
ABSTRACT
Obesity is a leading risk factor for post-menopausal breast cancer, and this is concerning as 40% of cancer diagnoses in 2014 were associated with overweight/obesity. Despite this epidemiological link, the underlying mechanism responsible is unknown. We recently published that visceral adipose tissue (VAT) releases FGF2 and stimulates the transformation of skin epithelial cells. Furthermore, obesity is differentially associated with many epithelial cancers, and this mechanistic link could be translational. As FGF2 and FGFR1 are implicated in breast cancer progression, we hypothesize that VAT-derived FGF2 plays a translational role in promoting adiposity-associated mammary epithelial cell transformation. In this brief report, data suggest that FGF2/FGFR1 signaling is a potential mechanistic link in VAT-stimulated transformation of breast epithelial cells.
Introduction
Obesity is a well-established risk factor for post-menopausal breast cancer. [Citation1] A greater waist to hip ratio [indicative of a higher content of visceral adipose tissue (VAT)] increases the risk of post-menopausal breast cancer [Citation2–Citation5]. In pre-menopausal breast cancer, when adjusted for weight or body mass index (BMI), women with the smallest waist to hip ratios have a 37% lower risk [Citation6]. Thus, visceral obesity, an increase in adipose tissue surrounding the intra-abdominal organs, directly relates to the magnitude of obesity-related breast cancer risk [Citation2–Citation5]. However, the underlying mechanisms responsible for the VAT- breast cancer link are not fully elucidated [Citation7].
Anatomical and physiological differences between visceral adipose tissue (VAT) and subcutaneous adipose tissue (SAT) determine the extent of how these depots contribute to obesity, metabolic syndrome (MetS), and cancer [Citation8,Citation9]. In obese individuals, adipocytes become hypertrophic, which makes them dysfunctional and insulin resistant. The pathophysiology of obesity-induced insulin resistance has been attributed to ectopic fat deposition, increased inflammation, oxidative stress, adipose tissue hypoxia and mitochondrial dysfunction, and impaired adipocyte expansion and angiogenesis [Citation8,Citation9]. Excess VAT contains a greater number of large adipocytes in contrast to SAT, which contains smaller, insulin sensitive adipocytes [Citation8]. Moreover, expanding adipose tissue can induce hypoxia from insufficient vasculature and oxygen supply [Citation8]. This hypoxia can induce immune cell infiltration, causing low-grade chronic inflammation [Citation10]. Adipocyte hypertrophy and immune cell infiltration alters the release of adipokines (cytokines derived from adipose tissue) that can exacerbate the immune response and induce systemic release of adipokines that can act on neighboring and distant targets [Citation7,Citation10]. These obesity-related changes are associated with insulin resistance, which in turn leads to hyperglycemia, hypertension, dyslipidemia, and other metabolic abnormalities [Citation9,Citation11]. Similarly, MetS is characterized by a cluster of three or more metabolic abnormalities including visceral obesity, insulin resistance, dyslipidemia, hypertension, and hyperglycemia. However, these similar physiological aspects are not mutually exclusive; for example, not everyone who is obese has inflammation and metabolic syndrome [Citation12]. Regardless, many studies have concluded the rise in visceral obesity has led to an increase in MetS [Citation9]. Epidemiological studies show both obesity and MetS are breast cancer risk factors [Citation3,Citation13]. Bridging the link between obesity, MetS and breast cancer risk, Kabat et al. showed obesity is associated with increased breast cancer risk and metabolically unhealthy obese individuals had the highest risk [Citation12]. However, epidemiological associations and obesity-related changes fall short of explaining the biological mechanisms by which adiposity contributes to cancer promotion and malignant transformation (a change a cell undergoes to become malignant).
Animal models have given insight into the mechanistic link between visceral adiposity and cancer. In a rat model of intestinal cancer, removing VAT significantly attenuated obesity-associated intestinal tumorigenesis [Citation14]. In addition, we previously demonstrated removing VAT in HFD-fed mice significantly reduces UVB-induced squamous cell carcinomas by 75–80% when compared to sham-operated control animals [Citation15]. These data suggest VAT-derived factors are critical for carcinogenesis [Citation14,Citation15]. We also utilized an ex vivo model to evaluate the ability of VAT-derived growth factors to stimulate transformation of non-tumorigenic JB6 P+ mouse skin epithelial cells. Cellular transformation as indicated by anchorage-independent growth in soft agar is a well-established, stringent method for detecting the tumorigenic potential of transformed cells [Citation16–Citation18]. JB6 P+ cells cannot proliferate in an anchorage-independent manner but have the ability to transform upon treatment of tumor promoters [Citation16–Citation18]. Using this model, we identified fibroblast growth factor 2 (FGF2) as the critical VAT-derived factor in stimulating JB6 P+ growth in soft agar [Citation19]. JB6 P+ cells that lacked the fibroblast growth factor receptor 1 (FGFR1), FGF2's receptor, failed to transform in the presence of VAT, suggesting the FGF2/FGFR1 signaling axis is critical in VAT-stimulated transformation of epithelial cells at distant targets. How generalizable this mechanism is to other tissues and human cells is unknown. Therefore, we hypothesize that VAT-stimulation of skin carcinogenesis through the FGF2/FGFR1 signaling is translational to VAT-associated breast cancer.
The objective of this study is to determine the effects of human VAT on the transformation of MCF-10A human mammary epithelial cells. MCF-10A cells are non-tumorigenic and do not exhibit anchorage-independent growth in soft agar. We hypothesized that VAT will stimulate the transformation of MCF-10A cells and this activity will be dependent on FGFR1. Establishing a human model of VAT-stimulated transformation will strengthen support for the direct role of VAT in adiposity-promoted carcinogenesis. There are fundamental differences in the transformation susceptibility of human and mouse cells specifically in the greater number of events required to transform human cells than those required for non-tumorigenic mouse cells [Citation20,Citation21]. Consequently, establishing a human model of VAT-stimulated transformation of mammary epithelial cells shows adiposity-promoted carcinogenesis is relevant to both mouse and human models and is translational to obesity-associated breast cancer.
Results
To test the effects of VAT on mammary epithelial transformation, human fat tissue filtrate (HuFTF) was generated from VAT of six different human donors. VAT was obtained from omental tissue of cancer-free female obese human subjects purchased from Spectrum Health Universal Biorepository (SHUB, Grand Rapids, MI). These subjects were undergoing surgery for gastrointestinal conditions. describes the human donor characteristics including age, BMI, gender, and ethnicity. We were not able to obtain information on menopausal status, metabolic status or serum metabolites. To determine if HuFTF stimulates transformation of human mammary epithelial cells, MCF-10A cells were incubated with 200 μg/mL of HuFTF in soft agar and scored for colony formation. Colonies (8 cells or greater) were visually counted and a percent of colony formation was obtained by relating the number of colonies with the number of cells plated (750 cells/well). While MCF-10A cells are non-tumorigenic epithelial cells, they have a low level of spontaneous transformation in contrast to tumorigenic epithelial cells, which have almost 100% transformation. HuFTF significantly stimulated colony formation above the control with statistical significance (p<0.05) ().
Table 1. Clinical characteristics of donors.
Figure 1. Inhibition of FGFR1 attenuates HuFTF-stimulated transformation of MCF-10A cells. HuFTF significantly stimulated colony formation above the untreated control. Cells were treated with 200 μg/mL of HuFTF protein. HuFTF-stimulated growth in soft agar was significantly attenuated by the FGFR1 Ab. MCF-10A ells were treated with a FGFR1 neutralizing antibody (FGFR1 Ab) at 2 μg/mL and treated with HuFTF from six different donors. The percent of colony formation was calculated as described in Methods, MCF-10A cells were cultured as described in Methods. Data is presented as mean ± six biological replicates. Each biological replicate had three technical replicates. Confidence intervals (CI) were calculated for HuFTF treated MCF-10A cells (95% CI 3.024–7.658) and for HuFTF+Ab (95% CI 0.093–2.929). Statistical significance between HuFTF and control and HuFTF and HuFTF+FGFR1 Ab was determined by unpaired t-test (*p < 0.05).
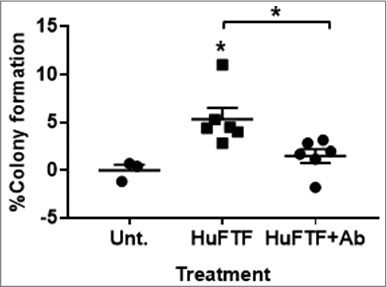
To determine the role of FGFR1 signaling in HuFTF-stimulated transformation, MCF-10A cells were incubated with a tyrosine kinase fibroblast growth factor receptor 1 antibody antagonist (FGFR1 Ab). Inhibiting FGFR1 receptor activity attenuated HuFTF-stimulated transformation of MCF-10A cells (). The FGFR1 Ab (2 μg/mL) significantly decreased HuFTF-stimulated colony formation, indicating FGFR1 signaling is required for optimal HuFTF-stimulated transformation.
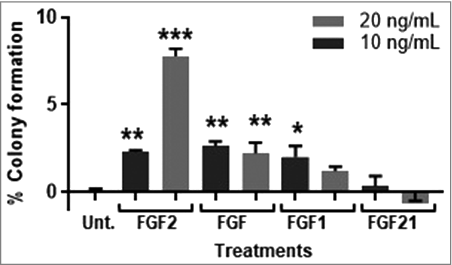
FGFR1 is a receptor for many FGF ligands and to determine the optimal ligand for stimulating MCF-10A transformation, MCF-10A cells were incubated with FGF1, FGF2, FGF18, and FGF21. FGF1 binds to all four FGFR receptors, FGF18 has the highest affinity for FGFR3 IIIc, with some affinity for FGFR4, FGF21 has the highest affinity for FGFR4, followed by FGFR2 IIb and FGFR IIIc, and FGF2 has the highest affinity for FGFR1 IIIc, FGFR3 IIc, and FGFR4 with some affinity for FGFR2 IIIc [Citation22,Citation23]. While FGF1, FGF2, and FGF18, significantly increased colony formation in MCF-10A cells, FGF2 was the only ligand to induce a concentration response at 10 and 20 ng/mL ().
Figure 2. FGF2 transforms MCF-10A cells in a concentration-dependent manner. FGF1 and FGF2 significantly stimulated transformation of MCF-10A cells at 10 and 20 ng/mL. FGF18 significantly stimulate transformation at 10 ng/mL but not 20 ng/mL and FGF21 was not statistically significant. MCF-10A cells were cultured as described in Methods, control cells were untreated. Data is presented as mean ± of triplicate values. Statistical significance was determined by one-way ANOVA with multiple comparisons (*p < 0.05, **p < 0.01, ***p < 0.001). The percent of colony formation was calculated as described in Methods.
A colony of FGF2-stimulated MCF-10A cells was isolated from soft agar and grown in traditional cell culture plates. The FGF2-transformed MCF-10A cells demonstrated a fibroblastic-spindle morphology compared to the parent MCF-10A cells that are more epithelial-like with a polygonal shape (). After several passages this spindle morphology remained and the FGF2-transformed MCF-10A cells demonstrated an increased and irreversible ability to grow in soft agar.
Figure 3. FGF2-tranformed MCF-10A cells are morphologically and functionally distinct from parent MCF-10A cells. (A) MCF-10A cells have epithelial-like morphology and transformed MCF-10A cells have spindle-like morphology. Transformed MCF-10A cells were obtained by treating MCF-10A cells with FGF2 in soft agar. After 14 days, a colony was isolated and cultured in traditional cell culture conditions for several passages, making transformed MCF-10A cells. Untreated transformed MCF-10A cells formed over 44% more colonies in soft agar compared to untreated parent MCF-10A cells. The percent of colony formation was calculated ([colonies counted × 100] 750 cells).
![Figure 3. FGF2-tranformed MCF-10A cells are morphologically and functionally distinct from parent MCF-10A cells. (A) MCF-10A cells have epithelial-like morphology and transformed MCF-10A cells have spindle-like morphology. Transformed MCF-10A cells were obtained by treating MCF-10A cells with FGF2 in soft agar. After 14 days, a colony was isolated and cultured in traditional cell culture conditions for several passages, making transformed MCF-10A cells. Untreated transformed MCF-10A cells formed over 44% more colonies in soft agar compared to untreated parent MCF-10A cells. The percent of colony formation was calculated ([colonies counted × 100] 750 cells).](/cms/asset/7e90dc0e-aa38-41b0-9a03-1feed2579bc3/kadi_a_1445889_f0003_b.gif)
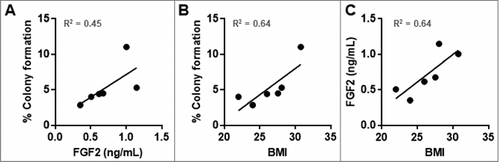
The concentration of FGF2 in each HuFTF donor was determined by ELISA and related to the percent of colony formation and BMI (). The transforming activity of each HuFTF in the soft agar assay was moderately associated with FGF2 concentration in the filtrates (R2 = 0.45) () and with BMI (R2 = 0.64) (). Additionally, there was a moderate association between BMI and HuFTF FGF2 concentrations (R2 = 0.64) ().
Figure 4. HuFTF-stimulated transformation of MCF-10A cells is moderately associated with the HuFTF FGF2 concentration and BMI. (A) HuFTF with higher FGF2 concentrations more potently stimulated MCF-10A transformation compared with HuFTF with lower FGF2 concentrations (R2 = 0.45). (B) HuFTF from donors with a higher BMI more potently stimulated MCF-10A transformation compared to HuFTF from donors with a lower BMI (R2 = 0.64). (C) Higher HuFTF FGF2 concentrations is moderately associated with a higher BMI ((R2 = 0.64). The % colony formation, HuFTF FGF2 concentration, and BMI of six HuFTF were used. MCF-10A cells were cultured as described in Methods. Data were analyzed with Linear regression (performed in PRISM).
Discussion
Visceral adiposity is significantly associated with breast cancer risk, and despite this strong association, the mechanism is unknown [Citation24,Citation25]. We previously showed visceral obesity promoted skin tumor formation, with our mechanistic finding that VAT-derived FGF2 stimulates skin epithelial cell transformation through FGFR1 [Citation19]. As obesity is associated with many different cancers, we hypothesized our mechanistic finding may be translational to other obesity associated cancers, like breast cancer. Visceral obesity, as measured by large waist circumferences and waist-to-hip ratios, is strongly correlated with pre- and post-menopausal breast cancer risk [Citation2–Citation5]. Herein, we describe a translational role for VAT-derived FGF2 in stimulating mammary epithelial cell transformation through FGFR1. These findings highlight FGF2/FGFR1 signaling as a potential link between VAT and breast cancer risk.
Previous research implicates FGF2/FGFR1 signaling in breast cancer [Citation26]. Constitutive activation of FGFR1 in normal mouse mammary epithelium induced proliferation, invasive lesions, and antiapoptotic effects [Citation27]. In breast cancer cells, FGF2 is a strong mitogen and potent antiapoptotic and induces invasiveness while subverting various chemotherapeutic agents, leading to drug resistance [Citation28–Citation30]. In addition, FGFR1 activation increases proliferation and invasion of breast cancer cell lines [Citation31,Citation32]. Clinical studies have shown that FGF2 levels in serum, nipple aspirate fluid, and tumor samples are higher in patients with cancerous breast tumors as compared with benign breast diseases/tumors [Citation33–Citation36]. In breast cancer patients, FGFR1 amplification is seen in up to 10–15% of all breast cancers and is associated with early relapse and poor survival [Citation13,Citation37]. Likewise, tumors overexpressing FGFR1 exhibited increased proliferation and decreased distant metastasis-free survival [Citation37]. The role of FGF2/FGFR1 in breast cancer onset is less clear. One study demonstrated that genetic variants in FGFR1, FGFR3, or FGFR4 had no impact on breast cancer risk, [Citation38] whereas an intronic single-nucleotide polymorphism (SNP) in the FGFR2 gene was associated with an increased risk of breast cancer, particularly estrogen receptor (ER) positive disease [Citation38]. A separate study demonstrated no significant associations with SNPs in FGF2 and breast cancer risk [Citation39]. The functional relevance of these FGF2 polymorphism for function are unknown.
Our data demonstrate that FGFR1 activation is critical for optimal VAT-stimulated MCF-10A cell transformation. These data add additional relevance to the previous findings that FGFR1 activation by inducible dimerization of the receptor induced growth in soft agar of MCF-10A cells [Citation31]. Moreover, we showed VAT FGF2 concentrations were associated with VAT transforming activity (). Collectively, these data suggest that FGF2 from VAT stimulates mammary epithelial cell neoplastic transformation through FGFR1 activation. The downstream effects of FGFR1 activation of transformation are unknown, but it would be interesting to observe a potential subtype or breast cancer signature in tumors that arise from visceral adiposity-promotion. Breast cancer is characterized into different subtypes based on expression of ER, progesterone receptor (PR), and human epidermal growth factor receptor (HER). Studies show FGFR1-overexpressing tumors are frequently ER positive, the primary subtype associated with obesity [Citation13,Citation40]. Therefore, we would hypothesize that FGF2 activation of FGFR1 would promote ER positive tumors in viscerally obese individuals.
Evaluating individual characteristics including age, gender, and ethnicity were not associated with transforming capacity of the HuFTFs. As obesity is more strongly associated with post-menopausal breast cancer, we requested samples from individuals of post-menopausal age. Menopause occurs on average in women at 51 years old, however, we were not able to confirm the menopausal status of the individual donors as two donors were 52 and 50 years old at the time of surgery. It would be interesting to investigate if menopause influences the quality of VAT, in turn affecting FGF2 levels. In our study, there was no relationship between age and the transforming capacity of the HuFTF (R2 = 0.09) (data not shown). For BMI, there is a moderate association with BMI and the transforming capacity of the HuFTF, giving an R2 value of 0.64 (). This suggests BMI might be an indicator of VAT FGF2 levels. We were not able to investigate ethnicity as a variable. Additionally, we were not afforded any information on serum metabolites or the metabolic status of each donor.
There are 22 structurally similar FGF ligands that mediate effects through activation of receptor tyrosine kinases (RTK), fibroblast growth factor receptors (FGFR) 1–4. FGFs can have affinities for more than one receptor and each receptor can bind multiple FGFs. Similarities between receptors has resulted in receptor redundancy as they can converge on key downstream signaling cascades [Citation41,Citation42]. All four FGFRs activate PLCγ/PKC, PI3K/AKT, RAS/MAPK, and STAT pathways [Citation41]. Activation of these pathways play important roles in migration, survival, differentiation, and proliferation [Citation42]. However, studies suggest the strength and specificity of each signaling cascade can vary depending on the type of FGFR and FGF [Citation41]. Additionally, pharmacologically inhibiting FGFR1 partially attenuated VAT-stimulated mammary epithelial transformation (p = 0.02) (). This partial attenuation suggests either ligands, receptors, and/or signaling cascades are influencing colony formation or the FGFR1 Ab does not have complete inhibition of FGFR1.
Furthermore, we found FGF1 and FGF18 induces colony formation, but not in the concentration-dependent manner as seen with FGF2 (). One study using MCF-10A cells revealed phenotypic distinctions in 3D growth stimulated by different RTKs, including EGFR and MET [Citation43]. Other FGFRs or RTKs activated by other ligands could be inducing signaling pathways that attribute to VAT-stimulated transformation [Citation42]. Therefore, while other RTKs could contribute to transformation, these data suggest biased agonism associated with FGF2/FGFR1 is optimal for VAT-stimulated MCF-10A transformation.
HuFTF from donor 02 had the highest level of FGF2 at 1.14 pg/mL. Independently FGF2 required at least 10 ng/mL to stimulate transformation of MCF-10A cells. This could be due to a potential difference in the FGF2 isoform in the HuFTF compared to the recombinant protein. FGF2 exists in five different isoforms that are divided into two groups low molecular weight (LMW) and high molecular weight (HMW) proteins. Studies have suggested there are distinct biological activities of LMW and HMW proteins. For example, one study showed that overexpressing LMW FGF2 enhanced bone mineral density (BMD) whereas overexpressing HMW FGF2 lowered BMD [Citation44] and another study showed LMW FGF2 suppressed hepatic fibrosis and HMW enhanced hepatic fribrosis [Citation45]. In contrast, other studies have showed FGF2 isoforms exhibit different potencies. Kole et al. showed that all FGF2 isoforms exhibited mitogenic activity in dermal fibroblasts, however, HMW isoforms were less efficient [Citation46]. Additionally, a study by Mydlo et al. showed that FGF2 derived from omental VAT demonstrated greater mitogenic and angiogenic activity than FGF2 derived from either benign and cancerous renal tissue [Citation47]. The recombinant protein used [in this study] is a LMW FGF2 (18 kDa), and the ELISA used to detect FGF2 is nonspecific regarding FGF2 isoforms. Therefore, the recombinant protein might not be representative of the most active isoform of FGF2 in the HuFTF. Additionally, HMW FGF2 could be more potent than LMW FGF2, accounting for the difference in dose of FGF2 in HuFTF and the recombinant FGF2 used. Currently, only LMW FGF2 is commercially available, and isolating FGF2 from HuFTF would provide a more accurate representation of the transformative capabilities of VAT-derived FGF2. In addition, in HuFTF, FGF2 may be synergizing with other growth factors.
FGF2 is classically considered to function in both an autocrine and paracrine manner, however, our research suggests FGF2 functions in an endocrine manner acting on distant targets. Our previous study showed an induction of serum FGF2 in HFD-fed mice compared to LFD-fed mice [Citation19]. This serum induction was depleted following lipectomy of VAT suggesting the circulating levels of FGF2 are of adipose tissue origin [Citation19]. Circulating levels of FGF2 in these animals were associated with UVB-induced squamous cell carcinomas, suggesting that FGF2 secreted from VAT influences tumor promotion at distant sites [Citation19]. In tandem, one study found FGF2 concentrations in serum increased with higher BMIs [Citation48], and another found plasma FGF2 levels of obese Chinese men were correlated with adipose tissue mass [Citation49]. Our recent study demonstrates that human serum samples with elevated FGF2 had greater efficacy in stimulating JB6 P+ cell growth in soft agar. Future studies are needed to assess circulating FGF2 concentrations in relation to visceral obesity and breast cancer risk to ascertain potential associations and a role for FGF2 as an endocrine growth factor and as a biomarker for at risk individuals.
Obesity and breast cancer are independently complex diseases with multiple factors potentially influencing their etiology. FGF can be secreted from many different tissues along with other FGFR1 ligands. For example, FGF2 is secreted from skin [Citation50], heart [Citation51], liver [Citation45], lungs [Citation52], and SAT [Citation53] and could contribute to circulating FGF2 levels. The contribution of FGF2 from VAT and other sources to mammary tumorigenesis will be determined in vivo in future investigations. Although VAT is more strongly correlated with breast cancer risk than its subcutaneous counterpart, there is an intimate and bidirectional interaction between mammary epithelium and adjacent subcutaneous mammary adipose tissue (MAT). The total absence of MAT in transgenic mice prevents non-tumorigenic mammary gland development and MAT supports and amplifies breast cancer progression [Citation54]. Dialog between MAT and mammary epithelium might persist and influence breast cancer onset as a potential source of FGF2 [Citation54,Citation55]. This exposes a limitation in our study as assessing VAT and mammary epithelial cells in our in vitro model does not evaluate whole body effects in vivo.
In summary, we demonstrate FGF2 from human VAT stimulates transformation of non-tumorigenic mammary epithelial cells. Our data suggests differences the transformative ability of human VAT is associated with FGF2 levels and that inhibiting FGFR1 attenuated this transformation. These findings highlight FGF2/FGFR1 signaling as a potential link between visceral adiposity and elevated breast cancer risk. Future studies will use in vivo mouse models to determine the tumorigenicity of transformed MCF-10A cells, the ability of HFD to promote mammary tumorigenesis, and the effect of lipectomy on mammary tumorigenesis. FGF2/FGFR1 signaling could be a therapeutic target for breast cancer prevention strategies and/or a biomarker for identifying at risk individuals.
Methods
Cell Culture: MCF-10A cells (human mammary epithelial cells) were obtained from ATCC (Manassas, VA, USA). Cells were cultured in DMEM/ Ham's F12 media supplemented with 5% horse serum (HS), 1% penicillin/streptomycin, 100 ng/mL cholera toxin, 20 ng/mL epidermal growth factor (EGF), 10 µg/mL insulin, 0.5 mg/mL hydrocortisone, 7.5% sodium bicarbonate, 15mM HEPES, and 2 mM L-Glutamine (growth media). MCF-10A cells were trypsinized with 0.05% trypsin and quenched in DMEM/ Ham's F12 media with 20% horse serum and antibiotics (resuspension media). The FGFR1 Ab was purchased from R&D Systems (Minneapolis, MN, USA #MAB765).
Human FTF: VAT was homogenized in equal volume of serum free media on ice for 30 seconds using Tissue Ruptor (Qiagen, Hilden, Germany) on medium speed. Homogenates were filtered through a hanging transwell insert 15-mm wide 0.4 um filter (Millicell, cat# MCHT06H48) positioned in 6-well plates filled with 750 μL of serum free media. Plates were incubated on a rocker for 1 hour to allow fat derived factors to diffuse into the media. After incubation, filtrates were centrifuged at 4500 rpm for 5 minutes and the supernatant was collected. Protein concentrations were quantified using BCA Assay. An aliquot of 200 μg/mL concentration of HuFTF was used for respective experiments.
Anchorage-Independent Colony Formation Soft Agar Assay: MCF-10A cells were seeded at 750 cells per well in a 24-well plate in 200 µL of DMEM/Ham's F12, 5% HS, and 0.33% agar with or without HuFTF and/or inhibitors which was overlaid onto 350 µL of DMEM/Ham's F12, 5% HS, and 0.5% agar. Soft agar plates were left at room temperature for 30 minutes before 200 µL of growth media was gently added to each well and then stored at 37°C. Every 3–4 days, the growth media was removed from each well and replenished with 200 µL of growth media. After two weeks, the colonies were fixed in 70% ethanol (EtOH) and stained with 150 µL of 0.01% crystal violet. Colonies were visually counted and used to calculate the percent of colony formation from the number of cells plated ([Colonies counted x 100] / 750 cells). The % colony formation was normalized to the untreated control to determine the increase in % of colony formation. (% Colony formation of treatment - the % colony formation of untreated control).
FGFR1 Ab Treatment: FGFR1 Ab was added directly into the top layer of the soft agar assay. MCF-10A cells were pre-incubated with the monoclonal FGFR1 Ab (2 µg/mL) in 37°C for 1.5 hours before being added to the top soft agar layer.
Statistics: Six biological and three technical replicates were used to ensure adequate power to detect a significant change in growth in soft agar. Data are presented as mean ± s.e. Unpaired t-test and one-way ANOVA for multiple comparisons were used appropriately. For all statistical tests, 0.05, 0.01, and 0.001 level of confidence, were accepted for statistical significance.
FGF2 Quantification: FGF2 concentrations in HuFTF was measured by ELISA kit according to the manufacturer's protocol using R&D Systems Quantikine ELISA kit' (Cat# DFB50). The lowest detectable FGF2 concentration was 0.625 pg/mL.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Ahn J, Schatzkin A, Lacey JV, et al. Adiposity, adult weight change, and postmenopausal breast cancer risk. Arch Intern Med. 2007;167(19):2091–2102. doi:10.1001/archinte.167.19.2091.
- Connolly BS, Barnett C, Vogt KN, et al. A meta-analysis of published literature on waist-to-hip ratio and risk of breast cancer. Nutr Cancer. 2002;44(2):127–38. doi: 10.1207/S15327914NC4402_02
- Gaudet MM, Carter BD, Patel AV, et al. Waist circumference, body mass index, and postmenopausal breast cancer incidence in the Cancer Prevention Study-II Nutrition Cohort. Cancer Causes Control. 2014 June 01;25(6):737–745. doi:10.1007/s10552-014-0376-4
- Hajian-Tilaki KO, Gholizadehpasha AR, Bozorgzadeh S, et al. Body mass index and waist circumference are predictor biomarkers of breast cancer risk in Iranian women. Med Oncol. 2011 Dec;28(4):1296–301. doi:10.1007/s12032-010-9629-6
- Huang Z, Willett W, Coldfe G, et al. Waist circumference, waist:hip ratio, and risk of breast cancer in the nurses' health study. Am J Epidemiol. 1999 04/08/1999;150(2):8.
- Harvie M, Hooper L, Howell AH. Central obesity and breast cancer risk: a systematic review. Obes Rev. 2003 Aug;4(3):157–73. doi:10.1046/j.1467-789X.2003.00108.x
- Schaffler A, Scholmerich J, Buechler C. Mechanisms of disease: adipokines and breast cancer – endocrine and paracrine mechanisms that connect adiposity and breast cancer. Nat Clin Pract Endocrinol Metab. 2007 Apr;3(4):345–54. doi:10.1038/ncpendmet0456
- Ibrahim MM. Subcutaneous and visceral adipose tissue: structural and functional differences. Obes Rev. 2010 Jan;11(1):11–8. doi:10.1111/j.1467-789X.2009.00623.x
- Kwon H, Kim D, Kim JS. Body fat distribution and the risk of incident metabolic syndrome: a longitudinal cohort study. Sci Rep. 2017 Sep 08;7(1):10955. doi:10.1038/s41598-017-09723-y
- Esser N, Legrand-Poels S, Piette J, et al. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract. 2014 Aug;105(2):141–50. doi:10.1016/j.diabres.2014.04.006
- Jung UJ, Choi MS. Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int J Mol Sci. 2014 Apr 11;15(4):6184–223. doi:10.3390/ijms15046184
- Kabat GC, Kim MY, Lee JS, et al. Metabolic obesity phenotypes and risk of breast cancer in postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2017 Sep 22;26(12):1730–1735. doi:10.1158/1055-9965.EPI-17-0495
- Elbauomy Elsheikh S, Green AR, Lambros MB, et al. FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast Cancer Res. 2007;9(2):R23. doi:10.1186/bcr1665
- Huffman DM, Augenlicht LH, Zhang X, et al. Abdominal obesity, independent from caloric intake, accounts for the development of intestinal tumors in Apc(1638N/+) female mice. Cancer Prev Res (Phila). 2013 Mar;6(3):177–87. doi:10.1158/1940-6207.CAPR-12-0414
- Lu YP, Lou YR, Bernard JJ, et al. Surgical removal of the parametrial fat pads stimulates apoptosis and inhibits UVB-induced carcinogenesis in mice fed a high-fat diet. Proc Natl Acad Sci U S A. 2012 Jun 05;109(23):9065–70. doi:10.1073/pnas.1205810109
- Wittwer JA, Robbins D, Wang F, et al. Enhancing mitochondrial respiration suppresses tumor promoter TPA-Induced PKM2 expression and cell transformation in skin epidermal JB6 cells. Cancer Prevention Research. 2011;4(9):1476–1484. doi:10.1158/1940-6207.capr-11-0028.
- Chang A, Yeung S, Thakkar A, et al. Prevention of skin carcinogenesis by the beta-blocker carvedilol. Cancer Prev Res (Phila). 2015 Jan;8(1):27–36. doi:10.1158/1940-6207.CAPR-14-0193
- Lu FJ, Tseng TH, Lee WJ, et al. Promoting neoplastic transformation of humic acid in mouse epidermal JB6 Cl41 cells. Chem Biol Interact. 2006 Sep 25;162(3):249–58. doi:10.1016/j.cbi.2006.07.007
- Chakraborty D, Benham V, Bullard B, et al. Fibroblast growth factor receptor is a mechanistic link between visceral adiposity and cancer [Original Article]. Oncogene. 2017; 36(48):6668–6679. doi:10.1038/onc.2017.278.
- Pang Y, Li W, Ma R, et al. Development of human cell models for assessing the carcinogenic potential of chemicals. Toxicol Appl Pharmacol. 2008 Nov 01;232(3):478–86. doi:10.1016/j.taap.2008.08.009
- Balmain A, Harris C. Carcinogenesis in mouse and human cells- parallels and paradoxes. Carcinogenesis. 2000;12(3):371–377. doi:10.1093/carcin/21.3.371.
- Zhang X, Ibrahimi OA, Olsen SK, et al. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006 Jun 09;281(23):15694–700. doi:10.1074/jbc.M601252200
- Ornitz DM, Xu J, Colvin JS, et al. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996 June 21, 1996;271(25):15292–15297. doi:10.1074/jbc.271.25.15292.
- Schapira D, Clar R, Wolff P, et al. Visceral obesity and breast cancer risk. Cancer. 1994;74(2):632. doi:10.1002/1097-0142(19940715)74:2%3c632::AID-CNCR2820740215%3e3.0.CO;2-T.
- Rankinen T, Kim SY, Pérusse L, et al. The prediction of abdominal visceral fat level from body composition and anthropometry: ROC analysis. Int J Obes. 1999;23(8):801–809. doi:10.1038/sj.ijo.0800929.
- Brady N, Chuntova P, Bade LK, et al. The FGF/FGFR axis as a therapeutic target in breast cancer. Expert Rev Endocrinol Metab. 2013 Jul;8(4):391–402. doi:10.1586/17446651.2013.811910
- Welm BE, Freeman KW, Chen M, et al. Inducible dimerization of FGFR1: development of a mouse model to analyze progressive transformation of the mammary gland. J Cell Biol. 2002 May 13;157(4):703–14. doi:10.1083/jcb.200107119
- Vandermoere F, El Yazidi-Belkoura I, Adriaenssens E, et al. The antiapoptotic effect of fibroblast growth factor-2 is mediated through nuclear factor-kappaB activation induced via interaction between Akt and IkappaB kinase-beta in breast cancer cells. Oncogene. 2005 Aug 18;24(35):5482–91. doi:10.1038/sj.onc.1208713
- Hu Y, Qiu Y, Yague E, et al. miRNA-205 targets VEGFA and FGF2 and regulates resistance to chemotherapeutics in breast cancer. Cell Death Dis. 2016 Jun 30;7(6):e2291. doi:10.1038/cddis.2016.194
- Lui J, Crepin M, Liu J, et al. FGF-2 and TPA induce matrix metalloproteinase-9 secretion in MCF-7 cells through PKC activation of the Ras-ERK pathway. Biochem Biophys Res Commun. 2002;293:8.
- Xian W, Pappas L, Pandya D, et al. Fibroblast growth factor receptor 1-transformed mammary epithelial cells are dependent on RSK activity for growth and survival. Cancer Res. 2009 Mar 15;69(6):2244–51. doi:10.1158/0008-5472.CAN-08-3398
- Xian W, Schwertfeger KL, Vargo-Gogola T, et al. Pleiotropic effects of FGFR1 on cell proliferation, survival, and migration in a 3D mammary epithelial cell model. J Cell Biol. 2005 Nov 21;171(4):663–73. doi:10.1083/jcb.200505098
- Rykala J, Przybylowska K, Majsterek I, et al. The −553 T/A polymorphism in the promoter region of the FGF2 gene is associated with increased breast cancer risk in Polish women. Arch Med Sci. 2015 Jun 19;11(3):619–27. doi:10.5114/aoms.2013.35996
- Rykala J, Przybylowska K, Majsterek I, et al. Angiogenesis markers quantification in breast cancer and their correlation with clinicopathological prognostic variables. Pathol Oncol Res. 2011 Dec;17(4):809–17. doi:10.1007/s12253-011-9387-6
- Sauter ER, Scott S, Hewett J, et al. Biomarkers associated with breast cancer are associated with obesity. Cancer Detect Prev. 2008;32(2):149–55. doi:10.1016/j.cdp.2008.06.002
- Hsiung R, Zhu W, Klein G, et al. High basic fibroblast growth factor levels in nipple aspirate fluid are correlated with breast cancer. Cancer J. 2002;8(4):7. doi:10.1097/00130404-200207000-00006.
- Turner N, Pearson A, Sharpe R, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010 Mar 01;70(5):2085–94. doi:10.1158/0008-5472.CAN-09-3746
- Agarwal D, Pineda S, Michailidou K, et al. FGF receptor genes and breast cancer susceptibility: results from the Breast Cancer Association Consortium. Br J Cancer. 2014 Feb 18;110(4):1088–100. doi:10.1038/bjc.2013.769
- Slattery ML, John EM, Stern MC, et al. Associations with growth factor genes (FGF1, FGF2, PDGFB, FGFR2, NRG2, EGF, ERBB2) with breast cancer risk and survival: the Breast Cancer Health Disparities Study. Breast Cancer Res Treat. 2013 Aug;140(3):587–601. doi:10.1007/s10549-013-2644-5
- Maehle B, Tretly S, Skjaerven R, et al. Premorbid body weight and its relations to primary tumour diameter in breast cancer patients; its dependence on estrogen and progesterone receptor status [Primary]. Breast Cancer Res Treat. 2001;68:10. doi:10.1023/A:1011977118921.
- Ornitz DM, Itoh N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol. 2015 May-Jun;4(3):215–66. doi:10.1002/wdev.176
- Harbinski F, Craig VJ, Sanghavi S, et al. Rescue screens with secreted proteins reveal compensatory potential of receptor tyrosine kinases in driving cancer growth. Cancer Discov. 2012 Oct;2(10):948–59. doi:10.1158/2159-8290.CD-12-0237
- Shaw KR, Wrobel CN, Brugge JS. Use of three-dimensional basement membrane cultures to model oncogene-induced changes in mammary epithelial morphogenesis. J Mammary Gland Biol Neoplasia. 2004 Oct;9(4):297–310. doi:10.1007/s10911-004-1402-z
- Homer-Bouthiette C, Doetschman T, Xiao L, et al. Knockout of nuclear high molecular weight FGF2 isoforms in mice modulates bone and phosphate homeostasis. J Biol Chem. 2014 Dec 26;289(52):36303–14. doi:10.1074/jbc.M114.619569
- Pan RL, Xiang LX, Wang P, et al. Low-molecular-weight fibroblast growth factor 2 attenuates hepatic fibrosis by epigenetic down-regulation of Delta-like1. Hepatology. 2015 May;61(5):1708–20. doi:10.1002/hep.27649
- Kole D, Grella A, Dolivo D, et al. High molecular weight FGF2 isoforms demonstrate canonical receptor-mediated activity and support human embryonic stem cell self-renewal. Stem Cell Res. 2017 May;21:106–116. doi:10.1016/j.scr.2017.04.006
- Mydlo JH, Kral JG, Macchia RJ. Preliminary results comparing the recovery of basic fibroblast growth factor (FGF-2) in adipose tissue and benign and malignant renal tissue. J Urol. 1998 Jun;159(6):2159–2163. doi:10.1016/S0022-5347(01)63298-1.
- Kuhn MC, Willenberg HS, Schott M, et al. Adipocyte-secreted factors increase osteoblast proliferation and the OPG/RANKL ratio to influence osteoclast formation. Mol Cell Endocrinol. 2012 Feb 26;349(2):180–8. doi:10.1016/j.mce.2011.10.018
- Hao RH, Guo Y, Dong SS, et al. Associations of Plasma FGF2 levels and polymorphisms in the FGF2 Gene with obesity phenotypes in Han Chinese Population. Sci Rep. 2016 Feb 16;6:19868. doi:10.1038/srep19868
- Grazul-Bilska A, Reynolds L, Bilski J, et al. Effects of basic fibroblast growth factor (FGF-2) on proliferation of human skin fibroblasts in type II diabetes mellitus. Exp Clin Endocrinol Diabetes. 2002 2002;110(4):176–181. doi:10.1055/s-2002-32149.
- Itoh N, Ohta H. Pathophysiological roles of FGF signaling in the heart. Front Physiol. 2013 Sep 06;4:247. doi:10.3389/fphys.2013.00247
- Matsui R, Brody J, Yu Q. FGF-2 induces surfactant protein gene expression in foetal rat lung epithelial cells through a MAPK-independent pathway. Elsevier. 1999;11(3):221–228.
- Gabrielsson B, Johansson J, Jennische E, et al. Depot-specific expression of fibroblast growth factors in human adipose tissue. Obes Res. 2002;10(7):608–17. doi:10.1038/oby.2002.83.
- Wang YY, Lehuede C, Laurent V, et al. Adipose tissue and breast epithelial cells: a dangerous dynamic duo in breast cancer. Cancer Lett. 2012 Nov 28;324(2):142–51. doi:10.1016/j.canlet.2012.05.019
- Zhao M, Sachs PC, Wang X, et al. Mesenchymal stem cells in mammary adipose tissue stimulate progression of breast cancer resembling the basal-type. Cancer Biol Ther. 2012 Jul;13(9):782–92. doi:10.4161/cbt.20561