
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Researchers contest the importance of gene flow in bacterial core genomes, as traditionalists view microbes as predominantly clonal, asexually reproducing organisms. Contrary to the traditional perspective, Escherichia coli core genes vary greatly in their levels of synonymous genetic diversity. This observation indicates that the relative importance of evolutionary forces such as mutation, selection, and recombination varies from gene to gene. In this paper, I highlight why the synonymous diversity observation is broadly relevant to researchers interested in the evolutionary dynamics of microbial populations and communities. I explain how a model of evolution called the coalescent relates neutral diversity (i.e. mutations with negligible fitness effects) to mutation rates, evolutionary time, and a parameter called effective population size. I then describe the possible ways in which mutation, selection, and recombination can explain observed patterns of synonymous diversity in E. coli. Finally, I describe a model for E. coli genome evolution in which different loci are subject to varying levels of gene flow among co-occurring microbes and viruses in the environment. Researchers can falsify the gene flow hypothesis by sequencing genes and strains isolated from stable microbiomes or by carrying out evolution experiments that trace gene genealogies in real-time.
Evolutionary dynamics of the Escherichia coli genome
As for many microbes, gene content across Escherichia coli strains is quite variable. The E. coli genome comprises a core set of genes shared by all E. coli isolates, and a set of flexible genes found in some but not all E. coli isolates. A commonplace assumption is that core genes share a common history of vertical descent. Over time, E. coli lineages accumulate mutations that have negligible effects on fitness. The rate at which these neutral mutations accrue is roughly proportional to the mutation rate. Synonymous mutations are a reasonable proxy for truly neutral mutations, because their fitness effects are usually (but not always) negligible compared to nonsynonymous mutations that change amino acid sequence.Citation1 From this line of reasoning it follows that levels of synonymous genetic diversity in core genes should be roughly proportional to the mutation rate at those core genes.
However, levels of synonymous genetic diversity vary by more than an order of magnitude over core E. coli genes.Citation1,2 Such variation in levels of synonymous diversity causes the branch lengths of some gene trees to be uniformly longer than the branches of other gene trees without affecting tree topology. Trees for highly expressed, important housekeeping genes tend to have shorter branch lengths (less synonymous diversity) than less important core genes. The implication is that either the mutation rate unexpectedly varies over orders of magnitude over core E. coli genes, or that there is a serious flaw in the preceding argument linking synonymous diversity to mutation rates. The rest of this paper delves into the evolutionary theory behind synonymous diversity, and examines the evolutionary forces that could cause synonymous diversity to vary over E. coli core genes. I argue that it is a mistake to assume that core genes in the same E. coli genome share the same history of vertical descent, when in fact recombination and gene transfer can cause the history of core genes (or pieces of core genes) present in the same genome to differ substantially without affecting the topologies of bacterial phylogenies.Citation3
The Wright-Fisher model and the coalescent: Neutral models of molecular evolution
In this section, I explain how neutral models of evolution help in understanding patterns of synonymous diversity. The neutral theory of molecular evolution makes clear predictions for how genetic drift, in the absence of all other evolutionary forces, shapes genetic diversity.Citation4 Neutral theory has become an essential tool for studying genome evolution because it is the null hypothesis that must be rejected before considering more complicated explanations for patterns of molecular variation.Citation5 I highly recommend refs. 6-8 to readers who are interested in a broader overview as well as a deeper exposition of the following ideas.
The Wright-Fisher model of neutral evolution describes an idealized population of N organisms (). In the absence of natural selection, all organisms are equally fit. We measure time in discrete generations, and the population size is fixed at N. Every generation, we randomly pick organisms from the current generation to leave offspring in the next generation. As in all neutral models, evolution reduces to random sampling of a finite population.
Figure 1. The population size in a neutral model of evolution also describes the average time for 2 lineages to coalesce in that model. A) One run of the Wright-Fisher model over 4 generations for a population of 4 individuals. B) The coalescent for the run of the Wright-Fisher model in part A). C) The probability that it takes t generations for 2 lineages to coalesce is identical to the probability of flipping t – 1 tails before flipping heads using a biased coin that has a probability of flipping heads (i.e., coalescence) of 1/N. A geometric distribution with mean N describes both processes.
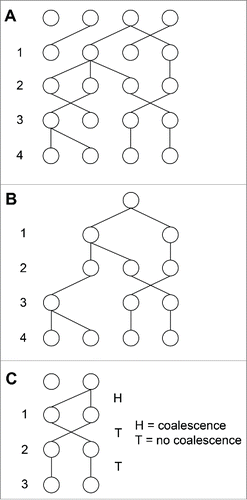
Due to random sampling, eventually the whole population descends from a single organism. If we trace the ancestry of a population backward in time, eventually we come to this individual: the most recent common ancestor (MRCA) of the population. The basic premise of the coalescent is that we run a model of neutral evolution backward in time to the MRCA (). The history of 2 given individuals coalesces in the generation in which they share a common ancestor. At any point in time, the probability that a second organism has the same ancestor as a first organism is . Therefore, the probability that 2 specific individuals coalesce in one generation is
, and the probability that they do not coalesce is 1 –
. Eventually, the histories of all individuals in the population coalesce to that of the MRCA.
The probability that 2 specific individuals in the current generation coalesce t generations in the past is the probability that they do not coalesce for t – 1 generations backward in time and then coalesce in the tth generation: . The coalescence of a pair of organisms is thus described by a geometric random variable X with a mean of N generations. The mathematics is identical to flipping a coin until reaching a flip of heads (). Intuitively, it takes 2 coin flips on average to flip heads once. Flipping a long stretch of tails before flipping heads is unlikely with a fair coin, because the probability of flipping a long stretch of tails before flipping heads decreases geometrically
. Coalescence for 2 specific individuals is like flipping a biased coin where the probability of heads (coalescence) is
, and the probability of tails (no coalescence) is
.
Effective population size, coalescence times, and neutral diversity
It is important to remember that N is not the population size for organisms evolving in the real world, but the population size of organisms in an idealized model of neutral evolution. For this reason, researchers add a subscript to make it clear that Ne is the population size of the idealized model of neutral evolution that best fits molecular data. Much of the power of coalescent theory derives from the fact that more complicated models of evolution involving recombination, natural selection, and population structure make predictions for patterns of molecular variation that are identical to a neutral model with an appropriately scaled effective population size Ne.9 In general, effective population sizes are usually orders of magnitude smaller than actual census population sizes in nature.Citation9 For example, a population that has experienced a recent selective sweep or population bottleneck coalesces to the MRCA after a short period of time, causing a dramatically lower effective population size with regard to levels of neutral genetic diversity. Researchers interested in bacterial speciation have used computer simulations to demonstrate that recombination, mutation, and population structure (i.e., dividing a population into many subpopulations) can cause populations to cluster or diverge genetically in the absence of natural selection. In these models, effective population size is simply the number of organisms in the simulation, and levels of neutral genetic diversity depend on the relative importance of recombination, mutation, and population structure in the model. In neutral models, clusters of diverged genotypes (“species”) do not easily form in recombining populations, implying a strong role for either natural selection or strong population subdivision (or both) in bacterial speciation.Citation10,11
In clonal populations, neutral genetic diversity should accumulate uniformly across the genome because all genes in a genome are completely linked, and thus equally affected by evolutionary forces such as mutation or natural selection. Variation in synonymous genetic diversity among core genes allows us to reject the null hypothesis that core E. coli genes experience the same evolutionary forces. Neutral theory applies equally well to genes as to individuals, so on average, the MRCA for 2 neutrally evolving sequences existed Ne generations in the past. If the mutation rate µ is constant over the genome, then the number of neutral genetic differences between 2 sequences in the present day is θ = 2 µNe . If we use synonymous variation as a proxy for neutral genetic changes, then synonymous diversity θs = 2 µNe is a natural statistical estimator for both the effective population size as well as the coalescence time for pairs of sequences. In the next section, I discuss possible explanations why synonymous genetic diversity varies so much across the core genome of E. coli.
Explanations for variation in synonymous diversity in E. coli core genes
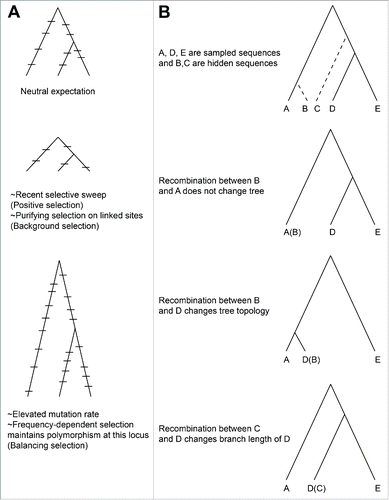
Many evolutionary forces, including mutation, selection, and recombination, have similar as well as correlated effects on both µ and Ne. Disentangling the contributions of these forces to patterns of natural variation remains challenging. I discuss the effects of these evolutionary processes on µ and Ne in turn ().
Figure 2. Mutation, selection, and recombination affect the branch lengths and topology of phylogenetic trees. A) Differing selection pressures or mutation rates can lengthen or shorten branch lengths. B) Recombination with an ingroup will not change the tree, while recombination with an outgroup always changes either the topology of the tree or disproportionately changes the length of some branches.
Mutation
One explanation for why some genes are more variable than others is mutation rate variation. While there is good evidence for local differences in the point mutation rate in bacterial genomes, explanations that solely rely on local mutation rate variation are implausible because no studies to date have found a correlation between mutation rates and patterns of synonymous variation in E. coli.Citation1 In short, variation in the mutation rate does not appear to be strong enough to explain orders of magnitude differences in synonymous genetic diversity across E. coli core genes.
Natural selection
Selection plays an important role in determining genetic variability across loci. When a highly beneficial mutation sweeps through a population (positive selection), it also reduces genetic variability at linked sites and decreases the time to coalescence to the MRCA. Because a selective sweep reduces variation at all linked sites, this explanation cannot account for patterns in synonymous genetic diversity in E. coli without sufficient recombination, because a selective sweep uniformly reduces standing genetic diversity in completely clonal populations.
Background selection is a more satisfying explanation for patterns of synonymous diversity in E. coli. Housekeeping core genes are more conserved on the amino acid level than other core genes, because mutations in these most essential core genes can have large effects on organismal fitness. This form of selection is known as purifying selection because it promotes sequence conservation. Purifying selection on deleterious mutations also decreases variability at nearby sites in the genome, and selection on neutral mutations due to purifying selection on nearby sites is called background selection. Background selection is the most parsimonious explanation for variation in synonymous diversity, although Martincorena et al.Citation2 rejected it as a sufficient explanation.
Negative frequency-dependent selection (balancing selection) on a locus preserves genetic diversity. Such beneficial mutations do not complete selective sweeps because the fitness advantage conferred by the mutation decreases as it increases in frequency in the population. Mutations conferring frequency-dependent advantages are common in evolution experiments,Citation12 and are probably even more common in complex and heterogeneous environments such as the animal gut. However, this explanation again requires recombination, otherwise frequency-dependent selection would maintain synonymous variation at similar levels across the genome.
Recombination
Many studies have estimated the relative contributions of recombination and mutation to E. coli diversity.Citation13,14 An important open question outside the scope of this paper is how and why diverse bacterial species and populations vary in their propensity toward freely-recombining and clonal lifestyles. Some natural populations of Synechococcus have enough homologous recombination to generate quasisexual evolutionary dynamics,Citation15 while some Pseudomonas populations appear to be largely clonal.Citation16
Recombination can affect synonymous diversity because a recombination event between diverged sequences causes multiple changes to appear simultaneously, while recombination between closely related or even identical sequences may not be detectable at all. If some genes have had a history of more successful recombination events with diverged homologs compared to other genes in the genome, then those genes will be more diverse than genes with a history of fewer successful recombination events. However, recombination with diverged homologs cannot explain observed patterns of synonymous diversity in E. coli. Any recombination event with an outgroup will either change the topology of the gene tree or cause anomalously long branches (), while observed patterns of synonymous diversity in E. coli core genes are inconsistent with these predictions.Citation2 Nonetheless, a combination of recombination and positive selection or negative frequency-dependent selection could account for some of the observed variation in synonymous diversity.
Mutagenic effects of recombination
Recent sequencing studies have found that new mutations correlate with the location of recent crossover events in human sperm as well as in plants and honeybees.Citation17,18 It is unclear whether the molecular mechanisms responsible for elevated mutated rates in these studies also occur in E. coli. Nonetheless, error-prone repair of double-strand breaks associated with recombination events could contribute to higher levels of synonymous diversity at loci with a history of many successful but undetected recombination events in E. coli.
Population structure
Population structure measures the degree to which populations are not well-mixed. A simple case is a metapopulation, or a population subdivided into a large number of subpopulations. Populations can be structured at multiple spatial scales (i.e. subpopulations of subpopulations), and population structure generally maintains genetic diversity by restricting the scope of selective sweeps. Population structure can also reduce effective population sizes and coalescence times due to local extinctions, colonization events and local population bottlenecks.Citation11
Gene flow could explain patterns of synonymous genetic variation in E. coli
In this section, I present a model that combines aspects of recombination, selection, and population structure to explain patterns of synonymous genetic variation in Escherichia coli. Although this model is not parsimonious, it is testable and consistent with existing molecular and ecological observations in the literature.Citation19
While it is well-known that flexible E. coli genes differ in their histories of recombination and selection across diverged microbial species in gut communities, the same may hold true for many E. coli core genes. Imagine a “wind” of diverse alleles blowing into a population of E. coli, this “wind” being the migration of alleles into the population from other E. coli populations, viral populations, or other microbes in the community. Resident genes under purifying selection can resist this “wind” more strongly, and they will have a shorter coalescence time than genes that cannot effectively resist replacement by diverse alleles. In terms of the Wright-Fisher model, gene flow between species within a community increases the effective population size of that gene compared to species-specific genes (). This argument is general in that it holds for subpopulations of a single bacterial species, or for populations of co-evolving phage and bacteria. For instance, imagine 2 subpopulations of E. coli, each adapted to different parts of an animal’s gut. Genes under stronger purifying selection in one subpopulation would better resist gene flow from the other subpopulation. The key point in this model is that gene flow within microbial communities can change effective population sizes and coalescence times at core genes without changing the topology of gene trees constructed with single isolates from diverse ecological sources. In the most extreme cases, between-species divergence and within-species polymorphism may be indistinguishable. One likely mechanism for gene flow in microbial communities are phage-bacteria infection networks in which generalized transducing phage infect multiple microbial species and act as viral vectors.Citation13,20 The gene flow model makes a strong prediction: genes with high synonymous diversity should tend to cluster according to microbial community, while genes with low synonymous diversity should tend to cluster by species (). Evolution experiments or appropriate sampling of microbiomes could test this prediction to falsify the gene flow model.
Figure 3. Different rates of gene flow at different loci causes effective population size to vary at these loci, in turn affecting gene tree coalescence times without changing tree topology for genes co-occurring in the same genome. A) Gene flow at this locus occurs between species within communities, increasing the effective population size of this locus. In this case, communities cluster in the gene tree. B) Gene flow does not occur between species at this second locus. The effective population size at this locus is the population size of the species in which it is found. In this case, species cluster in the gene tree.

The gene flow model has some support in the literature. Retchless et al.Citation21 proposed the fragmented speciation model in which different segments of bacterial chromosomes become genetically isolated at different times. Species-specific alleles become isolated first; alleles can sweep across species boundaries, and gene flow stops earlier at earlier diverging loci. This study came to the conclusion that in some cases, it may not be possible to make a clear distinction between intraspecific and interspecific variability in microbes. Sheppard et al.Citation22 found evidence of increasing gene flow between previously distinct Campylobacter species. Retchless et al.Citation23 argued that phylogenetic incongruence in gene trees made with genes found in Escherichia, Salmonella, and Citrobacter provides further evidence for the fragmented speciation model. Luo et al.Citation24 described the genomes of environmental isolates of E. coli and found little evidence of gene exchange with gut commensal E. coli due to plausible ecological barriers. Although they found within-clade transfer of core genes, this paper rejected the fragmented speciation model because fragmented speciation posits gene flow across E. coli clades except at niche-specific adaptive mutations or genetic incompatibilities restricting gene flow. Karberg et al.Citation25 found that recently acquired genes in Salmonella and Escherichia genomes have similar codon usage frequencies, while core genes in Salmonella and Escherichia have noticeably diverged in codon usage. Therefore, it appears that Salmonella and Escherichia strains acquire genes from a common pangenome shared among enterobacterial species. Smillie et al.Citation26 built a database of horizontally transferred sequences among 2,235 full bacterial genomes to explore the effects of phylogeny, geography, and ecology on horizontal gene transfer. This study found that shared ecology is far more important than phylogenetic relatedness in structuring networks of gene flow across bacterial species.
Conclusion
Synonymous genetic diversity depends on both the mutation rate and effective population size. In neutral models of evolution, effective population size has a second interpretation as the average time for 2 lineages to coalesce. Many evolutionary forces, including mutation, selection, and recombination can affect genome-wide variation in synonymous genetic diversity. While researchers recognize the importance of gene flow in structuring the flexible genome of microbes, gene flow may also affect the core genome of microbes. If so, gene flow could explain why highly important E. coli core genes have less synonymous genetic diversity than other core genes. While the importance of gene flow in microbial genome evolution depends strongly on ecological context, many important microbiomes, such as the animal gut, might be effectively described as metapopulations of genes that interact within and across genomes over multiple spatial and temporal scales.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
I thank Alita Burmeister and Michael Wiser for comments on the manuscript; and I thank John Wakeley, Sergey Kryazhimskiy, and Justin Meyer for critical comments and discussions; and I thank Richard Lenski for guidance and support.
Funding
This work was supported by the BEACON Center for the Study of Evolution in Action (National Science Foundation Cooperative Agreement DBI-0939454).
References
- Maddamsetti R, Hatcher PJ, Cruveiller S, Médigue C, Barrick JE, Lenski RE. Synonymous genetic variation in natural isolates of Escherichia coli does not predict where synonymous mutations occur in a long-term experiment. Mol Biol Evol 2015; 32:2897-904; PMID:26199375; http://dx.doi.org/10.1093/molbev/msv161
- Martincorena I, Seshasayee AS, Luscombe NM. Evidence of non-random mutation rates suggests an evolutionary risk management strategy. Nature 2012; 485:95-8; PMID:22522932; http://dx.doi.org/10.1038/nature10995
- Hedge J, Wilson DJ. Bacterial phylogenetic reconstruction from whole genomes is robust to recombination but demographic inference is not. MBio 2014; 5: e02158; PMID:25425237; http://dx.doi.org/10.1128/mBio.02158-14
- Kimura, M. The neutral theory of molecular evolution. Cambridge: Cambridge University Press; 1983. 384 p.
- Lynch M. The origins of genome architecture. Sunderland (MA): Sinauer Associates; 2007. 494 p.
- Wakeley J. Coalescent theory: an introduction. Greenwood Village (CO): Roberts & Company Publishers; 2009. 352 p.
- Rice SH. Evolutionary theory. Sunderland (MA): Sinauer Associates; 2004. 370 p.
- Hartl DL, Clark AG. Principles of population genetics. 4th ed. Sunderland (MA): Sinauer Associates; 2006. 545 p.
- Charlesworth B. Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat Rev Genet 2009; 10:195-205; PMID:19204717; http://dx.doi.org/10.1038/nrg2526
- Fraser C, Hanage WP, Spratt BG. Recombination and the nature of bacterial speciation. Science 2007; 315:476-80; PMID:17255503; http://dx.doi.org/10.1126/science.1127573
- Fraser C, Alm EJ, Polz MF, Spratt BG, Hanage WP. The bacterial species challenge: making sense of genetic and ecological diversity. Science 2009; 323:741-6; PMID:19197054; http://dx.doi.org/10.1126/science.1159388
- Maddamsetti R, Lenski RE, Barrick JE. Adaptation, clonal interference, and frequency-dependent interactions in a long-term evolution experiment with Escherichia coli. Genetics 2015; 200:619-31; PMID:25911659; http://dx.doi.org/10.1534/genetics.115.176677
- Dixit PD, Pang TY, Studier FW, Maslov S. Recombinant transfer in the basic genome of Escherichia coli. Proc Natl Acad Sci U S A 2015; 112:9070-5; PMID:26153419; http://dx.doi.org/10.1073/pnas.1510839112
- Bobay LM, Traverse CC, Ochman H. Impermanence of bacterial clones. Proc Natl Acad Sci U S A 2015; 112:8893-900; PMID:26195749; http://dx.doi.org/10.1073/pnas.1501724112
- Rosen MJ, Davison M, Bhaya D, Fisher DS. Fine-scale diversity and extensive recombination in a quasi-sexual bacterial population occupying a broad niche. Science 2015; 348:1019-23; PMID:26023139; http://dx.doi.org/10.1126/science.aaa4456
- Sarkar SF, Guttman DS. Evolution of the core genome of Pseudomonas syringae, a highly clonal, endemic plant pathogen. Appl Environ Microbiol 2004; 70:1999-2012; PMID:15066790; http://dx.doi.org/10.1128/AEM.70.4.1999-2012.2004
- Arbeithuber B, Betancourt AJ, Ebner T, Tiemann-Boege I. Crossovers are associated with mutation and biased gene conversion at recombination hotspots. Proc Acad Natl Sci U S A 2015; 112:2109-14; http://dx.doi.org/10.1073/pnas.1416622112
- Yang S, Wang L, Huang J, Zhang X, Yuan Y, Chen JQ, Hurst LD, Tian D. Parent-progeny sequencing indicates higher mutation rates in heterozygotes. Nature 2015; 523:463-7; PMID:26176923; http://dx.doi.org/10.1038/nature14649
- Polz MF, Alm EJ, Hanage WP. Horizontal gene transfer and the evolution of bacterial and archaeal population structure. Trends Genet 2013; 29:170-5; PMID:23332119; http://dx.doi.org/10.1016/j.tig.2012.12.006
- Modi SR, Lee HH, Spina CS, Collins JJ. Antibiotic treatment expands the resistance reservoir and ecological network of the phage metagenome. Nature 2013; 499:219-22; PMID:23748443; http://dx.doi.org/10.1038/nature12212
- Retchless AC, Lawrence JG. Temporal fragmentation of speciation in bacteria. Science 2007; 317:1093-6; PMID:17717188; http://dx.doi.org/10.1126/science.1144876
- Sheppard SK, McCarthy ND, Falush D, Maiden MC. Convergence of Campylobacter species: implications for bacterial evolution. Science 2008; 320:237-9; PMID:18403712; http://dx.doi.org/10.1126/science.1155532
- Retchless AC, Lawrence JG. Phylogenetic incongruence arising from fragmented speciation in enteric bacteria. Proc Natl Acad Sci U S A 2010; 107:11453-8; PMID:20534528; http://dx.doi.org/10.1073/pnas.1001291107
- Luo C, Walk ST, Gordon DM, Feldgarden M, Tiedje JM, Konstantinidis KT. Genome sequencing of environmental Escherichia coli expands understanding of the ecology and speciation of the model bacterial species. Proc Natl Acad Sci U S A 2011; 108:7200-5; PMID:21482770; http://dx.doi.org/10.1073/pnas.1015622108
- Karberg KA, Olsen GJ, Davis JJ. Similarity of genes horizontally acquired by Escherichia coli and Salmonella enterica is evidence of a supraspecies pangenome. Proc Natl Acad Sci U S A 2011; 108:20154-9; PMID:22128332; http://dx.doi.org/10.1073/pnas.1109451108
- Smillie CS, Smith MB, Friedman J, Cordero OX, David LA, Alm EJ. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 2011; 480:241-4; PMID:22037308; http://dx.doi.org/10.1038/nature10571