
ABSTRACT
The exocytosis of signaling molecules from neuronal, neuroendocrine and endocrine cells is regulated by membrane fusion involving SNAP-25 and associated SNARE proteins. The importance of this process for metabolic control recently became evident by studies of mouse mutants genetically engineered to only express one of 2 closely related, alternatively-spliced variants of SNAP-25. The results showed that even minor differences in the function of proteins regulating exocytosis are sufficient to provoke metabolic disease, including hyperglycaemia, liver steatosis, adipocyte hypertrophy and obesity. Thus, an imbalance in the dynamics of hormonal and/or neurotransmitter release can cause obesity and type 2 diabetes. This recent discovery highlights the fact that metabolic health requires a perfectly operating interplay between the SNARE protein machinery in excitable cells and the organs responding to these messengers.
The findings in our recent publication “Replacing SNAP-25b with SNAP-25a expression results in metabolic disease” showed that minor alterations in the SNARE fusion machinery, expressed in neurons, neuroendocrine and endocrine cells, provide a novel link with metabolic disease.Citation1 Importantly, we proposed that impaired release of hormones such as insulin may not always represent symptoms of metabolic disease. Instead, a genetic mutation that changes the expression or function of proteins, which directly or indirectly regulate stimulus-dependent membrane fusion, can be the cause of illness. Thus, our discovery suggests that new therapeutic targets and treatment strategies for metabolic disease should be explored.
Stimulation of excitable cells and subsequent Ca2+-triggered exocytosis forms the basis for not only synaptic transmission but also for the regulated secretion of hormones and neuropeptidesCitation2,3 (). One of the last steps in the events which lead to vesicle fusion with the plasma membrane is the formation of a transmembrane soluble N-ethylmaleimidesensitive factor (NSF) attachment protein receptor (SNARE) complex.Citation2 The core trans-SNARE complex consists of 3 compartmentally defined proteins: vesicle-associated VAMP2; syntaxin, a protein possessing a plasma membrane anchor; and the 25 kDa cytoplasmic synaptosomal-associated protein, SNAP-25, which associates with membranes through palmitoylation. The three SNARE proteins are held together by strong protein-protein interactions in which their cytoplasmic domains form a 4 α-helix coiled-coiled bundleCitation4 (). SNAP-25 contributes 2 of the 4 α-helices, which include both the amino- and the carboxyl-terminal of the protein, whereas syntaxin and VAMP2 add one α-helix each. In non-excitable cells, secretion is mediated by cellular homologues to SNAP-25/syntaxin/VAMP proteins.
Figure 1. Regulated exocytosis and role of SNAP-25 (synaptosomal-associated protein of 25 kDa). (A) Regulated exocytosis, or regulated membrane fusion, is the process by which excitable cells release signaling molecules such as hormones or neurotransmitters through the fusion of intracellular vesicles with the plasma membrane. The final stages of exocytosis are classified into docking, priming and fusion. Vesicles are recruited and tethered to the plasma membrane, where they are docked. During priming, SNARE complexes of proteins including SNAP-25 are formed, after which the vesicles are pulled closer to the plasma membrane bilayers and fuse with them; the vesicle contents are then released into the extracellular space. Fusion is initiated by an influx of calcium via specific channels. (B) The gene for SNAP-25 is located on chromosome 20 p12-12p11.2. (C) This gene encodes a presynaptic protein with two differentially expressed splicing variants, SNAP-25a and SNAP-25b. The two mutually exclusive splice variants result from a duplication of exon 5 in the Snap25 gene. Four single nucleotide polymorphisms (SNPs) or mutations in the human Snap25 gene have been found to be associated with metabolic traits (C). The rs363050 polymorphism in exon 1 of the Snap25 gene results in reduced expression of SNAP-25 protein, and when this SNP coincides with T2D, patients demonstrates higher levels of fasting glucose and HbA1c (39). Diabetic individuals carrying the rs363050 polymorphism also exhibit lower insulinaemia, possibly due to an impaired exocytosis machinery in beta cells (39). The rs3746544 and TaiI SNPs located in the 3' UTR region of the human SNAP-25 mRNA have been associated with weight gain after antipsychotic treatment (37,38). Furthermore, SNP genotypes of rs362551 located 7 kb downstream of exon 8 of the Snap25 gene have been associated with the severity of the syndrome (40). (D) SNAP-25a and SNAP-25b differ only in 9 out of 206 amino acids. The two alternative exon 5 sequences both encode 39 amino acids spanning positions 56 to 94 in the SNAP-25 polypeptide. Abbreviations: VGCC, voltage-gated calcium channel; 3'UTR, 3'untranslated region.
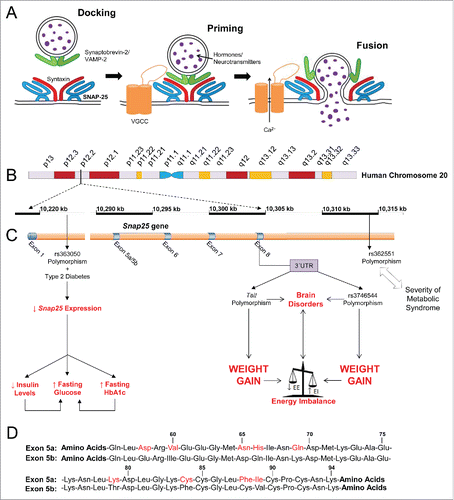
SNAP-25 is expressed as 2 developmentally regulated and complementary distributed splice variants from a single copy gene Citation5-9 (). The discovery of alternative splicing, or split genes, explained how there can be more than one protein generated from one gene.Citation10,11 Alternative splicing increases the possibility for diversity and can cause distinct adjustments in a specific protein's function.Citation12,13 Alternative splicing of the primary Snap25 gene transcript is an obligate choice between 2 closely spaced exon 5 sequences that are arranged in tandem, so that 9 of the 206 amino acids in the polypeptide differ in a isoform-specific manner (). Five of the amino acid differences are located in a submerged region within the SNARE structure: Asp58, Val60, Asn70, His71, and Gln74 in SNAP-25a and Glu58, Ile60, Asp70, Gln71, and Lys74 in SNAP-25b (). The other 4 amino acid sequence differences are not part of the α-helical structure of the protein and include both a Lys to Thr change at amino acid 84, and also the reorganization of a ´quartet of cysteines´ within the protein sequence. This quartet includes the only cysteines present in both SNAP-25a and SNAP-25b and these cysteine are substrates for the post-translational palmitoylation events that are required for membrane targeting of the protein ().
SNAP-25 is essential for stimulated neuroexocytosis. Targeted disruption of the mouse Snap25 gene abolishes evoked synaptic transmission and mutant mice die at birth.Citation14 Indeed, other mouse mutants of SNAP-25 have revealed that alterations in the total levels of SNAP-25 protein expression appear to affect neuronal function, especially if the SNAP-25a/SNAP-25b ratio is changed.Citation15 The functional difference between SNAP-25a and SNAP-25b is still not fully understood, since the splice variants appear to be interchangeable in SNARE-mediated vesicle fusion. In neuronal cells, SNAP-25a seems to be the splice variant which is predominantly expressed during early development, whereas SNAP-25b is the major variant in adult neuronal populations.Citation8 In other excitable cells, such as endocrine and neuroendocrine cells, SNAP-25a appears to be the dominant isoform throughout life.Citation8 However, even in adulthood, the neuroanatomical distribution of SNAP-25a and SNAP-25b appears not to be strictly and permanently distributed among cell populations. Overexpression of the SNAP-25 isoforms in primary embryonic adrenal chromaffin cells from SNAP-25 null embryos demonstrated that SNAP-25b has a greater capacity to stabilize primed vesicles than SNAP-25a, leading to changes in the burst of Ca2+-evoked release.Citation16 Taken together, it appears that alternative splicing between SNAP-25a and SNAP-25b fine-tunes the kinetics of regulated membrane fusion.
Obesity and type2 diabetes (T2D) are complex pathophysiological disorders that can be associated with impairments in both energy metabolism and feeding behavior. These conditions possess a multifactorial pathophysiology and are most frequently not caused by single genetic mutations, but instead are triggered by ‘external risk factors', such as increased calorie intake and decreased physical activity. Physiological mechanisms regulating feeding behavior are integral to the pancreas-gut-adipose tissue-brain axis (). Therefore, adiposity and circulating hormones such as leptin, cholecystokinin, ghrelin and insulin are involved in the regulation of both energy balance and appetite/satiety via their direct or indirect actions on the brainstem and hypothalamic nuclei, as well as in peripheral metabolic organs.Citation17-19 However, as it is considered that regulated membrane fusion is at the heart of the stimulus-activated information flow from excitable cells, we have speculated that even small differences in the dynamics of signaling molecule release can predispose to metabolic disease, particularly if combined with increased calorie intake.
Figure 2. (A) SNAP-25b deficiency and/or Western Diet (WD) intervention can impair the peripheral organ-brain circuitry in glucose-sensing organs. SNAP-25b deficiency and/or a WD intervention impair hypothalamic function, disrupting the balance between satiety and hunger signals which are integrated in this brain area (1). These pathophysiological conditions are exacerbated when combining a WD intervention with genetic manipulations affecting the alternative splicing of SNAP-25 (1). These events can also have an impact on sympathetic and parasympathetic efferents. Thus, we speculate that both SNAP-25b-deficiency and WD have a negative effect on sympathetic efferents from the hypothalamus that project to the intermediolateral cell column (in blue) and also impair the inputs from the PBN and the BLM. Parasympathetic efferents (in red) comprise projections from the BLM and the NTS. In our model, we hypothesize that alterations in the parasympathetic efferents affect the network of interactions between the hypothalamus, the BLM and the NTS. Sympathetic innervation triggers glucagon secretion from α cells, inhibits insulin secretion from β cells, and activates epinephrine secretion from the adrenal gland. By contrast, parasympathetic efferents stimulate insulin secretion and inhibit hepatic glucose production (50). It is also possible that SNAP-25b-deficiency, by directly affecting insulin secretion from pancreatic β cells, initiates the metabolic phenotype. Also note a possible involvement of sensory neurons. Abbreviations: BLM, basolateral medula; CMGP, celiac/mesenteric ganglion plexus; HTH, hypothalamus; NTS,Figure 2. Continued nucleus tractus solitarii; PBN, parabrachial nucleus. 2B. Synoptic model describing the impact of SNAP-25b deficiency in the development of metabolic disease. SNAP-25b deficiency impairs the dynamics of secretion of both insulin and of other hormones, resulting in disrupted glucose homeostasis, including defective glucose sensing in β cells and the triggering of insulin resistance in peripheral tissues. These events, together with the increased food intake detected in our mutant mice, induce a severe accumulation of fat not only in natural depots (white adipose tissues), but also ectopically in the liver. As a consequence, severe dyslipidemia develops which impacts upon adipocyte turnover, leading to hypertrophic adipose cells. This increased fat accumulation favors the development of hyperleptinemia. The main consequence of higher leptin levels in our obese SNAP-25b-deficient mice is the desensitization of the hypothalamic leptin receptor, leading to inactivation of leptin signaling pathways in this brain area. Thus, the lack of satiety signals in the hypothalamus, together with decreased levels of the orexigenic hormone ghrelin in SNAP-25b deficient mice, might contribute to disrupted feeding behavior, leading to obesity, T2D and the metabolic syndrome. Abbreviations: CHO, cholesterol; FA, fatty acids; ObRb, long isoform of leptin receptor; pAMPK, phosphorylated AMP-activated protein kinase; pERK, phosphorylated extracellular-signal-regulated kinases; pSTAT3, phosphorylated signal transducer . Synoptic model describing the impact SNAP-25b-deficiency has in the development of metabolic disease.
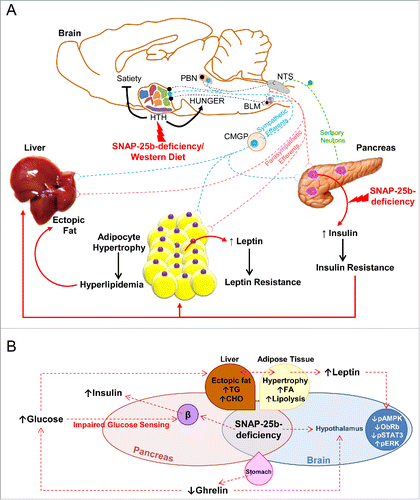
To test our hypothesis, we placed male and female SNAP-25b-deficient mutant mice on a Western Diet (WD) rich in fat and simple carbohydrates.Citation1 We discovered that, by interfering with the alternative splicing of SNAP-25, this mutation alone triggered dyslipidemia, adipocyte hypertrophy, impaired glucose homeostasis, hepatic steatosis and hypothalamic dysfunction, all of which are features of metabolic syndrome Citation1,20,21 (). These features were worsened when the mutation was combined with WD. Furthermore, the mutant mice had an impaired regulation of energy expenditure which resulted in body weight gain.
It is well known that the hypothalamus is involved in the control of food intake and energy balance, and its dysfunction is associated with obesity and T2D.Citation1,22-25 Indeed, we found that in the hypothalamus of mutant mice, the active form of AMP-activated protein kinase (AMPK), a master metabolic regulator, was decreased compared to WT mice. We also noticed that activation of molecules involved in the leptin and insulin signaling pathways such as STAT3 and ERK1/2, was impaired in mutants, suggesting hypothalamic leptin and insulin resistance. Interestingly, the hypothalamic leptin receptor was found to be downregulated only in WD-fed animals.Citation1
Furthermore, we observed dramatic sex differences between male and female mice, both due to the mutation but also in the response to WD.Citation1 In mutant females on control diet (CD), the body weight gain and increases in triglyceride levels appeared earlier than in males, most likely due to increased food intake.Citation1 In male mutants, however, obesity was dependent on the failure of circadian feeding behavior, which resulted in constant eating throughout the day.Citation1 Sex hormones are likely playing an important role in the susceptibility and development of the metabolic syndrome, and further scientific investigations should address this question.
A critical issue is, of course, the nature of the ‘weakest point’ that initially triggers the metabolic phenotype in our mouse mutants. Our data support the idea that it is a defect in the exocytosis machinery in those neurons, neuroendocrine and endocrine cells which regulate energy balance, rather than the diet itself, that drives dyslipidemia and derangements in glucose homeostasis.Citation1 If this condition is originating from such excitable cells, then there are many possibilities: for example, a direct effect on insulin secretion from pancreatic β cells, or on other circulating hormones that are also released by regulated membrane fusion, such as cholecystokinin or ghrelin (). Also, deleterious processes could be triggered by impaired signals from cells in the brainstem and hypothalamic nuclei that normally regulate energy balance and appetite/satiety (). Another possibility is the presence of defects in peripheral nerves innervating target organs such as the pancreas, liver and adipose tissue ().
The effect caused by the small mutation introduced into the mouse Snap25 gene is remarkable indeed. It is tempting to speculate that many individuals carrying an even more trivial genetic mutation that directly or indirectly affects hormonal or transmitter release in excitable cells have a predisposition for metabolic disease. In this respect it is intriguing that most genes that were identified as risk genes for T2D in GWAS were genes affecting β cell function and/or insulin release.Citation26 Such a minor genetic vulnerability might be ‘silent’ and not noticed, until the individual increases the disease risk by combining it with an unhealthy lifestyle; this may include lack of physical activity or increased food intake. Many of the features of metabolic illness are manifested in cells such as adipocytes and hepatocytes that do not express SNARE proteins, including SNAP-25, that are the trademarks for neurons, neuroendocrine and endocrine cells. Therefore these injuries have to be secondary to the mutation, and a result of the intricate interplay between the liver, pancreas, adipose tissue, hypothalamus and/or nerves innervating these target organs ().
Numerous polymorphisms have been identified in the human gene for Snap25, and they have so far been associated with neuropsychiatric disorders and/or cognitive function.Citation27-36 However, little is known about how these polymorphisms affect transcription of the Snap25 gene, or if they interfere with splicing, micro-RNA binding or other post-transcriptional modifications that affect mRNA stability. Interestingly, recent findings show that some of these polymorphisms are associated with ‘non-neuronal’ traits, such as weight gain after antipsychotic treatment, and altered levels of serum triglycerides,Citation37,38 as well as with glycaemic parameters or with the severity of the metabolic syndrome in T2D Citation39,40 (). Furthermore, genes encoding proteins which directly or indirectly interact with SNAP-25 have been linked to childhood obesity, impaired glucose metabolism and obesity in adults, the requirement for insulin treatmentor, or the age of T2D onset.Citation41-46 As neurons and endocrine cells are closely related, it is not surprising that, for example, mutations found in the Snap25 gene are also linked to metabolic traits such as weight gain. These mutations, which are located in the 3′ untranslated region (UTR) of the mRNA and which have previously been identified as being associated with ADHD, and possibly span the binding site for a micro-RNA, mir-641 Citation46 (). In addition, a polymorphism previously associated with IQ Citation34 has now been linked to higher levels of fasting glucose and lower serum insulin levels in T2D patients.Citation39 It is intriguing that the same mutations in the gene for SNAP-25 demonstrate both central and peripheral phenotypes. This further strengthens our hypothesis that mutations in SNARE or SNARE interacting proteins can cause metabolic disease.
As T2D and obesity are increasing world-wide, there is an urgent need to find additional therapies to combat these disorders. Today, almost 382 million people are afflicted by T2D,Citation47 and, in 2014, approximately 600 million people were obese, with almost 2 billion adults worldwide being overweight, according to the WHO.Citation48 Obesity in itself causes a general, chronic low grade inflammation which not only increases the risk for T2D but also predisposes for other complications, such as cardiovascular disease and cancer. Other risk factors for T2D are physical inactivity, ethnicity, increasing age, high blood pressure and genetic predispositions.Citation47 Metabolic disorders usually develop when several risk factors coincide, although there are exceptions. One example of such an exception is monogenic diabetes, or “Maturity Onset Diabetes of the Young” (MODY), which mainly affects β cell function.Citation49 We have now demonstrated that manipulating the expression of the splicing variants of SNAP-25, a protein controlling the efficacy of regulated membrane fusion in neurons, neuroendocrine and endocrine cells, can create a phenotype which includes all of the features of metabolic syndrome. Further research will reveal, if there are corresponding monogenic mutations in SNARE or SNARE-regulating proteins in humans that can cause similar conditions. In conclusion, our recent discoveries have revealed additional mechanisms that may underlie the onset of metabolic disease. This seems encouraging for the future, as it opens up new avenues for therapy.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Dr. Neil Portwood for comments on the manuscript.
Funding
This work was supported by grants from The Swedish Research Council, The Family Erling-Persson Foundation, Karolinska Institutet Funds, Magnus Bergvall's Foundation, Gun and Bertil Stohne's Foundation, Längmanska Kulturfonden, Fogelström's Foundation and Sven Mattsson's Foundation.
References
- Valladolid-Acebes I, Daraio T, Brismar K, Harkany T, Ögren SO, Hökfelt TG, Bark C. Replacing SNAP-25b with SNAP-25a expression results in metabolic disease. Proc Natl Acad Sci USA 2015; 112(31): E4326-35; PMID:26195742; http://dx.doi.org/10.1073/pnas.1511951112
- Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 1993; 75(3): 409-18; PMID:8221884; http://dx.doi.org/10.1016/0092-8674(93)90376-2
- Südhof TC. The molecular machinery of neurotransmitter release (Nobel lecture). Angew Chem Int Ed Engl 2014; 53(47): 12696-717; PMID:25339369; http://dx.doi.org/10.1002/anie.201406359
- Sutton RB1, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 1998; 395(6700): 347-53; PMID:9759724; http://dx.doi.org/10.1038/26412
- Bark IC. Structure of the chicken gene for SNAP-25 reveals duplicated exon encoding distinct isoforms of the protein. J Mol Biol 1993. 233:67-76; PMID:8377193; http://dx.doi.org/10.1006/jmbi.1993.1485
- Bark IC, Wilson MC. Human cDNA clones encoding two different isoforms of the nerve terminal protein SNAP-25. Gene 1994a; 139:291-2; PMID:8112622; http://dx.doi.org/10.1016/0378-1119(94)90773-0
- Bark IC, Hahn KM, Ryabinin AE, Wilson MC. Differential expression of SNAP-25 protein isoforms during divergent vesicle fusion events of neural development. Proc Natl Acad Sci USA 1995;92(5):1510-4. PMID:7878010; http://dx.doi.org/10.1073/pnas.92.5.1510
- Bark C, Bellinger FP, Kaushal A, Mathews JR, Partridge LD, Wilson MC. Developmentally regulated switch in alternatively spliced SNAP-25 isoforms alters facilitation of synaptic transmission. J Neurosci 2004. 24:8796-805; PMID:15470145; http://dx.doi.org/10.1523/JNEUROSCI.1940-04.2004
- Johansson JU, Ericsson J, Janson J, Beraki S, Stanić D, Mandic SA, Wikström MA, Hökfelt T, Ögren SO, Rozell B, et al. An ancient duplication of exon 5 in the Snap25 gene is required for complex neuronal development/function. PLoS Genetics 2008; 4: e1000278; PMID:19043548; http://dx.doi.org/10.1371/journal.pgen.1000278
- Carr K. Nobel goes to discoverers of ‘split genes’. Nature 1993; 365(6447):597; PMID:8413620; http://dx.doi.org/ 10.1038/365597a0
- Sharp PA. Split Genes and RNA Splicing Nobel Lecture. Cell 1994; 77:805-815; PMID:7516265; http://dx.doi.org/10.1016/0092-8674(94)90130-9
- Jangi M, Sharp PA. Building robust transcriptomes with master splicing factors. Cell 2014; 159(3):487-98; PMID:25417102; http://dx.doi.org/10.1016/j.cell.2014.09.054
- de Klerk E, ´t Hoen PA. Alternative mRNA transcription, processing: and translation: insights from RNA sequencing. Trends Genet. 2015; 31(3):128-39; PMID:25648499; http://dx.doi.org/10.1016/j.tig.2015.01.001
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Benditó G, Molnár Z, Becher MW, Valenzuela CF, Partridge LD, et al. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 2002; 5(1):19-26; PMID:11753414; http://dx.doi.org/10.1038/nn783
- Bark C. SNAP-25 and gene-targeted mouse mutants. Ann N Y Acad Sci 2009; 1152:145-53; PMID:19161385; http://dx.doi.org/10.1111/j.1749-6632.2008.04009.x
- Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell 2003; 114:75-86; PMID:12859899; http://dx.doi.org/10.1016/S0092-8674(03)00477-X
- Gao Q, Horvath TL. Neurobiology of feeding and energy expenditure. Annu Rev Neurosci 2007; 30:367-98; PMID:17506645; http://dx.doi.org/10.1146/annurev.neuro.30.051606.094324
- Morton GJ, Meek TH, Schwartz MW. Neurobiology of food intake in health and disease. Nat Rev Neurosci 2014; 15(6): 367-378; PMID:24840801; http://dx.doi.org/10.1038/nrn3745
- Müller TD, Tschöp MH. Ghrelin - a key pleiotropic hormone-regulating systemic energy metabolism. Endocr Dev 2013; 25:91-100; PMID:23652395; http://dx.doi.org/10.1159/000346590
- Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014; 384(9945): 766-81; PMID:24880830; http://dx.doi.org/10.1016/s0140-6736(14)60460-8
- Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults. Findings from the third National Health and Nutrition Examination Survey. JAMA 2001; 287(3):356-59; PMID:11790215; http://dx.doi.org/10.1001/jama.287.3.356
- Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest 2006; 116(7):1776-83; PMID:16823475; http://dx.doi.org/10.1172/JCI29044
- Grahame Hardie D. AMP-activated protein kinase. A key regulator of energy balance with many roles in human disease. J Intern Med 2014;276(6): 543-59; PMID:24824502; http://dx.doi.org/10.1111/joim.12268
- Friedman J. 20 years of leptin. Leptin at 20: An overview. J Endocrinol 2014; 223(1):T1-T8; PMID:25121999; http://dx.doi.org/10.1530/JOE-14-0405
- Tanti JF, Jager J. Cellular mechanisms of insulin resistance. Role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr Opin Pharmacol 2009; 9(6):753-62; PMID:19683471; http://dx.doi.org/10.1016/j.coph.2009.07.004
- Herder C, Roden M. Genetics of type 2 diabetes. pathophysiologic and clinical relevance. Eur J Clin Invest 2011; 41(6):679-92; PMID:21198561; http://dx.doi.org/10.1111/j.1365-2362.2010.02454.x
- Barr CL, Feng Y, Wigg K, Bloom S, Roberts W, Malone M, Schachar R, Tannock R, Kennedy JL. Identification of DNA variants in the SNAP-25 gene and linkage study of these polymorphisms and attention-deficit hyperactivity disorder. Mol Psychiatry 2000; 5:405-409; PMID:10889551; http://dx.doi.org/ 10.1038/sj.mp.4000733
- Brophy K, Hawi Z, Kirley A, Fitzgerald M, Gill M. Synaptosomal-associated protein 25 (SNAP-25) and attention deficit hyperactivity disorder (ADHD): evidence of linkage and association in the Irish population. Mol Psychiatry 2002; 7:913-17; PMID:12232787; http://dx.doi.org/10.1038/sj.mp.4001092
- Kustanovich V, Merriman B, McGough J, McCracken JT, Smalley SL, Nelson SF. Biased paternal transmission of SNAP-25 risk alleles in attention-deficit hyperactivity disorder. Mol Psychiatry 2003; 8:309-15; PMID:12660803; http://dx.doi.org/10.1038/sj.mp.4001247
- Feng Y, Crosbie J, Wigg K, Pathare T, Ickowicz A, Schachar R, Tannock R, Roberts W, Malone M, Swanson J, et al. The SNAP25 gene as a susceptibility gene contributing to attention-deficit hyperactivity disorder. Mol Psychiatry 2005; 10:998-1005, 973; PMID:16088329; http://dx.doi.org/10.1038/sj.mp.4001722
- Mill J, Xu X, Ronald A, Curran S, Price T, Knight J, Craig I, Sham P, Plomin R, Asherson P. Quantitative trait locus analysis of candidate gene alleles associated with attention deficit hyperactivity disorder (ADHD) in five genes. DRD4, DAT1, DRD5, SNAP-25, and 5HT1B. Am J Med Genet B Neuropsychiatr Genet 2005; 133B: 68-73; PMID:15578613; http://dx.doi.org/10.1002/ajmg.b.30107
- Kim JW, Waldman ID, Faraone SV, Biederman J, Doyle AE, Purcell S, Arbeitman L, Fagerness J, Sklar P, Smoller JW. Investigation of parent-of-origin effects in ADHD candidate genes. Am J Med Gent B Neuropsychiatr Genet 2007a; 144B:776-80; PMID:17427194; http://dx.doi.org/10.1002/ajmg.b.30519
- Kim JW, Biederman J, Arbeitman L, Fagerness J, Doyle AE, Petty C, Perlis RH, Purcell S, Smoller JW, Faraone SV, et al. Investigation of variation in SNAP-25 and ADHD and relationship to co-morbid major depressive disorder. Am J Med Gent B Neuropsychiatr Genet 2007b; 144B:781-90; PMID:17455213; http://dx.doi.org/10.1002/ajmg.b.30522
- Gosso MF, de Geus EJ, Polderman TJ, Boomsma DI, Heutink P, Posthuma D. Common variants underlying cognitive ability. further evidence for association between the SNAP-25 gene and cognition using a family-based study in two independent Dutch cohorts. Genes Brain Behav 2008; 7:355-64; PMID:17908175; http://dx.doi.org/10.1111/j.1601-183X.2007.00359.x
- Guan L, Wang B, Chen Y, Yang L, Li J, Qian Q, Wang Z, Faraone SV, Wang Y. A high-density single-nucleotide polymorphism screen of 23 candidate genes in attention deficit hyperactivity disorder. suggesting multiple susceptibility genes among Chinese Han population. Mol Psychiatry 2009; 14:546-554; PMID:18180757; http://dx.doi.org/10.1038/sj.mp.4002139
- Söderqvist S, McNab F, Peyrard-Janvid M, Matsson H, Humphreys K, Kere J, Klingberg T. The SNAP25 gene is linked to working memory capacity and maturation of the posterior cingulate cortex during childhood. Biol Psychiatry 2010; 68:1120-25; PMID:20950795; http://dx.doi.org/10.1016/j.biopsych.2010.07.036
- Müller DJ, Klempan TA, De Luca V, Sicard T, Volavka J, Czobor P, Sheitman BB, Lindenmayer JP, Citrome L, McEvoy JP, et al. The SNAP-25 gene may be associated with clinical response and weight gain in antipsychotic treatment of schizophrenia. Neurosci Lett 2005; 379(2):81-89; PMID:15823421; http://dx.doi.org/10.1016/j.neulet.2004.12.037
- Musil R, Spellmann I, Riedel M, Dehning S, Douhet A, Maino K, Zill P, Müller N, Möller HJ, Bondy B. SNAP-25 gene polymorphisms and weight gain in schizophrenic patients. J Psychiatr Res 2008; 42(12):963-70; PMID:18191416; http://dx.doi.org/10.1016/j.jpsychires.2007.11.003
- Al-Daghri NM, Costa AS, Alokail MS, Zanzottera M, Alenad AM, Mohammed AK, Clerici M, Guerini FR. Synaptosomal Protein of 25 kDa (Snap25) Polymorphisms Associated with Glycemic Parameters in Type 2 Diabetes Patients. J Diabetes Res 2016; http://dx.doi.org/10.1155/2016/8943092
- Chen YL, Pei D, Hung YJ, Lee CH, Hsiao FC, Wu CZ, Lin JD, Hsu CH, Chang JB, Hsieh CH. Associations between genetic variants and the severity of metabolic syndrome in subjects with type 2 diabetes. Genet Mol Res 2015; 14(1): 2518-26; PMID:25867398; http://dx.doi.org/10.4238/2015.March.30.10
- Goldlust IS, Hermetz KE, Catalano LM, Barfield RT, Cozad R, Wynn G, Ozdemir AC, Conneely KN, Mulle JG, Dharamrup S, et al. Mouse model implicates GNB3 duplication in a childhood obesity syndrome. Proc Natl Acad Sci USA 2013; 110(37):14990-4; PMID:23980137; http://dx.doi.org/10.1073/pnas.1305999110
- Romeo S, Sentinelli F, Cavallo MG, Leonetti F, Fallarino M, Mariotti S, Baroni MG. Search for genetic variants of the SYNTAXIN 1A (STX1A) gene. The −352 A>T variant in the STX1A promoter associates with impaired glucose metabolism in an Italian obese population. Int J Obes 2008; 32(3):413-20; PMID:17912268; http://dx.doi.org/10.1038/sj.ijo.0803743
- Tsunoda K, Sanke T, Nakagawa T, Furuta H, Nanjo K. Single nucleotide polymorphism (D68D, T to C) in the syntaxin 1A gene correlates to age at onset and insulin requirement in Type II diabetic patients. Diabetologia 2001; 44(11):2092-97; PMID:11719842; http://dx.doi.org/10.1007/s001250100015
- Reinbothe TM, Alkayyali S, Ahlqvist E, Tuomi T, Isomaa B, Lyssenko V, Renström E. The human L-type calcium channel Cav1.3 regulates insulin release and polymorphisms in CACNA1D associate with type 2 diabetes. Diabetologia 2013; 56(2):340-49; PMID:23229155; http://dx.doi.org/10.1007/s00125-012-2758-z
- Olson TM, Terzic A. Human K(ATP) channelopathies. Diseases of metabolic homeostasis. Pflugers Arch 2010; 460(2):295-306; PMID:20033705; http://dx.doi.org/10.1007/s00424-009-0771-y
- Németh N, Kovács-Nagy R, Székely A, Sasvári-Székely M, Rónai Z. Association of impulsivity and polymorphic microRNA-641 target sites in the SNAP-25 gene. PLoS One 2013; 8(12): e84207; PMID:24391914; http://dx.doi.org/10.1371/journal.pone.0084207
- International Diabetes Federation (IDF) http://www.idf.org
- World Health Organization (WHO) http://www.who.int/mediacentre/factsheets/fs311/en
- McDonald TJ, Ellard S. Maturity onset diabetes of the young: identification and diagnosis. Ann Clin Biochem 2013; 50(5):403-15; PMID:23878349; http://dx.doi.org/10.1177/0004563213483458
- Steinbusch L, Labouèbe G, Thorens B: Brain glucose sensing in homeostatic and hedonic regulation. Trends Endocrinol Metab 2015; 26(9):455-66; PMID:26163755; http://dx.doi.org/10.1016/j.tem.2015.06.005