
ABSTRACT
In obesity, high levels of saturated fatty acids (SFAs) contribute to adipose tissue inflammation and dysfunction. Obesity-induced macrophage infiltration leads to insulin resistance, but the adipocyte itself may play a role in generating the inflammatory milieu. Given our recent findings of the role of TLR4 in myeloid biasing in obesity, we next investigated the role of TLR4 in adipocyte generated inflammatory responses to SFAs and lipopolysaccharides. We used WT and Tlr4−/- ear mesenchymal stem cell derived adipocytes (EMSC Ad) and bone marrow dendritic cells (BMDCs) to evaluate cell specific responses. Our work demonstrates a role for TLR4 in adipocyte- immune cell crosstalk and that SFA derived metabolites from adipocytes may induce proinflammatory stimulation of immune cells in a TLR4 independent manner.
Introduction
Obesity is associated with a wide variety of comorbidities, including type II diabetes, cardiovascular disease (CVD), and cancer [Citation1–6]. Obese adipose tissue is a site and source for inflammation [Citation7–10] that triggers tissue dysfunction such as impaired adipogenesis [Citation11–13] and excess free fatty acid (FFA) release [Citation14–17]. Adipose dysfunction is closely linked to the recruitment and activation of inflammatory leukocytes [Citation18–21], especially in visceral adipose tissue. In obesity, there is a profound increase in activated CD11 c+ pro-inflammatory adipose tissue macrophages (ATMs) within the white adipose tissue (WAT) [Citation22]. ATMs are a prominent source of inflammatory cytokines, such as IL1β, IL6 and tumour necrosis factor α (TNFα) [Citation21,Citation23,Citation24] and chemokines such as MCP1 [Citation25,Citation26] that are important contributors to insulin resistance and overall metabolic syndrome in obesity [Citation7,Citation27].
Among the toll-like receptor (TLR) family members, TLR4 has been recognized as particularly important in terms of adipose tissue inflammation [Citation21,Citation28]. Cytokine profiles in obesity have been observed to be similar to lipopolysaccharide (LPS)-induced TLR4 pathways. TLR4 has also been demonstrated to be involved in promoting alternative macrophage activation, with TLR4 deficient animals on a high fat diet (HFD) displaying a lower incidence of inflammatory CD11 c+ macrophages [Citation29]. Previous studies have also indicated a role for TLR4 in the development of adipose tissue fibrosis and insulin resistance in obesity [Citation30,Citation31]. Although TLR4 is typically thought of as a leukocyte receptor, it is also expressed on many non-immune cells, including adipocytes, hepatocytes, and muscle cells [Citation32–34]. Thus, TLR4 exhibits differential and cell-specific responses to a variety of stimulatory factors. The complex cellular makeup of adipose tissue raises an intriguing question regarding the role of TLR4 in promoting obesity-induced inflammation and adipose tissue dysfunction. Are the metabolic effects of TLR4 due to its activation in classical insulin-sensitive tissues or in immune cells? Or simultaneously in both?
Several experimental models, including the use of bone marrow transplants (BMTs) to generate TLR4 chimeras where TLR4 is either present or absent from the whole body or from the BM-derived cells, have been developed to explore the cell-specific effects of TLR4 in obesity [Citation21,Citation29,Citation31], but these studies have generated conflicting results. Saberi et al found that an absence of functional TLR4 in BM-derived cells conferred protection against HFD-induced insulin resistance [Citation31]. On the other hand, Orr et al and Griffin et al found that, while TLR4 on BM-derived cells controlled the inflammatory phenotype of macrophages infiltrating the adipose tissue, it was not sufficient to modulate insulin sensitivity in the body, and non-BM-derived TLR4 played a larger role in regulating fat and liver mass [Citation21,Citation29]. Given such discrepancies, further investigations are required to understand the cell-specific roles of TLR4 in adipose tissue.
Due to their close structural similarity to the LPS binding moiety, it is hypothesized that FFAs can bind and stimulate TLR4. Thus, elevated plasma FFAs in obesity have the potential to activate TLR4 [Citation14]. Shi et al found that Tlr4−/- mice are protected from the ability of elevated plasma FFAs to suppress insulin levels and reduce changes in glucose metabolism mediated by insulin [Citation35]. Along with their role in promoting insulin resistance, FFAs specifically, saturated fatty acids (SFAs), activated inflammatory transcription factors NF-κB and cyclooxygenase-2 (COX-2) expression in 293 T cells and RAW264.7 cells [Citation36]. The stimulatory effects of palmitic and stearic acid on MCP-1 secretion were observed in 3T3L1 adipocytes [Citation37]. Consistent with these findings, previous studies in our laboratory have also shown that TLR4 is necessary for the generation of activated bone marrow derived macrophages in response to SFA [Citation21], suggesting that the interaction between FFAs and TLR4 may play a role in macrophage polarization. In contrast to these results, other studies refute the idea that FFAs act as a direct binder and stimulator of TLR4. Studies by Lancaster et al suggest that palmitate, a SFA, is not a direct agonist of TLR4, instead proposing that palmitate-induced inflammation is dependent on TLR4 through its ability to alter cellular metabolism, gene expression, lipid metabolic pathways, and membrane lipid composition [Citation38]. Despite the structural similarity of FFAs to LPS, FFA activation of TLR4 has actually been shown to take many hours in contrast to LPS activation of TLR4, which happens on the time-scale of minutes [Citation38–41].
Palmitate-induced metabolic reprogramming in innate immune cells modulates inflammatory responses and contributes to disease progression [Citation42]. It is critical to investigate the cell type-specific responses of TLR4 to ligands such as palmitate to better understand TLR4’s role in SFA-mediated inflammation during obesity. In this study, we investigated the adipocyte-specific effects of TLR4 through a primary culture model using ear mesenchymal stem cells (EMSCs) that are multipotent and give rise to osteocytes, chondrocytes, and adipocytes. To determine the potential role of TLR4 in inhibiting proper adipogenesis during obesity, we stimulated adipocytes derived from WT and Tlr4−/- male mice EMSCs with LPS or palmitate during their differentiation in vitro and assessed adipogenic and inflammatory markers. To determine the potential effects of TLR4 in the crosstalk between adipocytes and immune cells, conditioned media from LPS or palmitate treated EMSC-derived adipocytes (EMSC Ad) was further used to stimulate bone marrow dendritic cells (BMDCs) derived from WT and Tlr4−/- male mice in culture. Our in vitro studies with WT and Tlr4−/- BMDCs and EMSC Ad suggest a role for TLR4 activation on both macrophages and adipocytes.
Materials and methods
Animal models
C57Bl/6 J (WT) and Tlr4−/- (B6.B10ScN-Tlr4lps-del/JthJ; 007227) male mice on C57Bl/6 J background were purchased from Jackson Laboratories at 5 weeks of age. Animals were housed in a specific pathogen-free facility with a 12-h light/12-h dark cycle and given free access to food and water. All mice were fed ad libitum a diet consisting of 4.5% fat (5001; LabDiet). Animal protocols were in compliance with the Institute of Laboratory Animal Research Guide for the Care and Use of Laboratory Animals and approved by the University Committee on Use and Care of Animals at the University of Michigan (animal welfare assurance number A3114-01).
EMSC isolation, differentiation and treatments
The primary cultures of rodent EMSC were prepared from ears of 7–8 week old wild-type and Tlr4−/- male mice [Citation43,Citation44]. Excised ears were washed with HBSS (Invitrogen #14,025-092) plus Primocin antibiotic (Invivogen #ant-pm-1). Minced ears were subjected to collagenase (2 mg/ml Collagenase I; Worthington Biochemical Corporation #LS004196) digestion for ½ h at 37°C. Following filtration, cells were collected via centrifugation (500 g; 7 mins at 4°C) and thereafter incubated with Red Blood Cell (RBC) Lysing Buffer for 5 mins (Sigma #R7757). Following RBC lysis, cells were resuspended in EMSC maintenance medium: Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12; Invitrogen #11,330-032) supplemented with 15% Foetal Bovine Serum (FBS; Invitrogen #10,082-147) and 1 ng/ml Fibroblast Growth Factor (FGF; Peprotech) and Primocin antibiotic. Mesenchymal stem cells were plated in maintenance medium at a density of 0.5 × 106 cells/well.
Cultured EMSC cells were differentiated into adipocytes with EMSC differentiation media I: DMEM/F12 supplemented with 15% FBS, 5 μg/ml Insulin (Sigma # I-5523), 1 μM Dexamethasone (Sigma # D-1756), 0.5 μM Methyl-isobutylxanthine (Sigma # I-5879), 5 μM Troglitazone (Cayman), and Penicillin Streptomycin Glutamine (Gibco # 10,378). On the third day, they were refed with EMSC differentiation media II: DMEM/F12 supplemented with 15% FBS, 5 μg/ml Insulin, 5 μM Troglitazone and Penicillin Streptomycin Glutamine. Cells were cultured for an additional 3 days, and then refed with maintenance medium and cultured for another 3–5 days.
Treatments with 10 ng/ml LPS or 200 μM of Palmitate were performed in differentiating EMSCs in differentiation media I and was replenished upon changing the media to the EMSC differentiation media II. EMSC Ad experimental groups consisted of WT EMSC Ad differentiated control, WT EMSC Ad + LPS, WT EMSC Ad + Palmitate, Tlr4−/- EMSC Ad differentiated control, Tlr4−/- EMSC Ad + LPS and Tlr4−/- EMSC Ad + Palmitate. At the end of differentiation and experimental treatment, conditioned EMSC Ad media was removed and stored at −20°C for further evaluations. Cells were then either used for Oil Red O Staining or for RNA isolation.
Preparation of fatty acids
Palmitic acid (Sigma, #P0500) was prepared in isopropanol at a stock concentration of 50 mM and then complexed with 10% BSA (endotoxin free, fatty acid free; Sigma #A8806) in isopropanol to make up 5 mM. Fatty acid free BSA was further used as control in experiments to rule out endotoxin contamination of BSA as the source of palmitic acid’s effects as reported earlier in some cases [Citation45,Citation46].
Oil Red O Staining and quantification
Oil Red O Stock Solution (ORO; 0.6%) was prepared in isopropanol. Differentiated WT and Tlr4−/- EMSC Ad were fixed in 10% Buffered Neutral Formalin (VWR, #89,370-094) for 1 h at RT. After fixation, cells were washed 2X with milliQ water and 1X with 70% ethanol. Cells were stained with ORO Working Solution (1.5 parts of ORO stock to 1-part distilled water, filtered) for 15 mins at RT. Cells were then washed 2X with 70% ethanol and 1X with water. Cells were imaged for ORO staining using brightfield imaging. To measure lipid accumulation and the extent and success of adipogenesis, ORO was quantified. Isopropanol was added to stained cells and incubated for 5 mins at RT to extract ORO. ORO absorbance was measured at 520 nm on an Epoch spectrophotometer and values were plotted.
Primary mouse BMDCs and treatment
Bone marrow cells were isolated from 7–8 week old WT and Tlr4−/- male mice femur and tibia aseptically after euthanization and cultured as described previously [Citation47]. Cells were plated at a density of ~ 1 × 106 cells per well in 12 well plates. Cells were considered to be fully differentiated to BMDCs after 6 days of incubation in DMEM (Invitrogen) containing 10% FBS (Invitrogen), penicillin/streptomycin, and GM-CSF (10 ng/ml; PeproTech). On Day 6, the media was replaced with conditioned EMSC derived adipocyte media (diluted 1:1 with BMDC media without GM-CSF) and incubated for 24 h. For both the WT and the Tlr4−/- BMDCs, 6 experimental groups were created with conditioned EMSC Ad media. WT BMDC stimulated with WT EMSC Ad Vehicle/Control Conditioned Media (WT EMSC Ad Veh → WT DC), Tlr4−/- BMDC stimulated with Tlr4−/- EMSC Ad Vehicle/Control Conditioned Media (Tlr4−/- EMSC Ad Veh → Tlr4−/- DC), WT BMDC stimulated with Tlr4−/- EMSC Ad treated with LPS (Tlr4−/- EMSC Ad LPS → WT DC), Tlr4−/- BMDC stimulated with WT EMSC Ad treated with LPS (WT EMSC Ad LPS → Tlr4−/- DC), WT BMDC stimulated with Tlr4−/- EMSC Ad treated with Palmitate (Tlr4−/- EMSC Ad Palm → WT DC), and Tlr4−/- BMDC stimulated with WT EMSC Ad treated with Palmitate (WT EMSC Ad Palm → Tlr4−/- DC). After 24 h of experimental treatment, media was removed from the BMDCs and stored at −80°C. BMDC cells were processed for RNA isolation.
ELISA
ELISA was performed for cytokine determination of IL6, IL1β and MCP1 levels with EMSC Ad and BMDC media after respective treatments. Testing was performed by the Cancer Centre Immunology core at the University of Michigan.
Real-time PCR
RNA was extracted from EMSC Ad and BMDC cells using RLT Buffer plus β-mercaptoethanol (Life Technologies) and cDNA was generated using a high capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). SYBR Green PCR Master Mix (Applied Biosystems) and the StepOnePlus system (Applied Biosystems) were used for real-time quantitative PCR. Gapdh expression was used as an internal control for data normalization. Primers used in the studies are provided in supplementary table 1.
Statistical analysis
All values are reported as mean ± SEM unless otherwise stated. Statistical analyses were performed in Prism (GraphPad). One-way ANOVA was performed using Brown-Forsythe test and post hoc analysis with the unpaired two-tailed Student’s t-test. p < 0.05 was considered as statistically significant.
Results
WT and Tlr4−/- EMSC differentiation into adipocytes
EMCSs derived from the outer ear of adult mice are multipotent stem cells and have the capacity to differentiate into osteocytes, chondrocytes, and adipocytes. The facile differentiation of EMSCs into adipocytes has previously been demonstrated [Citation43,Citation48], making them a good model system for our studies. The phenotype of the differentiated EMSCs with the adipogenic medium resembled the mature adipocyte with the presence of lipid accumulation, that is lacking in osteocytes and chondrocytes as confirmed in previous studies [Citation43,Citation44]. We characterized any intrinsic differences between the differentiation of WT and Tlr4−/- EMSCs into adipocytes. Brightfield microscope images showed no obvious difference between differentiated WT and Tlr4−/- EMSC Ad (), left panels). Cells began to adapt a more rounded morphology from their original spindle-like shape, and the formation of initial lipid droplets could be observed. ORO staining was used to evaluate adipogenesis in differentiated cells. Imaging showed expanded and rounded adipocyte cells with abundant numbers of lipid droplets (), left panels). No obvious differences in lipid content or cellular morphology could be distinguished between differentiated WT and Tlr4−/- EMSC Ad () left panel and 1B).
Figure 1. Adipogenesis in EMSC derived adipocytes. (a) Bright field images of WT EMSC derived adipocytes – TOP ROW, left- Differentiated; middle- LPS treatment (10 ng/ml); right- Palmitate treatment (200 μM). Bright field images of Tlr4−/- EMSC derived adipocytes- BOTTOM ROW, left- Differentiated; middle- LPS treatment (10 ng/ml); right- Palmitate treatment (200 μM). (b) Graph depicting ORO absorbance in WT (blue bar) and Tlr4−/- (red bar) EMSC Ad treated with LPS and palmitate. N = 6 per condition; Experiments were performed in duplicates and data is representative of three independent experiments. One-way ANOVA and Student’s t-test was performed for (B). Statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; error bars are SEM. Comparisons of WT Diff vs WT LPS are shown as #p < 0.05, WT diff vs WT Palmitate as p = 0.0763, Tlr4−/-Diff vs Tlr4−/- Palmitate are shown as $$p < 0.01. Diff = Differentiated; Ad = adipocyte
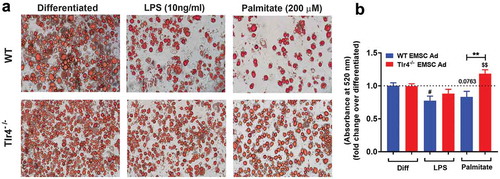
LPS and saturated fatty acids fail to inhibit EMSC adipogenesis in Tlr4−/- cells
Although some prior studies have suggested a role for TLR4 in promoting FFA-induced inflammation during obesity, many of the findings in recent literature are contradictory [Citation29,Citation31]. Previous investigations [Citation36,Citation49,Citation50] and past studies in our lab demonstrate the ability of SFAs to drive myelopoiesis in a TLR4-dependent manner [Citation21]. Non-haematopoietic TLR4 has been implicated in mediating glucose and insulin intolerance [Citation21,Citation29], and since adipose tissue dysfunction is known to be involved in metabolic syndrome [Citation2,Citation10,Citation13,Citation51], we sought to assess if treatment of EMSCs during differentiation to adipocytes with LPS or palmitate would disrupt normal adipogenesis. Consistent with previous literature and our hypothesis, the adipogenesis of WT EMSC Ad stimulated with LPS appeared to be inhibited as shown by the reduction of adipocytes and lipid droplets in comparison to untreated control. LPS significantly inhibited adipocyte differentiation, as demonstrated by the changes in morphological differentiation (), middle and left panels and 1B). However, the extent of inhibition by LPS was not significantly evident in the Tlr4−/- EMSC Ad as compared to control and WT and when assessed for relative lipid content by ORO (,)). Palmitate stimulation resulted in decreased relative lipid content by ORO absorbance and adipogenesis in WT EMSC Ad compared to Tlr4−/- EMSC Ad (,)). In contrast, palmitate stimulation increased lipid content in Tlr4−/- EMSC Ad (**p < 0.01).
Adipogenic markers were next assessed to investigate the effects of LPS and palmitate on WT and Tlr4−/- EMSCs differentiation. Adipogenic markers – Pparγ, Cebpα, and Pgc1α were upregulated significantly in Tlr4−/- differentiated adipocytes compared to WT () suggesting a possible role for Tlr4 in limiting adipogenesis in WT EMSC Ad. As observed earlier with ORO (,)), inhibition of adipogenesis by LPS and palmitate treatment led to decreased expression of Pparγ, Cebpα, and Pgc1α expression in WT EMSC Ad compared to WT differentiated control (). Pparγ, Cebpα, and Pgc1α expression were not significantly affected in Tlr4−/- EMSCs treated with LPS (). However, palmitate treatment led to a significant upregulation in the expression of the adipogenic marker Cebpα ()) as observed by increased ORO absorbance in Tlr4−/- EMSC Ad ()).
Figure 2. Assessment of adipogenic and inflammatory markers in WT and Tlr4−/- EMSC derived adipocytes. Relative gene expression of adipogenic markers – (a) Pparγ (b) Cebpα (c) Pgc1α. Relative gene expression of inflammatory markers – (d) Il1β (e) Il6 (f) Mcp1. ELISA estimation of (g) IL1β (h) IL6 (i) MCP1. Graph depicts WT (grey bar) and Tlr4−/- (black bar) EMSC Ad treated with LPS and palmitate. N = 5–6 per condition; Experiments were performed in duplicates and data is representative of three independent experiments. One-way ANOVA and Student’s t-test was performed for (a-i). Statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; error bars are SEM. Comparisons of WT Differentiated control vs Tlr4−/- Differentiated control are shown as #p < 0.05. Ad = adipocyte
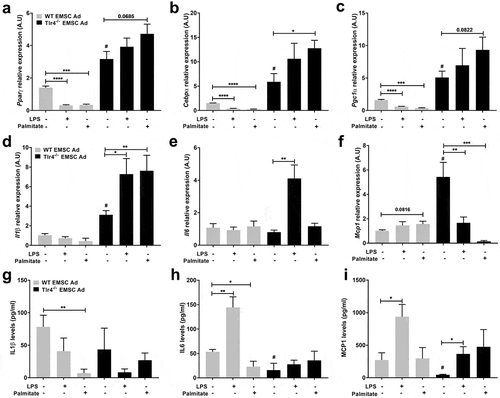
These results indicate that in WT adipocytes derived from EMSCs, both LPS and palmitate disrupt functional adipogenesis. In the absence of TLR4 however, LPS and palmitate fail to inhibit adipogenesis, suggesting a TLR4 dependent role in the impairment of adipogenesis.
Tlr4 deficiency failed to inhibit inflammatory gene expression in Tlr4−/- EMSC Ad
We next investigated proinflammatory gene expression as a possible cause for adipogenesis inhibition in WT EMSC Ad. Il1β, Il6 and Mcp1 gene expressions were evaluated in differentiated EMSCs. Interestingly, Il1β, Il6 and Mcp1 genes were not significantly affected by LPS or palmitate in WT EMSC Ad )). Contrastingly, in Tlr4−/- EMSC Ad treated with LPS, Il1β and Il6 were upregulated, while Mcp1 gene expression decreased. However, in Tlr4−/- EMSC Ad, with palmitate treatment, Mcp1 expression was significantly downregulated, while Il1β was upregulated (). Palmitate treatment did not show any significant changes in Il6 expression in Tlr4−/- EMSCs ()). ELISA was also performed for inflammatory markers in media obtained from these treatment conditions given that the timings of the treatments may differentially influence protein and gene expression (). While IL6 and MCP1 cytokine levels were lower in media from untreated WT EMSC Ad, LPS treatment led to a significant increase (,)). On the other hand, in Tlr4−/- EMSC Ad, IL6 and IL1β levels were not significantly affected even with LPS and palmitate treatments (,)). MCP1 levels increased with LPS treatment but still remained lower than WT EMSC Ad ()).
These results suggest that adipocytes may not be the most responsive to TLR4 binding ligands compared to macrophages. Also, there might exist a TLR4 independent mechanism to upregulate inflammatory genes in Tlr4−/- adipocytes.
TLR4 activation is not required in BMDCs to respond to adipocyte factors after palmitate exposure Treating cultured bone-marrow-derived macrophages or dendritic cells with SFAs recapitulates many features of macrophage polarization that are observed in the ATMs of mice consuming diets high in saturated fat, which implies a role for adipocyte-macrophage crosstalk [Citation52,Citation53]. We sought to investigate the potential role of TLR4 in this inflammatory crosstalk and hypothesized that conditioned culture media from LPS or palmitate treated WT or Tlr4−/- EMSC Ad might affect the in vitro polarization of BMDCs from WT or Tlr4−/- bone marrow cells. We therefore investigated the gene expression of inflammatory markers to assess this inflammatory response. As controls, WT EMSC Ad media was placed on WT BMDCs, and Tlr4−/- EMSC Ad media was placed on Tlr4−/- BMDCs (WT EMSC Ad→WTDC, Tlr4−/- EMSC Ad→Tlr4−/- DC), ().
Figure 3. Assessment of inflammatory markers in WT and Tlr4−/- BMDCs treated with EMSC derived adipocyte conditioned media. Relative gene expression of inflammatory markers – (a) Il1β (b) Il6 (c) Mcp1. ELISA estimation of (d) IL1β (e) IL6 (f) MCP1 in BMDC (DC) after treatment with EMSC derived adipocyte (ad) conditioned media. The experimental groups are: WT BMDC stimulated with WT EMSC Ad Vehicle/Control Conditioned Media (WT EMSC Ad Veh → WTDC), Tlr4−/- BMDC stimulated with Tlr4−/- EMSC Ad Vehicle/Control Conditioned Media (Tlr4−/- EMSC Ad Veh → Tlr4−/- DC), WT BMDC stimulated with Tlr4−/- EMSC Ad treated with LPS (Tlr4−/- EMSC Ad LPS → WTDC), Tlr4−/- BMDC stimulated with WT EMSC Ad treated with LPS (WT EMSC Ad LPS → Tlr4−/- DC), WT BMDC stimulated with Tlr4−/- EMSC Ad treated with Palmitate (Tlr4−/- EMSC Ad Palm → WTDC), and Tlr4−/- BMDC stimulated with WT EMSC Ad treated with Palmitate (WT EMSC Ad Palm → Tlr4−/- DC). N = 5–6 per condition; Experiments were performed in duplicates and data is representative of 3–4 independent experiments. One-way ANOVA and Student’s t-test was performed for (a-f). Statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; error bars are SEM. Ad = adipocyte; DC = dendritic cells, Veh = Vehicle, Palm = Palmitate
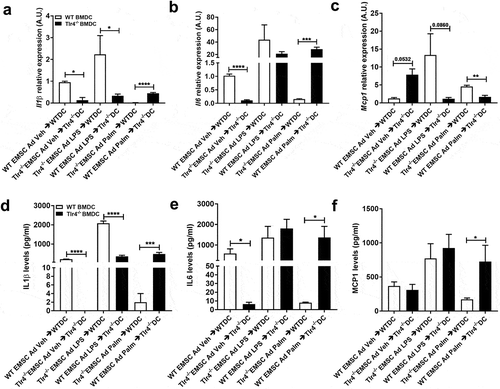
LPS conditioned media from WT adipocytes failed to upregulate Il1β and Mcp1 gene expression in Tlr4−/- BMDCs (WT EMSC Ad LPS → Tlr4−/- DC) compared to LPS conditioned media from Tlr4−/- adipocytes added to WT BMDCs (Tlr4−/- EMSC Ad LPS → WT DC) (–c)). The same treatment did not affect Il6 expression significantly ()). This suggests a vital requirement for TLR4 activation in BMDCs more than in adipocytes to promote an inflammatory response. These results were also confirmed from cytokine levels in the media (–f)).
Opposite effects were observed in Tlr4−/- BMDCs upon exposure to palmitate conditioned media from WT adipocytes. Il1β and Il6 gene expression were upregulated, while Mcp1 expression was downregulated in Tlr4−/- BMDCs compared to palmitate conditioned media from Tlr4−/- adipocytes exposed to WT BMDCs (–c)). Protein levels of these cytokines showed similar results (–f)). These results suggest a TLR4 independent mechanism in BMDCs to activate the inflammatory response in the case of SFAs. It is quite possible that Tlr4−/- BMDCs were able to respond to metabolic products and adipokines produced by WT adipocytes exposed to palmitate and induce proinflammatory cytokines.
Discussion
The synergistic interaction between adipocytes and macrophages is critical in maintaining obesity-induced adipose tissue inflammation and secondary insulin resistance. However, adipocytes and immune cells may exhibit cell-specific contributions. The aim of this study was to delineate cell type intrinsic differences in adipogenic, or inflammatory responses associated with TLR4. Our results show that the signals arising from the adipocyte compartment can influence the inflammatory status of BMDCs. Media from adipocytes exposed to LPS or palmitate promoted a distinct inflammatory response in the BMDCs.
The mechanism through which proinflammatory factors affect adipogenesis is speculated to be largely through the direct activation of NF-κB pathway, which in turn reduces PPARγ activity during the adipocyte differentiation process [Citation54,Citation55]. The inhibitory effects of inflammatory inducers on the adipocyte differentiation process have been studied extensively [Citation54–56]. The earliest [Citation57] and more recent evidence linking TLR4 to lipid-induced inflammation is through LPS [Citation31,Citation58,Citation59]. In obesity and metabolic syndrome, adipose tissue often displays impaired adipogenesis, which contributes to inflammation and metabolic dysfunction [Citation11–13]. In our approach, we mimicked an obese or inflammatory environment by performing TLR4 stimulation with LPS or palmitate in WT EMSC Ad throughout the differentiation process, and this attenuated adipogenesis as evidenced by the reduced expression of adipogenic genes. However, although WT EMSC Ad treated with LPS showed decreased lipid content by ORO absorbance compared to WT Control EMSCs, there were no significant differences between LPS treated WT and Tlr4−/- EMSC Ad in lipid content. LPS is an endotoxin, that may not necessarily increase lipid content in adipocytes but serves as a ligand for TLR4 induced inflammatory pathways [Citation32] that may eventually inhibit adipocyte differentiation. It could be speculated that only adipocyte differentiation was affected directly by TLR4 activation or through lipid metabolism which may be in contrast to some other studies due to differences in LPS treatment periods [Citation60]. However, the lack of cell count after the differentiation process might be a limitation in our study.
A major source of chronic inflammation in obesity is adipocyte dysfunction, which is associated with the interaction between adipocyte hypertrophy and macrophage infiltration of M1 polarized ATMs. In addition to the paracrine loop between adipocytes and macrophages, the intracellular metabolism of FFAs has also been found to control ATM function [Citation61]. While experimental evidence supports an important role for TLR4 in adipose inflammation [Citation31,Citation59], this is thought to directly involve TLR4 by SFAs for macrophage polarization and differentiation but several lines of evidence also argue against SFAs being direct TLR4 agonists [Citation38]. A previous study showed that 3T3L1 cells treated with 100 μM palmitate led to lipid accumulation but did not inhibit differentiation [Citation62]. In our study, palmitate stimulation with 200 μM decreased relative lipid content and inhibited adipogenesis in WT EMSC Ad but failed to induce impairment in Tlr4−/- EMSC Ad suggesting a suppression of lipid homoeostasis in mature adipocytes. In Tlr4−/- EMSC Ad, palmitate may be more readily incorporated into differentiating adipocytes, leading to heightened lipid content and enhanced adipogenic gene expression. However, in WT EMSC Ad, excess palmitate incorporation might not directly trigger inflammatory cytokine levels but might lead to increased apoptosis [Citation63] and de novo synthesis of ceramides [Citation64] thereby hampering efficient adipocyte differentiation. This suggests an indirect TLR4-dependent and independent role for palmitate in adipocytes. These findings are consistent with previous studies that have demonstrated a TLR4-dependent effect of FFA-induced inflammation [Citation21,Citation49,Citation65], and in contrast to those that have suggested that TLR4 does not play a direct role in FFA-induced inflammation [Citation38].
TLR2 has also been shown to recognize and respond to LPS, indicating that TLR2 and TLR4 may work synergistically to respond to inflammatory signals from the environment [Citation33,Citation65]. Further studies are required to address this question in adipocytes. A limitation of our study is that while we used fatty acid free BSA in order to rule out endotoxin contamination of BSA, this may still be an issue effecting results [Citation45,Citation46]. Additionally, other control groups might be required to further study the complexity of TLR4 signalling including shorter treatment times with LPS and palmitate, treatment pre- and post differentiation, treatment with unsaturated fatty acids and further combinations of WT and Tlr4−/- BMDCs and adipocytes.
Studies until now have relied on inflammatory changes in macrophages to modulate adipocyte function. However, adipocyte related dysfunction itself potentially influences macrophage phenotypes. With our evaluations of continuously treating adipocytes with TLR4 ligands during adipocyte differentiation, we tried to mimic the maintenance of an obesogenic environment. We hypothesized that the adipocyte micro‐environment, might be important regulators in shaping macrophage functions and phenotype. Palmitate induces lipotoxicity with the overproduction of reactive oxygen species (ROS) and when converted to palmitoyl-CoA also serves as a precursor for ceramides generation, which further enhance ROS production [Citation42]. Previous work from our lab showed that palmitic acid stimulated WT lineage negative bone marrow cells but not Tlr4−/- bone marrow cells when directly added to the cells [Citation21]. However, in the current study, we observed that conditioned media from palmitate treated WT adipocytes was able to stimulate inflammatory changes in Tlr4−/- BMDCs suggesting the contribution of adipocyte palmitate derived metabolites in the modulation of macrophage phenotype. A limitation is that although the conditioned media from the adipocytes was diluted prior to treatment of BMDCs, there might still be the presence of residual LPS or palmitate in our experiments. However, our data suggests that adipocyte associated cytokines and metabolites could alter the TLR4 response in a cell-specific manner either through a TLR4-dependent or independent pathway. In obesity, the level of circulating SFAs, particularly palmitate is elevated and is responsible for promoting tissue dysfunction and our studies suggest that the crosstalk between macrophages and surrounding cells are critical for this dysfunction.
Inflammatory cytokines may promote dysregulation of fatty acid metabolism in WAT. IL6 might interfere with adipocyte function by enhancing mesenchymal stem cell proliferation, maintaining the cells in an undifferentiated state and inhibiting adipogenesis [Citation66]. Additionally, IL6 can directly affect lipid metabolism and activate pathways to promote increased energy turnover. In humans, IL6 stimulates lipolysis, increases FFA concentrations and upregulates whole-body fat oxidation [Citation57,Citation67]. It is known that in obesity, adipose tissue represents a major source of increased circulating IL6 [Citation68], but the relevant adipose-tissue cell type is unclear because Il6 mRNA is expressed by adipocytes, ATMs, and other adipose-tissue cell types [Citation69]. Our observations highlight the complexity in determining cell intrinsic responses since the sources of inflammatory cytokines such as IL6 and IL1β are unclear. We observed varied expression of IL6 protein and mRNA that might be attributed to longer treatment times for LPS and palmitate. Therefore, a limitation of our study was the lack of assessment of cytokines at shorter time frames during the differentiation process making it challenging to interpret the protein and gene expression levels. A recent study showed that adipocyte IL6 potently promotes ATM accumulation in the absence of major changes in glucose or insulin tolerance. In contrast, myeloid cell IL6 suppressed M1 macrophage polarization, reduced ATM accumulation, and improved tolerance to both glucose and insulin [Citation70]. These studies and ours suggest the importance of assessing both cytokine protein and mRNA levels to understand the inflammatory response.
Our data demonstrates that adipose tissue inflammation is far more complex than simply a function of ATM accumulation. Overall, our studies demonstrate that TLR4 plays an important role in LPS and SFA-mediated adipocyte dysfunction through impairment of adipogenesis. Our studies show that adipocyte TLR4 might not elicit a potent inflammatory response to LPS or palmitate, but adipocyte metabolism associated mediators might be involved in the adipocyte-immune cell inflammatory cross-talk in a TLR4-dependent and -independent manner. Excess circulating palmitate is a potential dietary trigger resulting in the induction of chronic inflammation, such as that seen in type 2 diabetes. Further investigations are required to decipher the pathological role of palmitate in inflammatory associated diseases as palmitate uptake may control cellular lipid homoeostasis in physiological conditions. Thus, given the intricacy of adipose tissue inflammatory responses, this study emphasizes the need to delineate and clarify the role of various cell types and their interactions in obesity and associated metabolic diseases to strategize new potential therapeutic approaches in the future.
Supplemental Material
Download MS Word (15.4 KB)Acknowledgments
We thank Dr. Hiroyuki Mori for assistance with EMSC studies and Simin Abrishami for help with bone marrow studies.
Disclosure statement
The authors have declared that no conflicts of interest exist.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Stokes A, Preston SH. Deaths attributable to diabetes in the United States: comparison of data sources and estimation approaches. PLoS One. 2017;12(1):e0170219.
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867.
- Schelbert KB. Comorbidities of obesity. Prim Care. 2009;36(2):271–285.
- Fearon WF, Fearon DT. Inflammation and Cardiovascular Disease. Circulation. 2008;117(20):2577–2579.
- Jiang SZ, Lu W, Zong XF, et al. Obesity and hypertension. Exp Ther Med. 2016;12(4):2395–2399.
- Vucenik I, Stains JP. Obesity and cancer risk: evidence, mechanisms, and recommendations. Ann N Y Acad Sci. 2012;1271:37–43.
- Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017;542(7640):177–185.
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121(6):2111–2117.
- Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–445.
- Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest. 2017;127(1):1–4.
- Hammarstedt A, Gogg S, Hedjazifar S, et al. Impaired adipogenesis and dysfunctional adipose tissue in human hypertrophic obesity. Physiol Rev. 2018;98(4):1911–1941.
- Gustafson B, Gogg S, Hedjazifar S, et al. Inflammation and impaired adipogenesis in hypertrophic obesity in man. Am J Physiol Endocrinol Metab. 2009;297(5):E999–E1003.
- Gustafson B, Hedjazifar S, Gogg S, et al. Insulin resistance and impaired adipogenesis. Trends Endocrinol Metab. 2015;26(4):193–200.
- Boden G. Obesity and Free Fatty Acids (FFA). Endocrinol Metab Clin North Am. 2008;37(3):635–ix.
- Liu Z, Liu J, Jahn LA, et al. Infusing lipid raises plasma free fatty acids and induces insulin resistance in muscle microvasculature. J Clin Endocrinol Metab. 2009;94(9):3543–3549.
- Wolever TM, Bentum-Williams A, Jenkins DJ. Physiological modulation of plasma free fatty acid concentrations by diet. Metabolic implications in nondiabetic subjects. Diabetes Care. 1995;18(7):962–970.
- Reaven GM, Hollenbeck C, Jeng CY, et al. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes. 1988;37(8):1020–1024.
- Bouloumie A, Curat CA, Sengenes C, et al. Role of macrophage tissue infiltration in metabolic diseases. Curr Opin Clin Nutr Metab Care. 2005;8(4):347–354.
- Bruun JM, Helge JW, Richelsen B, et al. Diet and exercise reduce low-grade inflammation and macrophage infiltration in adipose tissue but not in skeletal muscle in severely obese subjects. Am J Physiol Endocrinol Metab. 2006;290(5):E961–967.
- Cancello R, Henegar C, Viguerie N, et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes. 2005;54(8):2277–2286.
- Griffin C, Eter L, Lanzetta N, et al. TLR4, TRIF, and MyD88 are essential for myelopoiesis and CD11c(+) adipose tissue macrophage production in obese mice. J Biol Chem. 2018;293(23):8775–8786.
- Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184.
- Crewe C, An YA, Scherer PE. The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J Clin Invest. 2017;127(1):74–82.
- Strissel KJ, Stancheva Z, Miyoshi H, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. 2007;56(12):2910–2918.
- Panee J. Monocyte chemoattractant protein 1 (MCP-1) in obesity and diabetes. Cytokine. 2012;60(1):1–12.
- Cranford TL, Enos RT, Velázquez KT, et al. Role of MCP-1 on inflammatory processes and metabolic dysfunction following high-fat feedings in the FVB/N strain. Int J Obes (Lond). 2016;40(5):844–851.
- Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116(6):1494–1505.
- Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med. 2010;14(11):2592–2603.
- Orr JS, Puglisi MJ, Ellacott KL, et al. Toll-like receptor 4 deficiency promotes the alternative activation of adipose tissue macrophages. Diabetes. 2012;61(11):2718–2727.
- Vila IK, Badin PM, Marques MA, et al. Immune cell Toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell Rep. 2014;7(4):1116–1129.
- Saberi M, Woods NB, de Luca C, et al. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009;10(5):419–429.
- Lin Y, Lee H, Berg AH, et al. The lipopolysaccharide-activated toll-like receptor (TLR)-4 induces synthesis of the closely related receptor TLR-2 in adipocytes. J Biol Chem. 2000;275(32):24255–24263.
- Matsumura T, Ito A, Takii T. Endotoxin and cytokine regulation of toll-like receptor (TLR) 2 and TLR4 gene expression in murine liver and hepatocytes. J Interferon Cytokine Res. 2000;20(10):915–921.
- Frantz S, Kobzik L, Kim YD, et al. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J Clin Invest. 1999;104(3):271–280.
- Shi H, Kokoeva MV, Inouye K, et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116(11):3015–3025.
- Lee JY, Ye J, Gao Z, et al. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem. 2003;278(39):37041–37051.
- Schaeffler A, Gross P, Buettner R, et al. Fatty acid-induced induction of Toll-like receptor-4/nuclear factor-kappaB pathway in adipocytes links nutritional signalling with innate immunity. Immunology. 2009;126(2):233–245.
- Lancaster GI, Langley KG, Berglund NA, et al. Evidence that TLR4 Is not a receptor for saturated fatty acids but mediates lipid-induced inflammation by reprogramming macrophage metabolism. Cell Metab. 2018;27(5):1096–1110.e1095.
- Galic S, Fullerton MD, Schertzer JD, et al. Hematopoietic AMPK beta1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J Clin Invest. 2011;121(12):4903–4915.
- Hernandez ED, Lee SJ, Kim JY, et al. A macrophage NBR1-MEKK3 complex triggers JNK-mediated adipose tissue inflammation in obesity. Cell Metab. 2014;20(3):499–511.
- Holzer RG, Park EJ, Li N, et al. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell. 2011;147(1):173–184.
- Tzeng HT, Chyuan IT, Chen WY. Shaping of innate immune response by fatty acid metabolite palmitate. Cells. 2019;8(12):12.
- Gawronska-Kozak B. Preparation and differentiation of mesenchymal stem cells from ears of adult mice. Methods Enzymol. 2014;538:1–13.
- Staszkiewicz J, Frazier T, Rowan B, et al. Cell growth characteristics, differentiation frequency, and immunophenotype of adult ear mesenchymal stem cells. Stem Cells Dev. 2010;19(1):83–92.
- Erridge C, Samani NJ. Saturated fatty acids do not directly stimulate Toll-like receptor signaling. Arterioscler Thromb Vasc Biol. 2009;29(11):1944–1949.
- Huang S, Rutkowsky JM, Snodgrass RG, et al. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J Lipid Res. 2012;53(9):2002–2013.
- Singer K, DelProposto J, Morris DL, et al. Diet-induced obesity promotes myelopoiesis in hematopoietic stem cells. Mol Metab. 2014;3(6):664–675.
- Rim JS, Mynatt RL, Gawronska-Kozak B. Mesenchymal stem cells from the outer ear: a novel adult stem cell model system for the study of adipogenesis. Faseb J. 2005;19(9):1205–1207.
- Weatherill AR, Lee JY, Zhao L, et al. Saturated and polyunsaturated fatty acids reciprocally modulate dendritic cell functions mediated through TLR4. J Immunol. 2005;174(9):5390–5397.
- Lee JY, Sohn KH, Rhee SH, et al. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276(20):16683–16689.
- Wood IS, de Heredia FP, Wang B, et al. Cellular hypoxia and adipose tissue dysfunction in obesity. Proc Nutr Soc. 2009;68(4):370–377.
- Suganami T, Tanimoto-Koyama K, Nishida J, et al. Role of the toll-like receptor 4/NF- B pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler Thromb Vasc Biol. 2007;27:84–91.
- Kratz M, Coats B, Hisert K, et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014;20:614–625.
- Wang L, Li L, Ran X, et al. Lipopolysaccharides reduce adipogenesis in 3T3-L1 adipocytes through activation of NF-kappaB pathway and downregulation of AMPK expression. Cardiovasc Toxicol. 2013;13:338–346.
- Chae G, Kwak S. NF-kappaB is involved in the TNF-alpha induced inhibition of the differentiation of 3T3-L1 cells by reducing PPARgamma expression. Exp Mol Med. 2003;431–437. DOI:10.1038/emm.2003.56
- Lacasa D, Taleb S, Keophiphath M, et al. Macrophage- secreted factors impair human adipogenesis: involvement of proinflammatory state in preadipocytes. Endocrinology. 2007;148:868–877.
- Liao F, Andalibi A, deBeer FC, et al. Genetic control of inflammatory gene induction and NF-kappa B-like transcription factor activation in response to an atherogenic diet in mice. J Clin Invest. 1993;91:2572–2579.
- Nagareddy P, Kraakman M, Masters S, et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 2014;19:821–835.
- Nguyen MT, Favelyukis S, Nguyen AK, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282:35279–35292.
- Zhao M, Chen X. Effect of lipopolysaccharides on adipogenic potential and premature senescence of adipocyte progenitors. Am J Physiol Endocrinol Metab. 2015;309(4):E334–344.
- Geric I, Tyurina Y, Krysko O, et al. Lipid homeostasis and inflammatory activation are disturbed in classically activated macrophages with peroxisomal beta‐oxidation deficiency. Immunology. 2018;153:342‐356.
- Madsen L, Petersen RK, Kristiansen K. Regulation of adipocyte differentiation and function by polyunsaturated fatty acids. Biochim Biophys Acta. 2005;1740(2):266–286.
- Guo W, Wong S, Xie W, et al. Palmitate modulates intracellular signaling, induces endoplasmic reticulum stress, and causes apoptosis in mouse 3T3-L1 and rat primary preadipocytes. Am J Physiol Endocrinol Metab. 2007;293(2):E576–586.
- Listenberger LL, Ory DS, Schaffer JE. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J Biol Chem. 2001;276(18):14890–14895.
- Lee JY, Plakidas A, Lee WH, et al. Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J Lipid Res. 2003;44(3):479–486.
- Pricola K, Kuhn N, Haleem-Smith H, et al. Interleukin-6 maintains bone marrow-derived mesenchymal stem cell stemness by an ERK1/2-dependent mechanism. J Cell Biochem. 2009;108(3):577–588.
- van Hall G, Steensberg A, Sacchetti M, et al. Interleukin-6 stimulates lipolysis and fat oxidation in humans. J Clin Endocrinol Metab. 2003;88(7):3005–3010.
- Carey A, Bruce C, Sacchetti M, et al. Interleukin-6 and tumor necrosis factor-α are not increased in patients with Type 2 diabetes: evidence that plasma interleukin-6 is related to fat mass and not insulin responsiveness. Diabetologia. 2004;47:1029–1037.
- Mauer J, Denson JL, Brüning JC. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol. 2015;36(2):92–101.
- Han MS, White A, Perry RJ, et al. Regulation of adipose tissue inflammation by interleukin 6. Proc Natl Acad Sci U S A. 2020;24:pii: 201920004.