), compared with cetirizine 10 mg in perennial allergic rhinitis: a randomized, double-blind, placebo-controlled trial. J Asthma Allergy. 2011;4:27–35.
- Colagiuri B, Morley K, Boakes R, et al. Expectancy in double-blind placebo-controlled trials: an example from alcohol dependence. Psychother Psychosom. 2009;78:167–171.