
Abstract
Statins lower cholesterol and are commonly prescribed for prevention and treatment of cardiovascular disease risk. Statins have pleotropic actions beyond cholesterol lowering, including decreased protein prenylation, which can alter immune function. The general anti-inflammatory effect of statins may be a key pleiotropic effect that improves cardiovascular disease risk. However, a series of findings have shown that statins increase the pro-inflammatory cytokine, IL-1β, via decreased protein prenylation in immune cells. IL-1β can be regulated by the NLRP3 inflammasome containing caspase-1. Statins have been associated with an increased risk of new onset diabetes. Inflammation can promote ineffective insulin action (insulin resistance), which often precedes diabetes. This review highlights the links between statins, insulin resistance and immunity via the NLRP3 inflammasome. We propose that statin-induced changes in immunity should be investigated as a mechanism underlying increased risk of diabetes. It is possible that statin-related insulin resistance occurs through a separate pathway from various mechanisms that confer cardiovascular benefits. Therefore, understanding the potential mechanisms that segregate statin-induced cardiovascular effects from those that cause dysglycemia may lead to improvements in this drugs class.
Abbreviations
| HMGCR | = | HMG-CoA reductase |
| LDL | = | Low density lipoprotein |
| GGPP | = | geranylgeranyl pyrophosphate |
| IL | = | interleukin |
| CRP | = | C-reactive protein |
| TNF | = | Tumor necrosis factor |
| SNP | = | single nucleotide polymorphisms |
| NLRP3 | = | NOD-like receptor family pyrin domain containing 3 |
| PRR | = | pattern recognition receptors |
| BMDM | = | bone marrow derived macrophages |
| AMPK | = | AMP-activated protein kinase |
| PTEN | = | phosphatase and tensin homolog |
| SREBP | = | sterol response element-binding protein |
| P2X7 | = | P2X purinoceptor 7 |
| PI3K | = | Phosphoinositol-3-kinase |
| PIP2 | = | phosphatidylinositol 4,5-bisphosphate |
| PIP3 | = | phosphatidylinositol 3,4,5-trisphosphate |
| PDK1 | = | phosphoinositide-dependent kinase-1 |
| MAPK | = | Mitogen activated protein kinase |
| JNK | = | c-Jun N-terminal kinase |
| ERK | = | extracellular regulated mitogen-activated protein kinase |
| mtROS | = | mitochondrial reactive oxygen species |
| ATP | = | adenosine triphosphate |
| FTase | = | Farnesyltransferase |
| GGTase-I and GGTase-II | = | Geranylgeranyltransferase I and II |
| IR | = | insulin receptor |
Statins, Cholesterol and Inflammation
Statins (such as LipitorTM, CrestorTM, LescolTM, PravacholTM, MevacorTM, ZocorTM and LivaloTM) are the most widely prescribed drug class in North America and are used for the prevention and treatment of cardiovascular disease risk.Citation1,2 Statins inhibit 3-hydroxy-3-methylglutaryl CoA reductase (HMGCR) a rate limiting step in the conversion of HMG-CoA to mevalonate, a precursor for several cellular processes. One result of HMGCR inhibition is reduced cholesterol biosynthesis and coupled with increased LDL-receptor mediated sequestration of LDL, statins are effective at lowering circulating LDL-cholesterol levels.Citation3,4 Statins have “pleotropic” actions beyond cholesterol lowering. Importantly, many cholesterol-independent, pleotropic actions of statins also involve inhibition of the mevalonate pathway.Citation5,6 Mevalonate is a precursor for the generation of farnesyl pyrophosphate and geranylgeranyl pyrophosphate (GGPP). These isoprenoids are required for ubiquinone, sterols, dolichol and for prenylation of proteins, an irreversible addition of isoprenyl lipids that occurs on ∼0.5% of cellular proteins.
Statins are generally anti-inflammatory. HMGCR inhibition and targeted decrease in protein prenylation predominantly result in skewing immune responses toward anti-inflammatory characteristics, an effect that can occur independently of cholesterol lowering.Citation5-9 This anti-inflammatory effect is thought to be a significant component of the efficacy of statin therapy and the reduction in C-reactive protein (CRP, an inflammatory marker) has been directly associated with a reduction of myocardial infarction risk.Citation10,11 This has fostered large scale efforts to understand the cholesterol- and inflammatory-mediated contributions to the efficacy of statins and testing of the “inflammatory hypothesis” to see if other non-statin, anti-inflammatory strategies can improve cardiovascular outcomes.Citation12 In addition to CRP, statins have been shown to reduce many inflammatory processes, including cell adhesion/migration and skewing cytokine profiles away from pro-inflammatory (Th1) toward Th2 characteristics.Citation7 Statins reduce pro-inflammatory cytokines such as Tumor necrosis factor (TNF) and Interleukin (IL)-6.Citation13,14 However, statins are not simply anti-inflammatory and a series of findings, including our own, have demonstrated the paradoxical statin-induced increase in IL-1β secretion as a result of decreased protein prenylation in immune cells such as macrophages.Citation15-20
Statins and Diabetes
Epidemiological evidence has revealed that several statins have been associated with small, but significant increased risk of new onset diabetes.Citation21,22 It is not yet clear if higher potency statins (often defined in terms of greater cholesterol lowering) equate to increased diabetes risk.Citation23,24 Also, there is not yet clear consensus if the use of specific statins confer greater risk.Citation21,25,26 The importance and relevance of statin-induced diabetes is hotly debated. All drugs have side effects and there are many factors to consider in this debate, including the prodigious evidence regarding the benefits of statin therapy for reducing major cardiovascular events and all-cause mortality.Citation27,28 It is also important to carefully consider the message that should be given to patients regarding statin therapy.Citation29 Much of the debate pertains to the cost/benefit analysis of statins. It has been argued that the cumulative benefits of statins outweigh the potentially small rise in new onset diabetes or other side effects such as myopathy.Citation30,31 This is a worthwhile debate for clinical practice; however, it is surprising that a drug class that improves blood lipid profiles and is largely anti-inflammatory does not improve blood glucose control, but can actually worsen it.
Why not try to discover ways to remove the risk of statin-related rise in blood glucose and limit the increased risk of diabetes? Understanding the potential mechanisms that separate the benefits of statins from those that cause dysglycemia may lead to improvements in this drugs class. This is increasingly relevant since statin use in primary prevention is expanding and raising concerns.Citation32,33 Understanding the cholesterol-dependent and pleotropic effects of statins may allow therapeutic strategies that separate biological pathways that control blood glucose from those that provide cardiovascular benefits. Such strategies may reduce diabetes incidence and actually improve the efficacy of statins since dysglycemia is an independent risk factor for premature death.Citation34 How do we start to separate the actions of statins on glycaemia versus those on cholesterol and other potentially beneficial pleotropic actions? Genetic analysis of single nucleotide polymorphisms (SNP) in the HMGCR gene across several statin trials demonstrated that the actions of statins on HMGCR inhibition partially explain the increased risk of type 2 diabetes.Citation35 This reinforces the importance of the mevalonate pathway, but does not show that the cholesterol lowering effect of statins are linked to risk of diabetes. Interrogation of other effectors that are dependent on the mevalonate pathway and relevant to blood glucose control is warranted.
The balance between insulin sensitivity and insulin secretion controls blood glucose. If increasing insulin resistance (i.e. ineffective insulin action in target cells) is not matched by increased insulin secretion, blood glucose rises and this is typical of the progression to type 2 diabetes. Inflammation has emerged as a critical factor in promoting insulin resistance and often precedes the development of type 2 diabetes. The inflammatory underpinnings of insulin resistance (and commonalties with cardiovascular disease) have been reviewed.Citation36-38 Much of this work is focused on the immunometabolism of obesity, a major risk factor of insulin resistance and consequent type 2 diabetes. An important task in this area is to understand how shared elements of nutrient and bacterial/pathogen sensing systems propagate obesity-related insulin resistance. In addition to nutrients, and bacterial factors (i.e., the microbiota), we should consider how drugs may engage the immunometabolism responses relevant to type 2 diabetes. Pattern recognition receptors of the innate immune system, such as Toll-like receptors and Nod-like receptors are a key point of convergence that can link immune responses to various metabolic characteristics of insulin resistance.Citation39-43
Statins, Insulin Resistance and NLRP3 Inflammasome
The NOD-like receptor family, pyrin domain containing (NLRP)3 inflammasome has been identified as a key connection between obesity-related immune responses and type 2 diabetes.Citation44,45 Type 2 diabetes patients have increased NLRP3 activation in immune cells and blocking or deleting specific components of the inflammasome consistently reduces insulin resistance in obese animal models.Citation46-48 The NLRP3 inflammasome has been described as a metabolic danger sensor and its relevance as a bridge between immunity and insulin resistance may be based on the ability of this inflammasome to detect and respond to a wide range of stimuli (in all of the major tissues) associated with insulin resistance and dysglycemia.Citation49,50 The NLRP3 inflammasome is a complex of at least NLRP3, ASC, and Caspase-1 that is best known for regulating biologically active IL-1β and IL-18.Citation51 Generation of downstream effectors by this inflammasome requires adequate priming followed by a signal that promotes assembly/activation. Priming can occur through activation of other pattern recognition receptors (PRR) resulting in NF-κB mediated transcription of inflammasome components such as NLRP3 and effectors such as pro-IL-1β and IL-18.Citation52 Activation signals for this inflammasome can involve changes in cellular metabolism that produce reactive oxygen species, K+ efflux, and lysosomal leakage.Citation51 The links between cellular metabolites and the NLPR3-mediated inflammatory basis of many diseases has been reviewed.Citation53 Blocking this inflammasome or its key effectors has shown promise for several chronic diseases, including diabetes.Citation54 In fact, blocking IL-1β is the current strategy for directly testing the inflammatory hypothesis of cardiovascular disease risk, which was derived from work with statins.Citation55 Conversely, it is logical that drugs could activate the NLRP3 inflammasome, particularly those that alter the cellular metabolic status.
We recently hypothesized that statins could promote insulin resistance by activating the NLRP3 inflammasome.Citation20 Our first finding corresponded with previous research showing statins increase IL-1β secretion in bone marrow derived macrophages (BMDM's) given adequate priming.Citation56 Statin-induced IL-1β secretion was dependent on NLRP3 and reversed with (a high dose of) the known inflammasome inhibitor, glyburide.Citation57 We also showed that adding back the mevalonate pathway intermediate GGPP prevented IL-1β release from BMDMs demonstrating a role for prenylated proteins. Next, we provided new information showing the importance of this statin-NLRP3 effect in metabolic tissues that help control blood glucose. Feeding a statin for several weeks to obese mice reduced insulin-stimulated glucose uptake into adipose tissue. Statin exposure increased caspase-1 activity in explanted adipose tissue, which was NLRP3-dependent and also was reduced with glyburide. Most importantly, statin treatment impaired insulin signaling in adipose tissue explants, but insulin signaling was normal in adipose tissue from mice that had lacked NLRP3 or when explants were treated with glyburide. Finally, we showed that statins could impair insulin signaling via an adipocyte autonomous cell response, but this occurred despite undetectable IL-1β.
Future Directions: Downstream of the Inflammasome
These findings connected statin treatment to insulin resistance through the NLRP3 inflammasome. However, it is not known; (1) what effector(s) downstream of the inflammatory complex is/are responsible for perturbing insulin signaling and (2) where the defect in insulin signaling cascade originates. IL-1β is a likely culprit given that this pro-inflammatory cytokine has been heavily implicated in insulin resistance and type 2 diabetes. IL-1β signaling can impair insulin signaling through engagement of MAPKs.Citation58 A direct role for IL-1β has not yet been shown and the role of NLRP3-mediated regulation of biologically active IL-1β in statin-induced insulin resistance should be tested. This is particularly important because our recent paper showed that fluvastatin impaired insulin signaling in 3T3-L1 adipocytes in a cell autonomous manner, which coincided with increased caspase-1 activity, but without a detectable rise in IL-1β. Further, it is important to understand the molecular regulation of IL-1β. In addition to statins and other NLRP3 inflammasome activators that are known to increase levels of (17 kDa) biologically active IL-1β, simvastatin and cerivastatin have recently been shown to processes pro-IL-1β to a 28 kDa intermediate form of IL-1β through a caspase-1-independent mechanism in macrophages.Citation59 Simvastatin produced this intermediate 28 kDa form of IL-1β independent of HMG-CoA reductase inhibition. This appears to be a different type of pleiotropic action of statins compared to those that arise from inhibition of various arms of the mevalonate pathway. This intermediate 28 kDa form of IL-1β was reported to not properly activate IL-1 receptor signaling and could potentially attenuate the actions of mature IL-1β. For example, this intermediate form of IL-1β can reduce IL-6 induction by mature IL-1β in macrophages by presumably interfering with IL-1 receptor activation. Therefore, if IL-1β turns out to be a critical link between statins and diabetes or if this single cytokine is directly implicated in statin-induced insulin resistance, the caspase-1-dependent and independent molecular regulation of IL-1β should be determined in the context of insulin signaling. In particular, it would be interesting to know the relative importance and net effect of intermediate and mature forms of IL-1β in altering insulin action in the metabolic cells responsible for glucose homeostasis.
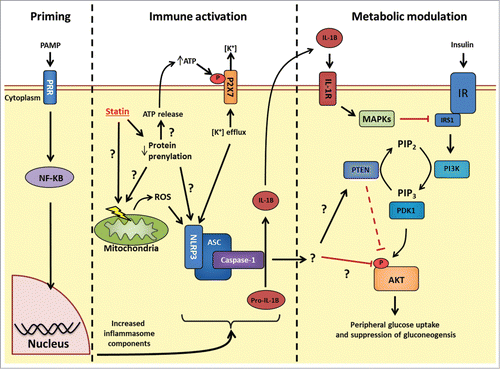
IL- 1β it is not the only candidate that could link the NLRP3 inflammasome to insulin action. The NLRP3 inflammasome also regulates biologically active IL-18. IL-18 tracks with obesity and insulin resistance, but IL-18 has recently been found to reduce weight gain and insulin resistance in mice through activation of AMP-activated protein kinase (AMPK).Citation60,61 Caspase-1 cleaves an array of proteins such as caspase-7, parkin, and enzymes of glycolysis.Citation62-64 It is not known if caspase-1 cleaves insulin signaling components or how it could facilitate crosstalk from the immune response to insulin signaling (). Recent findings from Birnbaum et al. demonstrated that rosuvastatin may relay signals through phosphatase and tensin homolog (PTEN) that inhibit AKT phosphorylation.Citation65 This builds on a very interesting connection between statin-induced increases in PTEN transcription via a sterol response element-binding protein (SREBP) pathway.Citation66 PTEN is a component in cell cycle regulation and inhibits transduction of the insulin signal by converting phosphatidylinositol (3,4,5)-trisphosphate (PIP3) back to PIP2 (), preventing activation of phosphoinositide-dependent kinase-1 (PDK1) and subsequent phosphorylation of AKT.Citation67 The relevant connections between statin-induced NLRP3 inflammasome activation, SREBPs, PTEN and insulin signaling should be investigated.
Figure 1. Possible mechanisms linking statin-induced NLRP3 inflammasome activation and insulin resistance. Priming: Following PRR stimulation, NF-κB stimulates transcriptional events that increase levels of the inflammasome (such as NLRP3) and inflammasome effectors (such as pro-IL-1β). Immune activation: HMGCR inhibition with statins causes pleotropic effects through decreased protein prenylation. Decrease in protein prenylation is a suspected cause for signals to promote increase NLRP3 inflammasome activity, but how these signals conspire to activate this inflammasome is not fully understood. Statins have been shown to cause mitochondrial membrane dysfunction, increase intracellular reactive oxygen species and also promote release of cellular ATP. Extracellular ATP can bind to the P2X7 receptor and promote potassium (K+) efflux, a key trigger for increased NLRP3 inflammasome activity. The identity of the prenylated protein(s) responsible for statin-induced inflammasome activation is not known. Following inflammasome activation, caspase-1 cleaves pro-IL-1β to biologically active IL-1β. Metabolic modulation: The connection between NLRP3 inflammasome activation and insulin signaling may occur through either IL-1β-mediated inflammation and activation MAPKs (JNK, ERK, p38) which inhibit insulin signaling at the level of receptor substrate-1 (IRS1); or through an unknown target of caspase-1 which may alter insulin signaling at the level of phosphatase and tensin homolog (PTEN) or another site such as AKT phosphorylation. PRR, pattern recognition receptor; NLRP, NOD-like receptor family, pyrin domain containing; HMGCR, HMG-CoA reductase; IL-1β, interleukin-1 β; P2 × 7, P2X purinoceptor 7; PI3K, Phosphoinositol-3-kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PDK1, phosphoinositide-dependent kinase-1; MAPK, Mitogen activated protein kinase; JNK, c-Jun N-terminal kinase; ERK, extracellular regulated mitogen-activated protein kinase; IR, insulin receptor.
Future Directions: Upstream of the Inflammasome
The statin-mediated cellular processes that elicit activation of the NLRP3 inflammasome in different cells are ill-defined. One common theme is the ability of statins to alter mitochondrial metabolism and cellular energy status in both immune and metabolic cells. Increased production of mitochondrial reactive oxygen species (mtROS), altered mitochondrial membrane potential and decreased intracellular ATP levels have been observed with concentrations of statin as low as 1 µM.Citation68-72 Statins have also been shown to promote a modest increase in cellular ROS and an increase of ATP release in THP-1 monocytes.Citation56 Statin-induced IL-1β production was also shown to be dependent on P2X purinoceptor 7 (P2X7) activation. Increased extracellular ATP can activate the P2X7 receptor, an ATP-gated ion channel resulting in cellular efflux of K+ ions. A drop in cellular K+ due to efflux has been shown to be necessary and sufficient for Caspase-1 activation and has been proposed as the unifying signal for NLRP3 inflammasome activation from various known stimuli.Citation73 It is still unclear how statin-related mitochondrial stress or changes in cellular nucleotides relate to K+ efflux and NLRP3 inflammasome activation. Statin-mediated inhibition of HMGCR, decreased mevalonate pathway intermediates and reduced protein prenylation may be a critical link, since addition of GGPP to statin-treated culture has been shown to restore ATP levels and inhibit IL-1β release.Citation17,20,56,59 However, it is not clear which prenylated protein(s) is/are involved. Three distinct prenyltransferases (Farnesyltransferase, FTase; Geranylgeranyltransferase I and II, GGTase-I and GGTase-II) are responsible for prenylation of an array of proteins including Ras, Rho and Rab family proteins.Citation74,75 Peripheral blood mononuclear cells isolated from healthy individuals when treated with an inhibitor of GGTase-I in the presence of LPS caused IL-1β secretion, but inhibition of FTase does not increase IL-1β.Citation17 Interestingly, blocking GGTase-II which prenylates Rab family proteins, prevents GGPP from restoring cellular ATP levels following addition of statin to C2C12 muscle cells.Citation69 This positions geranylgeranylated proteins, but not farnesylated proteins as key upstream signals mediating statin-induced inflammasome activation. Combining statins with potential NLRP3 inflammasome inhibitors that do not interfere with the ability of statins to reduce LDL-cholesterol appears worthy of testing. However, it is likely that such strategies should not interfere with a global reduction in protein prenylation. This may be critical in maintaining the efficacy of statins since reduced protein prenylation itself improves cardiovascular indices and lifespan -at least in flies.Citation76 Significant challenges arise in identifying which particular prenylated protein is responsible for linking statins and inflammasome activation as multiple proteins may be important and as one example there are over 60 known Rab proteins.Citation75
Priming of the NLRP3 inflammasome also should be investigated, particularly in vivo because it may reveal susceptibility to statin-induced NLRP3 activation and possibly statin-related insulin resistance or diabetes. We found no priming effect of statins alone in macrophages.Citation20 Cell or tissue culture models are typically primed with bacterial components such as LPS or peptidoglycan before an NLRP3 activating agent is tested. In theory, any stimuli that increases NF-kB-mediated transcription of inflammasome components (and/or pro-IL-1/18) is a potential priming agent for this inflammasome. It is not clear which in vivo environment contains sufficient priming signals for statin-induced inflammasome activation. An intriguing possibility would be if specific microenvironments such as expanded adipose tissue contain sufficient priming signals that are poised to be NLRP3 inflammasome activated. We speculate that there are numerous endogenous compounds that could prime the NLRP3 inflammasome including endogenous lipids, metabolic endotoxemia and/or neighboring cell death in specific tissues. All of these factors can regulate NF-κB signaling and have links to obesity and pre-diabetes. One could speculate that priming is associated with increased statin-induced diabetes risk in certain metabolic disease prone populations. Our data suggests this is worthwhile investigating because we have yet to find a protocol for feeding a statin to a healthy chow-fed mouse that alters glucose control or insulin sensitivity (unpublished), but we found that fluvastatin feeding to ob/ob mice promotes insulin resistance in adipose tissue.Citation20 Ob/ob mice have several features that could equate to increased priming of the NLRP3 inflammasome, including those related to increased adiposity (endogenous lipids, death of adipose resident cells) and substantial metabolic endotoxemia.Citation77,78 Hence, it is possible that increased priming of the NLRP3 inflammasome in ob/ob mice promotes statin-induced insulin resistance in the adipose tissue microenvironment.
Conclusion
Statins have proven effective in reducing cardiovascular events and all-cause mortality. All drugs have side effects. In addition to cost/benefit analysis, we believe it is important to understand the immunometabolism of statins in order to generate potential strategies to mitigate the side-effects of statins, including the increased risk of diabetes. Given this foundational knowledge, there may be opportunities to target the statin-mediated processes that regulate blood glucose, if they turn out to be separate from statin-mediated processes that provide cardiovascular benefits. Statin-mediated activation of the NLRP3 inflammasome is one possible immunometabolism link to the dysglycemia associated with statins. Whether this inflammasome can be targeted without altering the effectiveness of statin treatment is a key therapeutic question. Identifying the relative importance of the NLRP3 inflammasome in the face of the general anti-inflammatory effect of statins is an important biological question. These two questions are important because it is not clear why statins are effective at reducing cardiovascular disease, but increase diabetes risk. To the best of our knowledge, there has not been a definitive demonstration of an immune response that segregates the actions of statins on cardiovascular disease and insulin resistance. In fact, many elements of inflammation are shared between these 2 inter-related metabolic diseases.Citation79 One could speculate that divergent effects of statins on CVD and diabetes could be derived from the different cells types that instigate or propagate each disease process. For example, this could involve different effects of statins on endothelial cells that are more relevant to cardiovascular disease vs. the effects of statins on adipocytes, myocytes or hepatocytes that are more relevant to insulin resistance. In addition, the divergent effects of statins on cardiovascular disease versus diabetes may be the relative importance of cholesterol lowering. Statin-induced LDL-cholesterol-lowering may be far more important in preventing cardiovascular disease processes compared to those processes that drive insulin resistance or diabetes. Further, it will be important to position insulin resistance in context with statin-induced changes in insulin secretion, given that NLRP3 inflammasome in the pancreas and resident immune cells have been implicated in type 2 diabetes.Citation45,80,81 Many mechanistic questions remain about the connection between statins, NLRP3 and insulin action.Citation82 Understanding upstream and down-stream NLRP3 inflammasome effectors may foster new therapeutic or diagnostic strategies, including combination strategies or biomarkers for statin-induced disease risk.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
BDH was supported by an Ontario Graduate Scholarship (OGS). Supported by operating grants to JDS from the Canadian Institutes of Health Research (CIHR; MOP-130432) and the Canadian Diabetes Association (CDA; SC-5-12-3891-JS). JDS holds CDA Scholar (OG-3-12-3745-JS) and CIHR New Investigator awards (MSH-136665).
References
- Lindgren P, Jönsson B. Cost-effectiveness of statins revisited: lessons learned about the value of innovation. Eur J Health Econ 2012; 13:445-50; PMID:21528389; http://dx.doi.org/10.1007/s10198-011-0315-1
- Jackevicius CA, Cox JL, Carreon D, Tu JV, Rinfret S, So D, Johansen H, Kalavrouziotis D, Demers V, Humphries K, et al. Long-term trends in use of and expenditures for cardiovascular medications in Canada. CMAJ 2009; 181:E19-28; PMID:19581604; http://dx.doi.org/10.1503/cmaj.081913
- Hoeg JM, Brewer HB. 3-Hydroxy-3-methylglutaryl–coenzyme A reductase inhibitors in the treatment of hypercholesterolemia. JAMA 1987; 258:3532-6; PMID:3316727; http://dx.doi.org/10.1001/jama.1987.03400240064025
- Bilheimer DW, Grundy SM, Brown MS, Goldstein JL. Mevinolin and colestipol stimulate receptor-mediated clearance of low density lipoprotein from plasma in familial hypercholesterolemia heterozygotes. Proc Natl Acad Sci U S A 1983; 80:4124-8; PMID:6575399; http://dx.doi.org/10.1073/pnas.80.13.4124
- Greenwood J, Steinman L, Zamvil SS. Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat Rev Immunol 2006; 6:358-70; PMID:16639429; http://dx.doi.org/10.1038/nri1839
- Waiczies S, Bendix I, Zipp F. Geranylgeranylation but not GTP-loading of Rho GTPases determines T cell function. Sci Signal 2008; 1:pt3; PMID:18364514; http://dx.doi.org/10.1126/stke.112pt3
- Schönbeck U, Libby P. Inflammation, immunity, and HMG-CoA reductase inhibitors: statins as antiinflammatory agents? Circulation 2004; 109:II18-26; PMID:15173059
- Blank N, Schiller M, Krienke S, Busse F, Schatz B, Ho AD, Kalden JR, Lorenz H-M. Atorvastatin Inhibits T cell activation through 3-hydroxy-3-methylglutaryl coenzyme a reductase without decreasing cholesterol synthesis. J Immunol 2007; 179:3613-21; PMID:17785796; http://dx.doi.org/10.4049/jimmunol.179.6.3613
- Montecucco F, Mach F. Update on statin-mediated anti-inflammatory activities in atherosclerosis. Semin Immunopathol 2009; 31:127-42; PMID:19415282; http://dx.doi.org/10.1007/s00281-009-0150-y
- Albert MA, Danielson E, Rifai N, Ridker PM. Effect of statin therapy on C-reactive protein levels. JAMA 2001; 286:64; PMID:11434828; http://dx.doi.org/10.1001/jama.286.1.64
- Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, Pfeffer MA, Braunwald E. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20-8; PMID:15635109; http://dx.doi.org/10.1056/NEJMoa042378
- Ridker PM. Moving beyond JUPITER: will inhibiting inflammation reduce vascular event rates? Curr Atheroscler Rep 2013; 15:295; PMID:23225175; http://dx.doi.org/10.1007/s11883-012-0295-3
- Ando H, Takamura T, Ota T, Nagai Y, Kobayashi K. Cerivastatin improves survival of mice with lipopolysaccharide-induced sepsis. J Pharmacol Exp Ther 2000; 294:1043-6; PMID:10945857
- Rosenson RS, Tangney CC, Casey LC. Inhibition of proinflammatory cytokine production by pravastatin. Lancet 1999; 353:983-4; PMID:10459915; http://dx.doi.org/10.1016/S0140-6736(98)05917-0
- Kuijk LM, Mandey SH, Schellens I, Waterham HR, Rijkers GT, Coffer PJ, Frenkel J. Statin synergizes with LPS to induce IL-1beta release by THP-1 cells through activation of caspase-1. Mol Immunol 2008; 45:2158-65; PMID:18242710; http://dx.doi.org/10.1016/j.molimm.2007.12.008
- Lindholm MW, Nilsson J. Simvastatin stimulates macrophage interleukin-1beta secretion through an isoprenylation-dependent mechanism. Vascul Pharmacol 2007; 46:91-6; PMID:16942919; http://dx.doi.org/10.1016/j.vph.2006.07.001
- Mandey SHL, Kuijk LM, Frenkel J, Waterham HR. A role for geranylgeranylation in interleukin-1beta secretion. Arthritis Rheum 2006; 54:3690-5; PMID:17075828; http://dx.doi.org/10.1002/art.22194
- Montero MT, Matilla J, Gomez-Mampaso E, Lasuncion MA. Geranylgeraniol regulates negatively caspase-1 autoprocessing: implication in the Th1 response against Mycobacterium tuberculosis. J Immunol 2004; 173:4936-44; PMID:15470035; http://dx.doi.org/10.4049/jimmunol.173.8.4936
- Montero MT, Hernández O, Suárez Y, Matilla J, Ferruelo AJ, Martínez-Botas J, Gómez-Coronado D, Lasunción MA. Hydroxymethylglutaryl-coenzyme A reductase inhibition stimulates caspase-1 activity and Th1-cytokine release in peripheral blood mononuclear cells. Atherosclerosis 2000; 153:303-13; PMID:11164419; http://dx.doi.org/10.1016/S0021-9150(00)00417-2
- Henriksbo BD, Lau TC, Cavallari JF, Denou E, Chi W, Lally JS, Crane JD, Duggan BM, Foley KP, Fullerton MD, et al. Fluvastatin causes NLRP3 inflammasome-mediated adipose insulin resistance. Diabetes 2014; 63:3742-7; PMID:24917577; http://dx.doi.org/10.2337/db13-1398
- Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, de Craen AJM, Seshasai SRK, McMurray JJ, Freeman DJ, Jukema JW, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 2010; 375:735-42; PMID:20167359; http://dx.doi.org/10.1016/S0140-6736(09)61965-6
- Ma T, Tien L, Fang C-L, Liou Y-S, Jong G-P. Statins and new-onset diabetes: a retrospective longitudinal cohort study. Clin Ther 2012; xx:1-7.
- Carter AA, Gomes T, Camacho X, Juurlink DN, Shah BR, Mamdani MM. Risk of incident diabetes among patients treated with statins: population based study. BMJ 2013; 346:f2610; PMID:23704171
- Dormuth CR, Filion KB, Paterson JM, James MT, Teare GF, Raymond CB, Rahme E, Tamim H, Lipscombe L. Higher potency statins and the risk of new diabetes: multicentre, observational study of administrative databases. BMJ 2014; 348:g3244; PMID:24874977; http://dx.doi.org/10.1136/bmj.g3244
- Cho Y, Choe E, Lee Y-H, Seo JW, Choi Y, Yun Y, Wang HJ, Ahn CW, Cha BS, Lee HC, et al. Risk of diabetes in patients treated with HMG-CoA reductase inhibitors. Metabolism 2014:1-7.
- Freeman DJ, Norrie J, Sattar N, Neely RDG, Cobbe SM, Ford I, Isles C, Lorimer AR, Macfarlane PW, McKillop JH, et al. Pravastatin and the development of diabetes mellitus: rvidence for a protective treatment effect in the West of Scotland Coronary Prevention Study. Circulation 2001; 103:357-62; PMID:11157685; http://dx.doi.org/10.1161/01.CIR.103.3.357
- Ridker PM, Danielson E, Fonseca FAH, Genest J, Gotto AM, Kastelein JJP, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195-207; PMID:18997196; http://dx.doi.org/10.1056/NEJMoa0807646
- Protection H, Collaborative S. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7-22; PMID:12114036; http://dx.doi.org/10.1016/S0140-6736(02)09327-3
- Swerdlow DI, Sattar N. A dysglycaemic effect of statins in diabetes: relevance to clinical practice? Diabetologia 2014; 57:2433-5; PMID:25335441; http://dx.doi.org/10.1007/s00125-014-3409-3
- Goldfine AB. Statins: is it really time to reassess benefits and risks? N Engl J Med 2012; 366:1752-5; PMID:22533536; http://dx.doi.org/10.1056/NEJMp1203020
- Chatzizisis YS, Koskinas KC, Misirli G, Vaklavas C, Hatzitolios A, Giannoglou GD. Risk factors and drug interactions predisposing to statin-induced myopathy: implications for risk assessment, prevention and treatment. Drug Saf 2010; 33:171-87; PMID:20158283; http://dx.doi.org/10.2165/11319380-000000000-00000
- Reiner Ž. Statins in the primary prevention of cardiovascular disease. Nat Rev Cardiol 2013; 10:453-64; PMID:23736519; http://dx.doi.org/10.1038/nrcardio.2013.80
- Pencina MJ, Navar-Boggan AM, D'Agostino RB, Williams K, Neely B, Sniderman AD, Peterson ED. Application of new cholesterol guidelines to a population-based sample. N Engl J Med 2014; 370:1422-31; PMID:24645848; http://dx.doi.org/10.1056/NEJMoa1315665
- Seshasai SRK, Kaptoge S, Thompson A, Di Angelantonio E, Gao P, Sarwar N, Whincup PH, Mukamal KJ, Gillum RF, Holme I, et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N Engl J Med 2011; 364:829-41; PMID:21366474; http://dx.doi.org/10.1056/NEJMoa1008862
- Swerdlow DI, Preiss D, Kuchenbaecker KB, Holmes MV, Engmann JEL, Shah T, Sofat R, Stender S, Johnson PCD, Scott RA, et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet 2014:1-11.
- Fullerton MD, Steinberg GR, Schertzer JD. Immunometabolism of AMPK in insulin resistance and atherosclerosis. Mol Cell Endocrinol 2013; 366:224-34; PMID:22361321; http://dx.doi.org/10.1016/j.mce.2012.02.004
- Schertzer JD, Steinberg GR. Immunometabolism: the interface of immune and metabolic responses in disease. Immunol Cell Biol 2014; 92:303; PMID:24613974; http://dx.doi.org/10.1038/icb.2014.12
- Glass CK, Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab 2012; 15:635-45; PMID:22560216; http://dx.doi.org/10.1016/j.cmet.2012.04.001
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid–induced insulin resistance. 2006; 116:3015-25; PMID:17053832
- Könner AC, Brüning JC. Toll-like receptors: linking inflammation to metabolism. Trends Endocrinol Metab 2011; 22:16-23; PMID:20888253; http://dx.doi.org/10.1016/j.tem.2010.08.007
- Chi W, Dao D, Lau TC, Henriksbo BD, Cavallari JF, Foley KP, Schertzer JD. Bacterial peptidoglycan stimulates adipocyte lipolysis via NOD1. PLoS One 2014; 9:e97675; PMID:24828250; http://dx.doi.org/10.1371/journal.pone.0097675
- Schertzer JD, Tamrakar AK, Magalhães JG, Pereira S, Bilan PJ, Fullerton MD, Liu Z, Steinberg GR, Giacca A, Philpott DJ, et al. NOD1 activators link innate immunity to insulin resistance. Diabetes 2011; 60:2206-15; PMID:21715553; http://dx.doi.org/10.2337/db11-0004
- Tamrakar AK, Schertzer JD, Chiu TT, Foley KP, Bilan PJ, Philpott DJ, Klip A. NOD2 activation induces muscle cell-autonomous innate immune responses and insulin resistance. Endocrinology 2010; 151:5624-37; PMID:20926588; http://dx.doi.org/10.1210/en.2010-0437
- Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 2010; 11:136-40; PMID:20023662; http://dx.doi.org/10.1038/ni.1831
- Youm Y-H, Adijiang A, Vandanmagsar B, Burk D, Ravussin A, Dixit VD. Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology 2011; 152:4039-45; PMID:21862613; http://dx.doi.org/10.1210/en.2011-1326
- Lee H-M, Kim J-J, Kim HJ, Shong M, Ku BJ, Jo E-K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013; 62:194-204; PMID:23086037; http://dx.doi.org/10.2337/db12-0420
- Vandanmagsar B, Youm Y-H, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 2011; 17:179-88; PMID:21217695; http://dx.doi.org/10.1038/nm.2279
- Stienstra R, Joosten LAB, Koenen T, van Tits B, van Diepen JA, van den Berg SAA, Rensen PCN, Voshol PJ, Fantuzzi G, Hijmans A, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab 2010; 12:593-605; PMID:21109192; http://dx.doi.org/10.1016/j.cmet.2010.11.011
- Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science 2010; 327:296-300; PMID:20075245; http://dx.doi.org/10.1126/science.1184003
- Grant RW, Dixit VD. Mechanisms of disease: inflammasome activation and the development of type 2 diabetes. Front Immunol 2013; 4:50; PMID:23483669; http://dx.doi.org/10.3389/fimmu.2013.00050
- Haneklaus M, O'Neill LA, Coll RC. Modulatory mechanisms controlling the NLRP3 inflammasome in inflammation: recent developments. Curr Opin Immunol 2013:2-7.
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009; 183:787-91; PMID:19570822; http://dx.doi.org/10.4049/jimmunol.0901363
- Kono H, Kimura Y, Latz E. Inflammasome activation in response to dead cells and their metabolites. Curr Opin Immunol 2014; 30C:91-8; http://dx.doi.org/10.1016/j.coi.2014.09.001
- Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 2007; 356:1517-26; PMID:17429083; http://dx.doi.org/10.1056/NEJMoa065213
- Ridker PM. Closing the loop on inflammation and atherothrombosis: why perform the CIRT and CANTOS trials? Trans Am Clin Climatol Assoc 2013; 124:174-90; PMID:23874021
- Liao Y-H, Lin Y-C, Tsao S-T, Lin Y-C, Yang A-J, Huang C-T, Huang K-C, Lin WW. HMG-CoA reductase inhibitors activate caspase-1 in human monocytes depending on ATP release and P2´7 activation. J Leukoc Biol 2012; 92:1-11; PMID:22745457; http://dx.doi.org/10.1189/jlb.0312130
- Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM, Dixit VM. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol 2009; 187:61-70; PMID:19805629; http://dx.doi.org/10.1083/jcb.200903124
- Jager J, Grémeaux T, Cormont M, Le Marchand-Brustel Y, Tanti J-F. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 2007; 148:241-51; PMID:17038556; http://dx.doi.org/10.1210/en.2006-0692
- Davaro F, Forde SD, Garfield M, Jiang Z, Halmen K, Tamburro ND, Kurt-Jones E, Fitzgerald KA, Golenbock DT, Wang D. 3-Hydroxyl-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor (Statin)-induced 28-kDa interleukin-1β interferes with mature IL-1β signaling. J Biol Chem 2014; 289:16214-22; PMID:24790079; http://dx.doi.org/10.1074/jbc.M114.571505
- Bruun JM, Stallknecht B, Helge JW, Richelsen B. Interleukin-18 in plasma and adipose tissue: effects of obesity, insulin resistance, and weight loss. Eur J Endocrinol 2007; 157:465-71; PMID:17893261; http://dx.doi.org/10.1530/EJE-07-0206
- Lindegaard B, Matthews VB, Brandt C, Hojman P, Allen TL, Estevez E, Watt MJ, Bruce CR, Mortensen OH, Syberg S, et al. Interleukin-18 activates skeletal muscle AMPK and reduces weight gain and insulin resistance in mice. Diabetes 2013; 62:3064-74; PMID:23670974; http://dx.doi.org/10.2337/db12-1095
- Lamkanfi M, Kanneganti T-D, Van Damme P, Vanden Berghe T, Vanoverberghe I, Vandekerckhove J, Vandenabeele P, Gevaert K, Núñez G. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics 2008; 7:2350-63; PMID:18667412; http://dx.doi.org/10.1074/mcp.M800132-MCP200
- Kahns S, Kalai M, Jakobsen LD, Clark BFC, Vandenabeele P, Jensen PH. Caspase-1 and caspase-8 cleave and inactivate cellular parkin. J Biol Chem 2003; 278:23376-80; PMID:12692130; http://dx.doi.org/10.1074/jbc.M300495200
- Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J Biol Chem 2007; 282:36321-9; PMID:17959595; http://dx.doi.org/10.1074/jbc.M708182200
- Birnbaum Y, Nanhwan MK, Ling S, Perez-Polo JR, Ye Y, Bajaj M. PTEN upregulation may explain the development of insulin resistance and type 2 diabetes with high dose statins. Cardiovasc Drugs Ther 2014; 28(5):447-57; PMID:25106875
- Teresi RE, Planchon SM, Waite KA, Eng C. Regulation of the PTEN promoter by statins and SREBP. Hum Mol Genet 2008; 17:919-28; PMID:18065496; http://dx.doi.org/10.1093/hmg/ddm364
- Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 2006; 7:85-96; PMID:16493415; http://dx.doi.org/10.1038/nrm1837
- Wagner BK, Kitami T, Gilbert TJ, Peck D, Ramanathan A, Schreiber SL, Golub TR, Mootha VK. Large-scale chemical dissection of mitochondrial function. Nat Biotechnol 2008; 26:343-51; PMID:18297058; http://dx.doi.org/10.1038/nbt1387
- Wagner BK, Gilbert TJ, Hanai J, Imamura S, Bodycombe NE, Bon RS, Waldmann H, Clemons PA, Sukhatme VP, Mootha VK. A small-molecule screening strategy to identify suppressors of statin myopathy. ACS Chem Biol 2011; 6:900-4; PMID:21732624; http://dx.doi.org/10.1021/cb200206w
- Kaufmann P, Török M, Zahno A, Waldhauser KM, Brecht K, Krähenbühl S. Toxicity of statins on rat skeletal muscle mitochondria. Cell Mol Life Sci 2006; 63:2415-25; PMID:17013560; http://dx.doi.org/10.1007/s00018-006-6235-z
- Thuc LC, Teshima Y, Takahashi N, Nagano-Torigoe Y, Ezaki K, Yufu K, Nakagawa M, Hara M, Saikawa T. Mitochondrial K(ATP) channels-derived reactive oxygen species activate pro-survival pathway in pravastatin-induced cardioprotection. Apoptosis 2010; 15:669-78; PMID:20151195; http://dx.doi.org/10.1007/s10495-010-0473-0
- Sánchez CA, Rodríguez E, Varela E, Zapata E, Páez A, Massó FA, Montaño LF, Lóopez-Marure R. Statin-induced inhibition of MCF-7 breast cancer cell proliferation is related to cell cycle arrest and apoptotic and necrotic cell death mediated by an enhanced oxidative stress. Cancer Invest 2008; 26:698-707; http://dx.doi.org/10.1080/07357900701874658
- Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013; 38:1142-53; http://dx.doi.org/10.1016/j.immuni.2013.05.016
- Sinensky M. Recent advances in the study of prenylated proteins. Biochim Biophys Acta–Mol Cell Biol Lipids 2000; 1484:93-106; http://dx.doi.org/10.1016/S1388-1981(00)00009-3
- Stenmark H, Olkkonen VM. Protein family review The Rab GTPase family. 2001:1-7.
- Spindler SR, Li R, Dhahbi JM, Yamakawa A, Mote P, Bodmer R, Ocorr K, Williams RT, Wang Y, Ablao KP. Statin treatment increases lifespan and improves cardiac health in Drosophila by decreasing specific protein prenylation. PLoS One 2012; 7:e39581; PMID:22737247; http://dx.doi.org/10.1371/journal.pone.0039581
- Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced Inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008; 57:1470-81; PMID:18305141; http://dx.doi.org/10.2337/db07-1403
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007; 56:1761-72; PMID:17456850; http://dx.doi.org/10.2337/db06-1491
- Steinberg GR, Schertzer JD. AMPK promotes macrophage fatty acid oxidative metabolism to mitigate inflammation: implications for diabetes and cardiovascular disease. Immunol Cell Biol 2014; 92:340-5; PMID:24638063; http://dx.doi.org/10.1038/icb.2014.11
- Jourdan T, Godlewski G, Cinar R, Bertola A, Szanda G, Liu J, Tam J, Han T, Mukhopadhyay B, Skarulis MC, et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat Med 2013; 19:1132-40; PMID:23955712; http://dx.doi.org/10.1038/nm.3265
- Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol 2010; 11:897-904; PMID:20835230; http://dx.doi.org/10.1038/ni.1935
- Mitchell P, Marette A. Statin-induced insulin resistance through inflammasome activation: sailing between scylla and charybdis. Diabetes 2014; 63:3569-71; PMID:25342725; http://dx.doi.org/10.2337/db14-1059