
Abstract
Current approach to understanding bacteriophage transcription strategies during infection includes a combination of experimental and bioinformatics approaches, which is often time and resource consuming. Given the exponentially growing number of sequenced bacteriophage genomes, it becomes sensible asking to what extent one can understand bacteriophage transcription by using bioinformatics methods alone. We here argue that a suitable choice of computational methods may provide a highly efficient first-line approach for underst-anding bacteriophage transcription.
Regulation of gene expression is in prokaryotes, to a large degree, exhibited through the control of transcription initiation, which heavily relies on the sequence-specific recognition of promoter elements by sigma factors from RNAP holoenzyme.Citation1 Therefore, predicting promoters with reliable precision and uncovering the nature of interaction with a given sigma factor is an essential first step in understanding the control of transcription initiation. Moreover, for simple organisms, like bacteriophages, the understanding of gene expression regulation – i.e. transcription strategy, can be straightforwardly used for inferring their infection strategy. This becomes especially important given the potential utility of lytic bacteriophages, or their protein products, as highly specific weapons for fighting the growing number of bacterial pathogens resistant to antibiotics.Citation2
Promoter prediction in bacteria has long been a classical bioinformatics problem. Despite almost 3 decades of committed work, the existing methods for promoter prediction exhibit poor accuracy, i.e., they typically lead to a large number of false positivesCitation3 This mainly applies to the prediction of RpoD promoters, which are recognized by the bacterial housekeeping sigma factors from Sigma 70 Group I. In addition to the bacterial housekeeping promoters, bacteriophages also contain promoters that are recognized by their own sigma factors/RNA polymerases, which further complicates the promoter prediction in their genomesCitation4,5 The switching between sigma factors of different specificities allows transitioning through subsequent bacteriophage infection stages. This is accompanied by unequal temporal expression of distinct gene classes – therefore, the elucidation of temporal pattern of gene expression can facilitate promoter prediction, and vice versa, in an attempt to infer the phage infection strategy.
Standard Approach to Inferring Phage Infection Strategies
The standard approach to inferring phage infection strategies employs combined experimental and bioinformatics approach. A typical example of such approach is provided by the analysis of the transcription strategy of a lytic coliphage phiEco32 Citation6 Specifically, macroarray measurements of gene expression levels allowed clustering phiEco32 genes according to the temporal pattern of their expression, i.e. to early, middle and late genes. Since the transition between different temporal classes is accompanied by redirection of transcription machinery to promoters of different specificity, gene clustering by maccroarrays provided the information in which intergenic regions one should be looking for promoters of phage-encoded sigma factors – the promoters were subsequently revealed through primer extension analysis. The phage-specific promoters were first searched by bioinformatic means, but the unknown specificity combined with other unique features of phage promoters, which will be assessed further in the text, resulted in the failure of the applied unsupervised search; note that the unsupervised search had to be used, as the specificity of the phage-specific promoters was not known in advance. On the other hand, promoters of host sigma factors (bacterial RpoD) were predicted through bioinformatics procedure, though the search was now supervised – i.e., it used weight matrices based on alignments of experimentally confirmed RpoD promoters. Altogether, a combination of macroarray temporal gene clustering with biochemical and bioinformatics promoter prediction allowed defining different stages during phiEco32 gene expression, i.e. inferring the underlying infection process.
While this approach provides a wealth of information on bacteriophage transcription strategy, it is demanding both in time and resources. Accordingly, having in mind the availability of sequencing, and the resulting exponentially growing pace of bacteriophage genomes being sequenced, it becomes sensible to ask if there is a more efficient, less expensive, approach to analyzing phage genomes. This question raises even more interest from the perspective of significance and potential applications of phages in medicine and biotechnology, and the increasing number of incoming new isolates Citation7 Therefore, the ultimate goal of a prompt and efficient analysis of newly-sequenced genomes would be providing rapid assumptions on lifestyles and potential usefulness of novel phages, by shedding light on their transcription strategies. Provided the availability of a growing number of bioinformatics resources for analyzing biological data, the aim of this commentary is clarifying the extent to which one can understand bacteriophage transcription by using bioinformatics methods alone.
Bioinformatics Approach to Analyzing Newly-Sequenced Phage Genomes
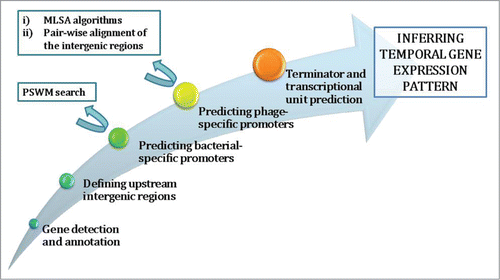
The starting step in analyzing phage genomes, and inferring the underlying infection strategy (Fig. 1), is gene prediction and annotation, which is a straightforward part of the analysis. The genes can be predicted by using some of the gene prediction algorithms, e.g. GeneMark Citation8 that reaches almost 99% accuracy when predicting ORFs. The predicted genes are usually annotated by looking for homologues in databases through BLAST. The gene prediction provides an overview of phage genome organization – i.e., inferring gene clusters in terms of their transcription orientation and presence of long intergenic regions; these long intergenic regions usually contain the most important cis-acting regulators of phage transcription – promoters and terminators. Further, the gene annotation reveals the functions of proteins that a given bacteriophage codes for, though only to a certain point, having in mind the extent of the phage genome mosaicism Citation9 However, in addition to already established genome organization, the gene annotation sometimes provides enough information for establishing the homology to some other, more thoroughly studied phage representative – this being of special importance for providing first clues on the regulatory pattern of transcription for a given phage. Finally, note that predicting phage infection strategies is reasonably robust to potential inconsistencies in the annotation of individual phage genes, as the process is based on inferring the gene classes (e.g., structural genes) that are transcribed in the same direction/cluster, for which annotations of a larger number of genes are used.
Detecting bacterial promoters in phage genomes
The promoter prediction comes as the next (and more complicated) part of the analysis, which includes both the phage-specific and host-bacterial promoter detection in the phage genome, as stated above. Even though bioinformatics predictions of RpoD promoters in bacterial genomes typically lead to many false-positives, this does not appear as a major obstacle in bacteriophage genomes; in a first place – since there is a notably less sequence quantity to be analyzed – typical total size of phage non-coding regions is of the order of 10Citation4 bp. More precisely, the total length of the intergenic regions in 7–11 phage genome, which has recently been analyzed entirely through bioinformatics methods, is ∼13 000 bp Citation10 Secondly, bacterial promoters in phage genomes are typically strong due to the need for elevated production of phage proteins in the host cell – this further simplifies the RpoD promoter detection.
As an illustration, we will now focus on the host promoter prediction in the phage 7–11 genome, which infects Salmonella enterica and has not been experimentally examined. The use of a PSWM (position specific weight matrix) approach – equivalent to the one in a coliphage phiEco32 analysis - led to discovery of RpoD promoters in the 7–11 genome, all localized on the reverse strand, where they govern the transcription of “-” orientated genes. Note that the predictions based on the weight matrices, in distinction to those based simply on the similarity to the consensus sequence, take into account that different positions within the binding motif, as well as different bases at a given position, can have different contributions to the binding affinity.
The majority of the detected promoter elements in 7–11 genome were found right upstream of the “-” gene cluster, with 2 additional copies – with weaker weight matrix scores – found within the cluster. This finding is in a good agreement with the conventional view of bacteriophage transcription, which assumes sharp grouping of phage genes into divergently transcribed clusters (one located on the reverse, and the other on the forward strand). Accordingly, promoters of different specificities – the phage-specific and bacterial RpoD, which govern the transcription of the opposite gene clusters, are also segregated to different strands – each promoter class right upstream of its respective cluster. In addition, a subset of phage-specific promoters can also be found on the same strand as the RpoD promoters, but these are localized in the downstream part of the respective RpoD cluster, where they are implicated in the transcription machinery takeover and expression of the phage middle genes.
Detecting phage-specific promoters in phage genomes
As opposed to the host promoter detection in the phage genomes, detection of the promoters recognized by the phage-encoded sigma factors/RNA polymerases is generally more complicated. The procedure corresponds to an unsupervised search of phage intergenic regions since, in the vast majority of cases, specificities of phage sigma factors/RNA polymerases are not known in advance. A whole set of specialized bioinformatics methods – multiple local sequence alignment algorithms (MLSA) were developed for the unsupervised search of regulatory elements, majority of which is based on the Gibbs search (BioProspector, Gibbs Motif Sampler) and EM – expectation maximization approach (MEME)Citation11-14.
EM methods are rigorous, deterministic algorithms which employ extensive process of integration in search for the most optimal solution. What may seem as a major advantage, can in fact become a serious shortcoming, since in parallel with the increase in the problem dimensionality (e.g., number of analyzed sequences) there is also a significant increase in the time needed to solve the problem – the employed integration may grow exponentially with the problem dimension. Hence, the method becomes computationally rather expansive which further imposes the limitation of successfully analyzing only a restricted set of sequences. Also, governed by the goal of finding the most optimal solution, EM methods suffer from the problem of getting trapped in the local maxima, when exploirng the probability landscape during integration. On the other hand, Gibbs search is a heuristic procedure, which increases the speed at the expense of sensitivity when detecting degenerated regulatory motifs. In addition to the increase in the search speed, which is a major advantage from the aspect of practical applications in bioinformatics, stochastic sampling brings another advantage over deterministic procedures – evading the problem of getting trapped in some of the local maxima. However, due to the stochastic nature of the sampling, the Gibbs-based algorithms often report suboptimal results which may differ between multiple runs of the algorithm on the same dataset. Also, as determining statistical significance of the obtained results is still an open problem, it is often needed to employ different implementations of the same basic algorithm (e.g., Gibbs Motif Sampler, BioProspector) to verifiy the reported motifs. Even though the underlying approach is different – whereby Gibbs algorithms represent stochastic, while EM methods deterministic sampling, these methods share the same, significant shortcoming – similar motifs, which randomly appear in the analyzed sequences, can mask the regulatory elements one is aimed identifying.
When analyzing bacteriophages, this shortcoming becomes even more obstructing, since phage-specific promoters are found in low copy numbers across the genome, which however often have pronounced strength. In a support of this notion, we return to the analysis of phages phiEco32 and 7–11,Citation10 where the authors indeed showed that the phage-specific promoters could not be detected through the standard bioinformatics methods (MLSA algorithms), when relying on the information provided solely by the sequence of the analyzed intergenic regions. More precisely, different implementations of the same basic algorithm (BioProspector and Gibbs Motif Sampler) instead of converging toward the same motif, report evidently unrelated sequence elements.
The established difficulty was so far addressed by relying on the experimental data (measurement of gene expression levels during infection), as provided by the example of phiEco32 genome analysis. The analogous approach was also used for analyzing the Xp10 phage, which infects a rice pathogen Xanthomonas oryzae and encodes its own RNA polymeraseCitation15 The simple method that enables clustering phage genes into different temporal expression classes can also be used to facilitate bioinformatics phage-specific promoter detection, since it reduces the search to a specified subset of intergenic regions, and, therefore, minimizes the chance for promoter masking by randomly appearing sets of motifs. More precisely, when the information on which groups of genes are transcribed in a certain stage of the infection (early, middle or late) is available, one can single out the intergenic regions located upstream of these genes as the ones expected to contain a certain class of promoter elements, and analyze them through bioinformatics methods. Due to the requirement for experimental data, this is no more than a halfway solution, so we will below discuss an alternative approach for direct bioinformatics prediction of phage-specific promoters.
Prediction of phage-specific promoters is also an interesting question from the bioinformatics aspect, since the problem corresponds to detecting several short motifs in a relatively long DNA sequence, which was addressed inCitation10 for phages 7–11, phiEco32 and Xp10. Failure of the MLSA algorithms in phage-specific promoter detection was partially accounted for in the text above. Moreover, the main reason for the occurrence of promoter masking by randomly repeating motifs, is that MLSA algorithms assume presence of searched motifs in a majority of the analyzed sequences, which is contrary to the rather small number of phage promoters as compared to the total number of the intergenic regions. By taking into account typically small variability of the sequences of the phage specific promoters, the authors of the previously mentioned study resorted to a simplified procedure that is based on a pair-wise alignment of phage intergenic regions - which can be implemented through e.g. BLAST, so that the method is readily available and well-known to a wide community of researchersCitation16 As a matter of caution, we should indicate that the specified pair-wise alignment can only detect promoters with a suficiently low level of the sequence variability, which, fortunately, is a widely shared feature of the phage-specific promoters. The study demonstrates that this pair-wise alignment procedure can detect the phage-specific promoters in all of the genomes analyzed. Results obtained for phiEco32 and Xp10 were verified through the comparison with their experimentally confirmed phage-specific promoters, whereby the 7–11 predictions were verified through the established homology with phiEco32 phage.
Predicting phage transcription strategy from the phage genome analysis
Predictions of the host and phage-specific promoters, present the main obstacle in defining the transcriptional units in bacteriophages, as the terminator predictions are much more straightforward – i.e. are based on detecting the stem-loop followed by poly-U tailCitation17 Consequently, the detected transcription signals - together with the information on gene homology - enables inferring plausible predictions on bacteriophage infection strategy, as demonstrated in the case of 7–11,Citation10 and also visually represented in the form of a transcriptional map provided by Figure 3 in reference 10. The analysis indicates that this phage, upon entering the bacterial cell, directs the host transcription machinery to its early promoters, localized upstream of the “-” gene cluster, from which the transcription of the early genes is initiated. For transcription regulation, the antisigma factor gene is the most important early gene, whose protein product disables bacterial RpoD sigma factor, and, therefore, enables core RNAP to form a complex with the phage-encoded sigma factor and initiate transcription of the phage middle genes from the phage-specific promoters. The annotation infers that the middle genes code for proteins, which are mainly involved in genome replication and therefore serve to ensure the production of a sufficient number of phage genome copies for upcoming viral progeny. As a last stage during infection, phage-specific late promoters, localized on the + strand, become predominantly active, most probably due to their longer motif that enables sufficient strength, and therefore, expression of structural genes for capsid formation and lysis of the bacterial cell. Herewith, all the conditions for releasing new phages to enter novel infections cycles are fulfilled; at the same time, putative expression patterns of all the key players involved in obstructing bacterial transcription and cell integrity are inferred. Among these, phage-encoded sigma and antisigma factors, along with RNA-polymerases, merit most of the attention.
Figure 1. Flow-chart of the underlying steps in the bioinformatics analysis of phage genomes. For promoter prediction, which is the most challenging part of the analysis, the bioinformatic methods that can be employed are specified (rectangle boxes in the upper left part of the figure). Note that MLSA stands for Multiple Local Sequence Alignment, while PSWM stands for Position Specific Weight Matrix – these bioinformatic methods will be further discussed in the text.
Outlook
The bioinformatics methods that we discussed above enable efficiently and systematically inferring the specificities for a variety of phage-encoded sigma factors and RNA polymerases. One such example is provided by the sigma factors encoded by phiEco32 and 7–11 genomes that belong to ECF subfamilyCitation10,18 This subfamily of sigma factors is very diverse and most abundant among alternative sigma factors, but poorly studiedCitation19 All the information available so far on the mechanism of promoter recognition comes from a few well examined bacterial representatives and new data are strongly influenced by the existing ones, which could eventually result in establishing incorrect/incomplete paradigms. To that end, bacteriophage ECF sigma factors, as outliers within this subfamily, could serve as suitable models for acquiring novel, self-contained data and shed light on previous discoveries, this being further encouraged by the easiness of bioinformatics phage analysis, due to a noteworthy simplicity of their genomes.
This presents only one example of how studying the regulation of bacteriophage gene expression can surpass the initial goals, which are mainly oriented toward efficiently inferring transcription strategies for a growing number of sequenced phage genomes, and provide for a successful crossover toward new, fundamental discoveries. Many others are known from the long-standing history of molecular biology, since the vast majority of fundamental paradigms on the genome structure and function were established through the research on bacteriophages as model systemsCitation20,21 We can, therefore, safely conclude that by combining the simplicity and peculiarity of phages as model organisms with the high efficiency of bioinformatics analyses, one can provide a plausible approach for quickly acquiring new insights, which could easily provide guidelines for applications in medicine and biotechnology or lead to establishing novel paradigms on fundamental processes in molecular biology.
To facilitate discovery of such novel paradigms, and to make the corresponding bioinformatic analysis available to a wide range of researchers, it would be quite useful to develop appropriate bioinformatic work-flows and pipilines that would authomate analysis of the newly sequenced phages. In fact, as we discussed here, the most challenging part of the bioinformatics analysis is transcription start site prediction, which is in general still an open problem, where in bacterial genomes a substantial number of false positives is generally obtained. However, as argued in the commentary, some of the methods which generaly lead to a large number of false positives in bacterial genomes (in particular PSWM searches), are, in fact, quite reliable in the case of bacteriophages – this being a consequence of short phage genome sizes and generally strong promoter signals. On the other hand, some of the well established methods for motif finding (in particular MLSA algorithms), are not well suited for bacteriophages, but can be substituted by more simple, yet robust, methods (in particular, pairwise alignment of the intergenic regions). We therefore hope that this commentary will motivate developing such work-flows in a reasonably near future, with the ingradient methods well suited for the specifics of the bacteriophage genome analysis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Work by MD and JG is supported by Marie Curie International Reintegration Grant within the 7th European community Framework Program (PIRG08-GA-2010-276996), by the Ministry of Education and Science of the Republic of Serbia under project number ON173052, and by the Swiss National Science foundation under SCOPES project number IZ73Z0_152297.
References
- Feklistov A, Sharon BD, Darst SA, Gross CA. Bacterial sigma factors: a historical, structural, and genomic perspective. Annu Rev Microbiol 2014; 68:357-76; PMID:25002089; http://dx.doi.org/10.1146/annurev-micro-092412-155737
- Viertel TM, Ritter K, Horz HP. Viruses versus bacteria-novel approaches to phage therapy as a tool against multidrug-resistant pathogens. J Antimicrob Chemother 2014; 69 (9):2326-36; PMID:24872344; http://dx.doi.org/10.1093/jac/dku173
- Djordjevic M. Integrating sequence analysis with biophysical modelling for accurate transcription start site prediction. J Integr Bioinform 2014; 11 (2):240; PMID:24953455
- Losick R, Pero J. Cascades of Sigma factors. Cell 1981; 25 (3):582-4; PMID:6793235; http://dx.doi.org/10.1016/0092-8674(81)90164-1
- Yuzenkova J, Nechaev S, Berlin J, Rogulja D, Kuznedelov K, Inman R, Mushegian A, Severinov K. Genome of Xanthomonas oryzae bacteriophage Xp10: an odd T-odd phage. J Mol Biol 2003; 330 (4):735-48; PMID:12850143; http://dx.doi.org/10.1016/S0022-2836(03)00634-X
- Pavlova O, Lavysh D, Klimuk E, Djordjevic M, Ravcheev DA, Gelfand MS, Severinov F, Akulenko N. Temporal regulation of gene expression of the Escherichia coli bacteriophage phiEco32. J Mol Biol 2012; 416 (3):389-99; PMID:22261232; http://dx.doi.org/10.1016/j.jmb.2012.01.002
- Haq IU, Chaudhry WN, Akhtar MN, Andleeb F, Qadri I. Bacteriophages and their implications on future biotechnology: a review. Virol J 2012; 9:9; PMID:22234269; http://dx.doi.org/10.1186/1743-422X-9-9
- Borodovsky M, Lomsadze A. Gene identification in Prokaryotic genomes, phages, metagenomes, and EST sequences with GeneMarkS suite. Curr Protoc Microbiol 2014; 32:1E 7 1-1E 7 17; PMID:24510847
- Hatfull GF, Hendrix RW. Bacteriophages and their genomes. Curr Opin Virol 2011; 1 (4):298-303; PMID:22034588; http://dx.doi.org/10.1016/j.coviro.2011.06.009
- Guzina J, Djordjevic M. Inferring bacteriophage infection strategies from genome sequence: analysis of bacteriophage 7-11 and related phages. BMC Evol Biol, 2015.15 Suppl 1:S1; PMID:25708710
- Lawrence C, Altschul S. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science 1993; 262 (5131):208; PMID:8211139; http://dx.doi.org/10.1126/science.8211139
- Bailey T, Williams N, Misleh C, Li WW. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res 2006; 34:W369-73; PMID:16845028; http://dx.doi.org/10.1093/nar/gkl198
- Liu X, Brutlag D, Liu J. BioProspector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac Symp Biocomput 2001:127-38; PMID:11262934.
- Thompson W, Rouchka EC, Lawrence CE, Gibbs recursive sampler: finding transcription factor binding sites. Nucleic Acids Res 2003; 31 (13):3580-5; PMID:12824370 ; http://dx.doi.org/10.1093/nar/gkg608
- Djordjevic M, Semenova E, Shraiman B, Severinov K. Quantitative analysis of a virulent bacteriophage transcription strategy. Virology 2006; 354 (2):240-51; PMID:16887164; http://dx.doi.org/10.1016/j.virol.2006.05.038
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215 (3):403-10; PMID:2231712; http://dx.doi.org/10.1016/S0022-2836(05)80360-2
- Ermolaeva MD, Khalak HG, White O, Smith HO, Salzberg SL. Prediction of transcription terminators in bacterial genomes. J Mol Biol 2000; 301 (1):27-33; PMID:10926490; http://dx.doi.org/10.1006/jmbi.2000.3836
- Savalia D, Westblade LF, Goel M, Florens L, Kemp P, Akulenko N, Pavlova O, Padovan JC, Chait BT, Washburn MP, et al. Genomic and proteomic analysis of phiEco32, a novel Escherichia coli bacteriophage. J Mol Biol 2008; 377 (3):774-89; PMID:18294652; http://dx.doi.org/10.1016/j.jmb.2007.12.077
- Staron A, Sofia HJ, Dietrich S, Ulrich LE, Liesegang H, Mascher T. The third pillar of bacterial signal transduction: classification of the extracytoplasmic function (ECF) sigma factor protein family. Mol Microbiol 2009; 74 (3):557-81; PMID:19737356; http://dx.doi.org/10.1111/j.1365-2958.2009.06870.x
- Gottesman M. Bacteriophage lambda: the untold story. J Mol Biol 1999; 293 (2):177-80; PMID:10550203; http://dx.doi.org/10.1006/jmbi.1999.3137
- Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, Rüger W. Bacteriophage T4 genome. Microbiol Mol Biol Rev 2003; 67 (1):86-156, table of contents; PMID:12626685; http://dx.doi.org/10.1128/MMBR.67.1.86-156.2003