
ABSTRACT
Obesity-induced adipose tissue inflammation is regulated by various immune cells for innate and adaptive immunity. Among adipose tissue immune cells, it has been proposed that invariant Natural Killer T (iNKT) cells play crucial roles in anti-inflammatory responses in obesity. iNKT cells recognize ‘lipid’ antigens loaded on CD1d of antigen presenting cells and modulate immune responses by secreting Th1 or Th2 type cytokines depending on species of lipid antigens, antigen presenting cell types, and environmental cytokine milieu. However, the regulatory mechanisms of antigen presenting cells for adipose iNKT cell stimulation have not been clearly elucidated. Recently, we have reported that CD1d expressing adipocytes could act as an antigen presenting cell for adipose iNKT cells by characterization of adipocyte-specific CD1d knockout (CD1dADKO) mice. Upon high-fat diet (HFD) feeding, CD1dADKO mice aggravated adipose tissue inflammation and insulin resistance compared with CD1df/f mice. In this commentary, we provide the additional data of adipocyte CD1d-dependent regulation of adipose iNKT cell responses as well as systemic insulin sensitivity. In addition, we discuss how the interaction between adipocytes and iNKT cells would be regulated with the progression of obesity.
Obesity is closely associated with numerous metabolic diseases such as type 2 diabetes, hyperlipidemia, hypertension, and cardiovascular diseases [Citation34]. In obesity, adipose tissue suffers from low-grade and chronic inflammation which are characterized by elevated expression of inflammatory genes such as TNF-α and IL-6 [Citation11,Citation19]. Furthermore, accumulating evidence has suggested that obesity-induced inflammation is one of the important causal factors that promote systemic health complications including insulin resistance and dysregulation of glucose and lipid metabolism [Citation37,Citation45]. Importantly, crosstalk between adipocytes and other cells residing in adipose tissue is critical for determining tones of adipose tissue inflammation in obesity [Citation5,Citation9,Citation14,Citation24]. Among various cells comprising stromal vascular cell (SVC) fraction of adipose tissues, immune cells sensitively respond to changes in nutritional status through modulating their compositions and characters which consequently lead to alteration of inflammatory tones in adipose tissue [Citation14,Citation46]. For instance, numerous anti-inflammatory immune cells such as M2 (alternatively activated macrophage), eosinophils, and regulatory T (Treg) cells are present in lean adipose tissues. On the contrary, the pro-inflammatory immune cells such as M1 (classically activated) macrophages, Th1 T cells, and CD8 T cells are significantly elevated in obese adipose tissue, concomitantly with the reduced number of anti-inflammatory immune cells [Citation6,Citation26,Citation36,Citation40,Citation49]. In particular, macrophages compose predominant proportion of adipose leukocytes, and they are one of the decisive cell types to control adipose tissue inflammation [Citation46]. Not only a total number of macrophages but also M1 polarized macrophages are increased in obese adipose tissues [Citation26]. Although the binary switch model for adipose tissue macrophages into M1 and M2 has been challenged due to their mixed marker profiles, adipose tissue macrophages are largely categorized into M1 as pro-inflammatory cytokine (TNF-α and IL-1β) secreting CD11c+ macrophages and M2 as anti-inflammatory and arginase-1 expressing CD11c−CD206high macrophages [Citation4,Citation29].
The key signaling components for M1/M2 macrophage polarization are Th1 (IFN-γ)/Th2 (IL-4, IL-5, IL-13) cytokines which are produced mostly by T cells [Citation4,Citation37]. Thus, it is likely that the balance between Th1 and Th2 responses plays a decisive role in the regulation of obese adipose tissue inflammation. Concomitant with increased M1 polarization, the predominance of Th1 response over Th2 response is one of the well-known characters of obesity-induced systemic inflammation as well as adipose tissue inflammation [Citation8,Citation14]. For example, increased IFN-γ secreting T helper cells in blood and adipose tissue support skewed Th1/Th2 response in obesity [Citation39,Citation40]. Additionally, C57BL6 mouse strain that exhibits Th1-biased response is more susceptible to obesity-induced inflammation compared with BALB/c mouse strain that exhibits Th2-biased response [Citation33,Citation35]. While IFN-γ is produced by Th1 cells, CD8 T cells, and NK cells in adipose tissue [Citation23,Citation36,Citation48], IL-4, one of the representative Th2 cytokines, is released from Th2 cells and eosinophils [Citation48,Citation49]. Furthermore, recent studies have demonstrated that adipose invariant Natural Killer T (iNKT) cells would be a significant contributor for Th2 type cytokines including IL-4 and IL-10 in adipose tissues [Citation12,Citation15,Citation16,Citation28,Citation29,Citation44].
NKT cells are innate-like lymphocytes which could connect innate and adaptive immune responses by recognition of lipid antigens [Citation1,Citation3]. NKT cells are largely classified into three types; invariant NKT (iNKT) cells (type I), diverse NKT (type II) cells, and NKT-like cells [Citation7]. Although both type I and type II NKT cells can recognize lipid antigens loaded on CD1d molecule, which is an MHC class I-like family protein, they recognize different species of lipid antigens [Citation1,Citation7]. iNKT cells have been identified as a predominant subset that is reactive to a marine sponge-derived glycolipid α-galactosylceramide (α-GC) [Citation20]. Upon activation signaling, iNKT cells can rapidly secrete a large amount of cytokines. Particularly, iNKT cells are able to differentially secrete either Th1-type or Th2-type cytokines depending on the species of lipid antigens, antigen presenting cell (APC) types, and cytokines [Citation3]. Along with these complex characters of iNKT cells, there are controversies for the role of iNKT cells in the regulation of obesity-related inflammation as well as adipose tissue inflammation [Citation12,Citation15,Citation16,Citation21,Citation29,Citation41,Citation42,Citation44,Citation50]. Whereas it has been reported that adipose iNKT cells might have pro-inflammatory roles or no significant roles for adipose tissue inflammation [Citation21,Citation41,Citation42,Citation50], many studies including our previous papers have shown that adipose iNKT cells harbor the anti-inflammatory functions in the regulation of obesity-related inflammation [Citation12,Citation15,Citation16,Citation28,Citation29,Citation44]. Of course, we can't exclude the possibility that several differences in high-fat diet composition, animal facility-related microbiota, and control group mice could affect different phenotypes of α-GC-treated mice or iNKT cell-deficient mice [Citation12,Citation13,Citation29]. These issues need to be clarified in the future study. Nonetheless, there are several pieces of evidence for the beneficial role of iNKT cells in obesity. Firstly, the number of adipose iNKT cells is decreased in obese adipose tissues of human and mouse models [Citation12,Citation16,Citation29]. Similar to iNKT cells, other anti-inflammatory cell types such as M2 macrophages, eosinophils, and Treg cells are also reduced in obese adipose tissues [Citation6,Citation26,Citation49]. Secondly, this numerical reduction in adipose iNKT cell population is one of the causal factors for obesity-related adipose tissue inflammation, which has been supported by the findings that increased susceptibility to obesity and its related metabolic complications in iNKT cell-deficient model mice (Jα18 KO mice) [Citation12,Citation29,Citation44]. It has been reported that HFD-fed Jα18 KO mice gain more body weight and fat mass as well as exhibit enhanced adipose tissue inflammatory responses including increased M1 macrophage number and pro-inflammatory gene expression [Citation12,Citation29]. Lastly, it has been shown that the adoptive transfer of iNKT cells and α-GC injection into obese mice improve insulin sensitivity and inflammation [Citation16,Citation29]. Moreover, IL-2, IL-4, and IL-10 have been suggested as the candidate mediators for anti-inflammatory roles of iNKT cells [Citation13,Citation15,Citation16,Citation28,Citation29].
Despite of these findings, the regulatory mechanisms for adipose iNKT cells in obesity have not been clearly elucidated. Very recently, we have reported that adipocytes could act as a crucial lipid APC for adipose iNKT cells in vivo [ Citation13]. This idea has been evolved with following findings. Firstly, we discovered that CD1d, which is a lipid antigen-presenting molecule for iNKT cells, is highly expressed in adipocytes than any other cell types including adipose tissue macrophages [Citation12,Citation13]. Secondly, it has been shown that α-GC-loaded adipocytes are able to activate iNKT cells in cell culture system [Citation12]. Thirdly, adipocytes are professional cells to sense and handle the alteration of dynamic lipid metabolism for whole body energy homeostasis [Citation24,Citation30]. Moreover, the level of pro-inflammatory gene expression is gradually and significantly elevated in adipocyte fraction as well as SVCs upon HFD feeding periods [Citation24]. These findings led us to hypothesize that adipocytes would be an effective APC to present lipid antigens for the fine-tuning of adipose iNKT cell responses depending on nutritional status.
We have observed that suppression of CD1d expression in 3T3-L1 adipocytes and primary adipocytes inhibits α-GC-induced iNKT cell activation [Citation12]. However, the in vivo role of adipocyte CD1d had not been clearly established because there are other classical APCs such as dendritic cells, macrophages, and B cells in adipose tissues [Citation2,Citation26,Citation47]. In addition, given that adipocytes actively process lipid metabolites which could stimulate iNKT cells, it is plausible to speculate that adipocyte could play important roles as an APC for adipose iNKT cells. To address these issues, we decided to generate adipocyte-specific CD1d KO (CD1dADKO) mice and have investigated adipose iNKT cells, HFD-induced inflammatory responses in adipose tissue, and systemic insulin sensitivity [Citation13]. Compared to control CD1df/f mice, CD1dADKO mice showed the decrement in adipose iNKT cells as well as in α-GC-induced iNKT cell activation in adipose tissue. Moreover, HFD-induced insulin resistance was further aggravated in CD1dADKO mice. In addition, the ratio of M1 to M2 macrophage number was significantly increased in long-term (8 weeks) as well as in short-term (1 week) HFD-fed CD1dADKO mice. Along with decreased proportion of M2 macrophages, the level of IL-4 was also down-regulated in adipose tissue of HFD-fed CD1dADKO mice. This reduction of IL-4 was attributable, at least in part, to decreased number of CD4+ iNKT cells which have more potency to produce IL-4 than CD4− iNKT cells in adipose tissue of HFD-fed CD1dADKO mice. Interestingly, one week of IL-4 supplementation improved insulin sensitivity and reduced adipose tissue inflammation in CD1dADKO mice, implying that reduced level of IL-4 production could be one of the key mediators of augmented insulin resistance and adipose tissue inflammation in HFD-fed CD1dADKO mice. Taken together, our finding suggests that adipocyte CD1d would play the defensive roles against HFD-induced adipose tissue inflammation by IL-4 production from adipose iNKT cell.
Notably, we also found that CD1d deletion in adipocytes reduced the number of iNKT cells in adipose tissue of normal chow diet (NCD)-fed mice. The levels of apoptosis and proliferation in adipose iNKT cells were not substantially altered by adipocyte CD1d deletion, indicating that reduced number of adipose iNKT cells in CD1dADKO mice might not be due to changes in apoptosis or proliferation. As CD1d knockdown in 3T3-L1 adipocytes significantly diminished the physical contact between iNKT cells and differentiated adipocytes [Citation12], it is possible that adipocyte CD1d would be involved in the retention signal for adipose iNKT cells. While LFA-1 and ICAM-1 are known as retention signals for iNKT cells in the liver, blocking of ICAM-1 and LFA-1 by neutralizing antibodies or ICAM-1 KO mice have shown the similar number of adipose iNKT cells compared to control mice [Citation28]. Collectively, our findings suggest that adipocyte CD1d would have distinct roles for the maintenance of iNKT cell numbers in adipose tissue and potentially could act as a retention signal.
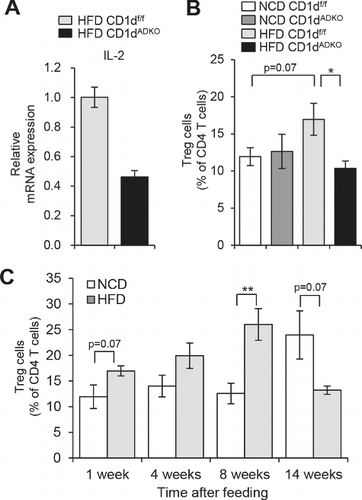
We have proposed that defective IL-4 production in adipose iNKT cells would be one of the crucial factors to deteriorate metabolic phenotypes in HFD-fed CD1dADKO mice [Citation13]. However, we also observed that IL-2 production was altered in adipose iNKT cells of HFD-fed CD1dADKO mice. IL-2 is a key cytokine for the generation, maintenance, and function of Treg cells [Citation25]. NKT cells can produce not only IFN-γ and IL-4 but also IL-2 upon TCR stimulation [Citation31]. Also, it has been shown that human CD4+ NKT cells promote moderate proliferation of Treg cells via IL-2 production [Citation17,Citation22]. Notably, when gene expression profiles have been compared between adipose iNKT cells and splenic iNKT cells, it has been revealed that adipose iNKT cells express higher level of IL-2 [Citation28]. It has been suggested that the low level of PLZF (Zbtb16) would be attributable to enhanced production of IL-2 in adipose iNKT cells [Citation28]. PLZF is a transcription factor known to be expressed by all iNKT cells [Citation43] but its expression in adipose iNKT cells is lower compared with iNKT cells in thymus and liver. Also, PLZF-deficient iNKT cells shows enhanced IL-2 production [Citation43]. In HFD-fed CD1dADKO mice, adipose iNKT cells expressed a lower level of IL-2 mRNA than those from HFD-fed CD1df/f mice (). Also, short-term (1 week) HFD-fed CD1dADKO mice impaired increment of Treg cell numbers (). Although it has been reported that the number of Treg cells is reduced in adipose tissue of long-term (29 weeks) HFD-fed mice or genetically obese mice such as ob/ob [Citation6], we repeatedly observed that frequency of Treg cells was slightly but substantially increased in short-term HFD-fed mice (). Thus, these data propose that adipocyte CD1d could modulate IL-2 expression from adipose iNKT cells, which would influence Treg cell-mediated anti-inflammatory responses upon excessive energy intake. On the other hand, given that IL-2 could act as an important regulator for type 2 cytokine production [Citation10,Citation38], it is possible that IL-2 might mediate the Treg cell-independent anti-inflammatory responses. Collectively, we observed that CD1dADKO mice impaired cytokine (IL-4 and IL-2) expression in adipose iNKT cells and downregulated anti-inflammatory responses including M2 macrophages and Treg cells in obese adipose tissue. Therefore, these data propose that adipocyte CD1d would play a pivotal role in the defensive process against excess nutrient-induced inflammatory responses by stimulating adipose iNKT cells.
Figure 1. Analysis of iNKT cells and Treg cells after 1 week of HFD feeding. A: The mRNA level of IL-2 in sorted iNKT cells (TCRβ+PBS57/CD1d tetramer+) from EATs of HFD-fed CDldf/f and CDldADKO mice. EATs were pooled from 15 mice per group. B: The percentage of Treg cells (TCRβ+CD4+Foxp3+) among CD4 T cells in EATs. n = 5. C: The percentage of Treg cells in EATs during NCD and HFD feeding period. N = 4–5. *p < 0.05 and **p < 0.0l. 8-week-old mice were fed NCD or 60% HFD for the indicated time periods.
It has been proposed that the interaction between adipocyte CD1d and adipose iNKT cells is critical in the regulation of anti-inflammatory responses, especially, in the early phase of obesity [Citation12,Citation13,Citation15]. For instance, many anti-inflammatory responses in adipose tissues are also further activated to inhibit excess nutrient-induced inflammation in the early stage of obesity rather than in the late stage of severe obesity. At the early stage of obesity, adipose iNKT cells are activated and produce anti-inflammatory cytokines including IL-4 and IL-2 by interacting with adipocyte CD1d even after one week of HFD feeding [Citation13] (). These cytokines contribute to the stimulation of anti-inflammatory immune cells including M2 macrophages and Treg cells. Concurrently, a subset of activated iNKT cells undergoes activation-induced cell death (AICD), which leads to the reduction of adipose iNKT cells [Citation12]. In the late stage of obesity, both mRNA and protein levels of adipocyte CD1d expression are considerably decreased [Citation12,Citation13]. The reduced CD1d expression in adipocytes could be mediated by PPARγ whose activity is significantly downregulated in adipocytes by enhanced inflammatory signaling cues such as TNF-α [Citation12,Citation13,Citation32]. In severely obese adipose tissue, adipocytes expressing reduced level of CD1d could not adequately mediate iNKT cell stimulation, resulting in enhanced adipose tissue inflammation and adipose tissue dysfunction (). The systemic effect of diminished iNKT cell responses induced by adipocyte CD1d deficiency could be explained by increased levels of circulating free fatty acids and hepatic triglyceride accumulation in long-term HFD-fed CD1dADKO mice () [Citation13]. These findings propose that adipocyte CD1d deletion-dependent adipose tissue dysfunction could increase free fatty acid release, which leads to ectopic lipid accumulation in liver and consequently augments systemic insulin resistance in obesity.
Figure 2. Proposed model for the interaction between adipocyte CDld and adipose iNKT cells in the regulation of adipose tissue inflammation with progressive obesity. In lean adipose tissue, adipocytes highly express CDld molecules which play a crucial role in the maintenance of adipose iNKT cell population. Upon HFD feeding, adipocytes present obesity-related lipid antigens via CDld molecules, which leads to iNKT cell activation and stimulates anti-inflammatory cytokine secretion from adipose iNKT cells. Concurrently, activation-induced cell death (AICD) is occurred in part of activated iNKT cells, which results in reduced number of iNKT cells in adipose tissues. On the other hand, the significantly reduced CDld expression on adipocytes from severely obese adipose tissues weakens adipose iNKT cell stimulation. Elevated inflammatory responses which are associated with the impairment of anti-inflammatory responses accelerate adipose tissue dysfunction including FFA release. Increased circulating FFAs could accumulate in the liver.
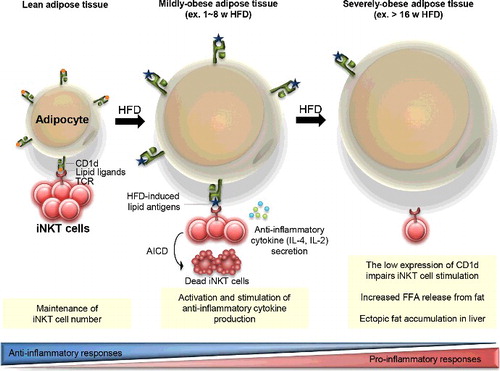
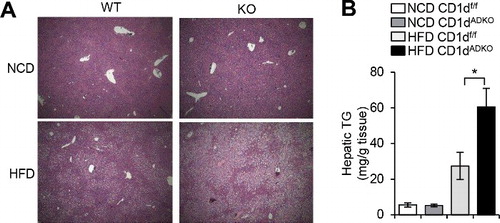
Figure 3. Increased hepatic triglyceride accumulation in HFD-fed CDtdADKO mice A: H&E staining images of liver from CDldf/f and CDldADKO mice upon NCD and 12 weeks of 60% HFD feeding. n = 5. B: Hepatic triglyceride concentration. n = 5 for NCD and n = 8 for 60% HFD. *P < 0.05.
On the contrary, Iwabuchi's group has reported that the interaction between adipocyte CD1d and NKT cells would mediate pro-inflammatory response upon HFD feeding [Citation42]. This report is consistent with their previous findings that adipose NKT cells would aggravate adipose tissue inflammation and insulin resistance in obesity [Citation41]. They have suggested that impaired IFN-γ secretion from adipose NKT cells in the absence of adipocyte CD1d might mediate the protective effects on diet-induced obesity. However, as we described in our research article, the expression of IL-4 rather than IFN-γ was induced in adipose iNKT cells upon HFD feeding [Citation13]. The contradictions between Iwabuchi's group and our group would result from several factors. For instance, two groups used different types of control mice (adiponectin cre/CD1dflox/+ mice vs. littermate CD1dflox/flox mice). Furthermore, the composition of HFD (tallow and safflower oil of high oleic type vs. lard) and environments of the animal facilities were different between Iwabuchi's group and our group. The precise causal factor(s) leading to different phenotypes need to be clarified. Although most phenotypes of HFD-fed CD1dADKO mice from Iwabuchi's group are opposite of our findings, there is one consistent conclusion that the activation marker (CD69) expression was reduced in adipose NKT cells in the absence of adipocyte CD1d. Such findings from both groups support the role of adipocyte as APC for adipose NKT cells.
We suggest that adipocytes, as an important ‘atypical’ APC type for adipose iNKT cells, would play pivotal roles in the regulation of adipose tissue inflammation. It has been known that not only professional APCs such as dendritic cells, macrophages, and B cells but also various ‘atypical’ APCs such as epithelial cells and stromal cells can present antigens to CD4+ T cells [Citation18]. Our findings from adipocyte-specific CD1d KO mice propose that excess nutrient-induced lipid metabolites could directly send a signal to iNKT cells for resolving pro-inflammatory responses. In other words, adipose iNKT cells patrol around the adipocytes and recognize altered lipid antigen(s) loaded on adipocyte CD1d, which could trigger anti-inflammatory responses to modulate adipose tissue homeostasis.
IL-4 has been known as one of the important cytokines to mediate thermogenic responses. More importantly, recent report indicated that α-GC induced-iNKT cell activation induces thermogenic browning of white fat [Citation27]. As we have demonstrated that adipocytes promptly drive functional changes of iNKT cells into IL-4 secreting cells, it would be interesting to investigate whether such interplay could be involved in iNKT cell-mediated thermogenesis and energy expenditure.
In obesity, a Th1 response is higher than a Th2 response. Thus, it has been considered that obesity-related inflammation would be resulted from a Th1-skewed imbalance. However, certain metabolic organs including adipose tissue appear to have a resolving process against this pro-inflammatory stress by induction of Th2 responses. The anti-inflammatory response which is mediated by the interaction between adipocytes and adipose iNKT cells is one of potential resolving processes. To understand the regulatory mechanism for adipose iNKT cell activation, the excess nutrient-induced lipid antigens as well as adipose iNKT cell-specific characters need to be figured out in the further study. Therefore, it seems that understanding regulatory mechanisms for anti-inflammatory response would be crucial to identify effective therapeutic targets for maintaining metabolic and immune homeostasis against obesity-related metabolic complications.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Jeu Park for critical reading of the manuscript.
Additional information
Funding
References
- Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–336. Available from https://www.ncbi.nlm.nih.gov/pubmed/17150027. doi:10.1146/annurev.immunol.25.022106.141711
- Bertola A, Ciucci T, Rousseau D, et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes. 2012;61(9):2238–2247. Available from https://www.ncbi.nlm.nih.gov/pubmed/22596049 .doi:10.2337/db11-1274
- Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol. 2013;13(2):101–117. Available from https://www.ncbi.nlm.nih.gov/pubmed/23334244. doi:10.1038/nri3369
- Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol. 2011;11(11):738–749. Available from https://www.ncbi.nlm.nih.gov/pubmed/21984069. doi:10.1038/nri3071
- Choe SS, Shin KC, Ka S, et al. Macrophage HIF-2α ameliorates adipose tissue inflammation and insulin resistance in obesity. Diabetes. 2014;63(10):3359–3371. Available from https://www.ncbi.nlm.nih.gov/pubmed/24947359. doi:10.2337/db13-1965
- Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15(8):930–939. Available from https://www.ncbi.nlm.nih.gov/pubmed/19633656. doi:10.1038/nm.2002
- Godfrey DI, MacDonald HR, Kronenberg M, et al. NKT cells: what's in a name? Nat Rev Immunol. 2004;4(3):231–237. Available from https://www.ncbi.nlm.nih.gov/pubmed/15039760. doi:10.1038/nri1309
- Guigas B, Molofsky AB. A worm of one's own: how helminths modulate host adipose tissue function and metabolism. Trends Parasitol. 2015;31(9):435–441. Available from https://www.ncbi.nlm.nih.gov/pubmed/25991556. doi:10.1016/j.pt.2015.04.008
- Ham M, Choe SS, Shin KC, et al. Glucose-6-Phosphate Dehydrogenase Deficiency Improves Insulin Resistance With Reduced Adipose Tissue Inflammation in Obesity. Diabetes. 2016;65(9):2624–2638. Available from https://www.ncbi.nlm.nih.gov/pubmed/27284106. doi:10.2337/db16-0060
- Hondowicz BD, An D, Schenkel JM, et al. Interleukin-2-Dependent Allergen-Specific Tissue-Resident Memory Cells Drive Asthma. Immunity. 2016;44(1):155–166. Available from https://www.ncbi.nlm.nih.gov/pubmed/26750312. doi:10.1016/j.immuni.2015.11.004
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. Available from https://www.ncbi.nlm.nih.gov/pubmed/7678183 doi:10.1126/science.7678183
- Huh JY, Kim JI, Park YJ, et al. A novel function of adipocytes in lipid antigen presentation to iNKT cells. Mole Cell Biol. 2013;33(2):328–339. Available from http://www.ncbi.nlm.nih.gov/pubmed/23149942. doi:10.1128/MCB.00552-12
- Huh JY, Park J, Kim JI, et al. Deletion of CD1d in Adipocytes Aggravates Adipose Tissue Inflammation and Insulin Resistance in Obesity. Diabetes. 2017;66(4):835–847. Available from https://www.ncbi.nlm.nih.gov/pubmed/28082459. doi:10.2337/db16-1122
- Huh JY, Park YJ, Ham M, et al. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol Cells. 2014;37(5):365–371. Available from https://www.ncbi.nlm.nih.gov/pubmed/24781408. doi:10.14348/molcells.2014.0074
- Ji Y, Sun S, Xia S, et al. Short term high fat diet challenge promotes alternative macrophage polarization in adipose tissue via natural killer T cells and interleukin-4. J Biol Chem. 2012;287(29):24378–24386. Available from https://www.ncbi.nlm.nih.gov/pubmed/22645141. doi:10.1074/jbc.M112.371807
- Ji Y, Sun S, Xu A, et al. Activation of natural killer T cells promotes M2 Macrophage polarization in adipose tissue and improves systemic glucose tolerance via interleukin-4 (IL-4)/STAT6 protein signaling axis in obesity. J Biol Chem. 2012;287(17):13561–13571. Available from https://www.ncbi.nlm.nih.gov/pubmed/22396530. doi:10.1074/jbc.M112.350066
- Jiang S, Game DS, Davies D, et al. Activated CD1d-restricted natural killer T cells secrete IL-2: innate help for CD4+CD25+ regulatory T cells? Eur J Immunol. 2005;35(4):1193–1200. Available from https://www.ncbi.nlm.nih.gov/pubmed/15770696. doi:10.1002/eji.200425899
- Kambayashi T, Laufer TM. Atypical MHC class II-expressing antigen-presenting cells: can anything replace a dendritic cell? Nat Rev Immunol. 2014;14(11):719–730. Available from https://www.ncbi.nlm.nih.gov/pubmed/25324123. doi:10.1038/nri3754
- Kern PA, Ranganathan S, Li C, et al. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab. 2001;280(5):E745–751. Available from https://www.ncbi.nlm.nih.gov/pubmed/11287357 doi:10.1152/ajpendo.2001.280.5.E745
- Kinjo Y, Wu D, Kim G, et al. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature. 2005;434(7032):520–525. Available from https://www.ncbi.nlm.nih.gov/pubmed/15791257. doi:10.1038/nature03407
- Kotas ME, Lee HY, Gillum MP, et al. Impact of CD1d deficiency on metabolism. PLoS One. 2011;6(9):e25478. Available from https://www.ncbi.nlm.nih.gov/pubmed/21980475. doi:10.1371/journal.pone.0025478
- La Cava A, Van Kaer L. Fu-Dong-Shi. CD4+CD25+ Tregs and NKT cells: regulators regulating regulators. Trends Immunol. 2006;27(7):322–327. Available from https://www.ncbi.nlm.nih.gov/pubmed/16735139. doi:10.1016/j.it.2006.05.003
- Lee BC, Kim MS, Pae M, et al. Adipose Natural Killer Cells Regulate Adipose Tissue Macrophages to Promote Insulin Resistance in Obesity. Cell Metab. 2016;23(4):685–698. Available from https://www.ncbi.nlm.nih.gov/pubmed/27050305. doi:10.1016/j.cmet.2016.03.002
- Lee YS, Li P, Huh JY, et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes. 2011;60(10):2474–2483. Available from http://www.ncbi.nlm.nih.gov/pubmed/21911747. doi:10.2337/db11-0194
- Liston A, Gray DH. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol. 2014;14(3):154–165. Available from https://www.ncbi.nlm.nih.gov/pubmed/24481337. doi:10.1038/nri3605
- Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184. Available from https://www.ncbi.nlm.nih.gov/pubmed/17200717. doi:10.1172/JCI29881
- Lynch L, Hogan AE, Duquette D, et al. iNKT Cells Induce FGF21 for Thermogenesis and Are Required for Maximal Weight Loss in GLP1 Therapy. Cell Metab. 2016;24(3):510–519. Available from https://www.ncbi.nlm.nih.gov/pubmed/27593966. doi:10.1016/j.cmet.2016.08.003
- Lynch L, Michelet X, Zhang S, et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat Immunol. 2015;16(1):85–95. Available from https://www.ncbi.nlm.nih.gov/pubmed/25436972. doi:10.1038/ni.3047
- Lynch L, Nowak M, Varghese B, et al. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity. 2012;37(3):574–587. Available from https://www.ncbi.nlm.nih.gov/pubmed/22981538. doi:10.1016/j.immuni.2012.06.016
- Masoodi M, Kuda O, Rossmeisl M, et al. Lipid signaling in adipose tissue: Connecting inflammation & metabolism. Biochim Biophys Acta. 2015;1851(4):503–518. Available from http://www.ncbi.nlm.nih.gov/pubmed/25311170. doi:10.1016/j.bbalip.2014.09.023
- Metelitsa LS, Naidenko OV, Kant A, et al. Human NKT cells mediate antitumor cytotoxicity directly by recognizing target cell CD1d with bound ligand or indirectly by producing IL-2 to activate NK cells. J Immunol. 2001;167(6):3114–3122. Available from https://www.ncbi.nlm.nih.gov/pubmed/11544296 doi:10.4049/jimmunol.167.6.3114
- Miles PD, Romeo OM, Higo K, et al. TNF-alpha-induced insulin resistance in vivo and its prevention by troglitazone. Diabetes. 1997;46(11):1678–1683. Available from https://www.ncbi.nlm.nih.gov/pubmed/9356012 doi:10.2337/diab.46.11.1678
- Mills CD, Kincaid K, Alt JM, et al. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164(12):6166–6173. Available from https://www.ncbi.nlm.nih.gov/pubmed/10843666 doi:10.4049/jimmunol.164.12.6166
- Moller DE, Kaufman KD. Metabolic syndrome: a clinical and molecular perspective. Annu Rev Med. 2005;56:45–62. Available from https://www.ncbi.nlm.nih.gov/pubmed/15660501. doi:10.1146/annurev.med.56.082103.104751
- Montgomery MK, Hallahan NL, Brown SH, et al. Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia. 2013;56(5):1129–1139. Available from https://www.ncbi.nlm.nih.gov/pubmed/23423668. doi:10.1007/s00125-013-2846-8
- Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15(8):914–920. Available from https://www.ncbi.nlm.nih.gov/pubmed/19633658. doi:10.1038/nm.1964
- Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. Available from https://www.ncbi.nlm.nih.gov/pubmed/20148674. doi:10.1146/annurev-physiol-021909-135846
- Olson MR, Ulrich BJ, Hummel SA, et al. Paracrine IL-2 Is Required for Optimal Type 2 Effector Cytokine Production. J Immunol. 2017;198(11):4352–4359. Available from https://www.ncbi.nlm.nih.gov/pubmed/28468971. doi:10.4049/jimmunol.1601792.
- Pacifico L, Di Renzo L, Anania C, et al. Increased T-helper interferon-gamma-secreting cells in obese children. Eur J Endocrinol. 2006;154(5):691–697. Available from https://www.ncbi.nlm.nih.gov/pubmed/16645016. doi:10.1530/eje.1.02138
- Rocha VZ, Folco EJ, Sukhova G, et al. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res. 2008;103(5):467–476. Available from https://www.ncbi.nlm.nih.gov/pubmed/18658050. doi:10.1161/CIRCRESAHA.108.177105
- Satoh M, Andoh Y, Clingan CS, et al. Type II NKT cells stimulate diet-induced obesity by mediating adipose tissue inflammation, steatohepatitis and insulin resistance. PLoS One. 2012;7(2):e30568. Available from https://www.ncbi.nlm.nih.gov/pubmed/22383967. doi:10.1371/journal.pone.0030568
- Satoh M, Hoshino M, Fujita K, et al. Adipocyte-specific CD1d-deficiency mitigates diet-induced obesity and insulin resistance in mice. Sci Rep. 2016;6:28473. Available from https://www.ncbi.nlm.nih.gov/pubmed/27329323. doi:10.1038/srep28473
- Savage AK, Constantinides MG, Han J, et al. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity. 2008;29(3):391–403. Available from https://www.ncbi.nlm.nih.gov/pubmed/18703361. doi:10.1016/j.immuni.2008.07.011
- Schipper HS, Rakhshandehroo M, van de Graaf SF, et al. Natural killer T cells in adipose tissue prevent insulin resistance. J Clin Invest. 2012;122(9):3343–3354. Available from https://www.ncbi.nlm.nih.gov/pubmed/22863618. doi:10.1172/JCI62739
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793–1801. Available from https://www.ncbi.nlm.nih.gov/pubmed/16823477. doi:10.1172/JCI29069
- Sun S, Ji Y, Kersten S, et al. Mechanisms of inflammatory responses in obese adipose tissue. Annu Rev Nutr. 2012;32:261–286. Available from https://www.ncbi.nlm.nih.gov/pubmed/22404118. doi:10.1146/annurev-nutr-071811-150623
- Winer DA, Winer S, Shen L, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17(5):610–617. Available from https://www.ncbi.nlm.nih.gov/pubmed/21499269. doi:10.1038/nm.2353
- Winer S, Chan Y, Paltser G, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15(8):921–929. Available from https://www.ncbi.nlm.nih.gov/pubmed/19633657. doi:10.1038/nm.2001
- Wu D. Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, Chawla A and Locksley RM. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332(6026):243–247. Available from https://www.ncbi.nlm.nih.gov/pubmed/21436399. doi:10.1126/science.1201475
- Wu L, Parekh VV, Gabriel CL, et al. Activation of invariant natural killer T cells by lipid excess promotes tissue inflammation, insulin resistance, and hepatic steatosis in obese mice. Proc Natl Acad Sci U S A. 2012;109(19):E1143–1152. Available from https://www.ncbi.nlm.nih.gov/pubmed/22493234. doi:10.1073/pnas.1200498109