
ABSTRACT
Adipocyte-mediated inflammatory signalling has been proposed to alter adipose physiology in obesity and Type 2 diabetes mellitus. Novel targets for alteration of inflammatory signalling are needed to improve obesity-related outcomes. The γ-secretase enzyme complex has been suggested to play a role both in adipocyte function as well as in immune regulation. We hypothesized that adipocyte-specific γ-secretase inhibition could alter the inflammatory makeup of adipose tissue. We found that genetic blockade of γ-secretase in adipocytes leads to a decrease in EMR1 (F4/80) expression, as a marker of macrophage presence, in adipose tissue without changes in expression of markers of other inflammatory cell types. To explore the mechanism by which adipocytes can alter macrophage function in vitro, fully differentiated 3T3-L1 adipocytes were treated with a γ-secretase inhibitor in the presence of lipopolysaccharide (LPS) and transcription of IL6 and ccl2 (MCP1) were quantified. IL-6 expression and secretion were significantly inhibited by γ-secretase blockade, with little effect on MCP1. Preconditioned media from 3T3-L1 adipocytes treated with a γ-secretase inhibitor also alters macrophage activation but did not affect macrophage translocation in vitro. Therefore, γ-secretase inhibition in fully differentiated adipocytes can alter IL-6 signalling to macrophages, consistent with our hypothesis that that γ-secretase is involved in adipocyte-initiated inflammatory signalling cascades.
Introduction
The proximal cause of obesity-induced insulin resistance is excessive adipose mass [Citation1] and one factor that can affect adipose function is inflammation. As there is a broad spread in pathophysiology, inflammatory signalling, and response to treatment in obesity, new signalling cascades and drug targets are needed to prevent the long-term morbidities that can arise in obese youth and persist into adulthood [Citation2–6]. The adipocyte itself has been implicated in direct regulation of adipose inflammation [Citation7–9]. Therefore, novel pathways regulating adipocyte-initiated inflammation could provide new avenues of treatment for obesity and type 2 diabetes (T2DM).
The γ-secretase enzyme complex is a heterotetramer of four obligate components – Nicastrin, PSEN1, APH-1, and PEN-2 – that regulates intramembranous cleavage of type-I transmembrane proteins termed regulated intramembrane proteolysis. This process releases the C-terminal intracellular domain of the transmembrane protein, which may go on to mediate downstream effects [Citation10]. Several immune signalling cascades utilize γ-secretase and regulated intramembrane proteolysis, including Notch, CD44, and TREM2 [Citation11–14]. Notch signalling, associated with cell-fate decisions [Citation14] and whole-body glucose metabolism [Citation15], is a classic example of both γ-secretase function and the consequences of its disruption [Citation16]. Upon activation, Notch undergoes regulated intramembrane proteolysis by γ-secretase, releasing the Notch intracellular domain (NICD). NICD translocates to the nucleus and activates Rbp-Jκ-dependent transcription of the Hes and Hey transcription factor families, which regulate the variety of cellular responses to Notch activation.
The roles of γ-secretase broadly and Notch specifically in adipose function are unclear. For example, Notch activation induces adipogenesis, but γ-secretase inhibition (GSI) leads to the same effect [Citation17,Citation18], suggesting either time-dependent and/or Notch-independent signalling events are required. Previous data directly implicated γ-secretase in the regulation of insulin signalling in adipose tissue, but in a Notch-independent manner [Citation19]. Adipocyte-specific blockade of γ-secretase activity was achieved by flox-mediated knockout of Nicastrin (A-Nicastrin) via cre-recombinase under the control of the adiponectin promoter. A-Nicastrin mice had no change in whole-body insulin sensitivity compared to cre-negative littermate controls, under both chow- and high-fat-fed conditions. However, non-esterified free fatty acids were increased following Nicastrin knockdown, consistent with decreased adipose insulin sensitivity. Other work has suggested inhibition of γ-secretase can lead to decreased obesity and increased browning of white adipose tissue [Citation20]. These data show that γ-secretase is active in adipocytes and may have diverse roles in both the development and functioning of mature adipocytes, in both Notch-dependent and Notch-independent cascades.
A novel function of γ-secretase could be in regulating immune signalling cascades initiated by adipocytes themselves in either Notch dependent or independent manners. For example, the Notch cascade is involved in the development and regulation of multiple immune cell types [Citation21]. Alternatively, Nicastrin knockdown directly decreases dendritic cell antigen presentation [Citation22]. Several inflammatory cytokines have been shown to be secreted by adipocytes, and have been tied to obesity or alterations in adipocyte function [Citation9,Citation23–25]. Among these, MCP1 and IL-6 have been well characterized in a variety of cell types. Both MCP1 and IL-6 signalling are regulated, at least in part, at the transcriptional level, but cytokines such as IL-6 can be regulated by their secretory machinery as well [Citation26]. Obesity is thought to lead to increased Toll-like receptor 4 (TLR4) stimulation in adipocytes via a variety of ligands [Citation27], and expression of both IL-6 and MCP1 by adipocytes can be induced by inflammation [Citation28]. Therefore, determining both the cell and cascade-specific effects and mechanisms of γ-secretase inhibition are needed to determine what role γ-secretase blockade might have in possible therapies.
We hypothesized that γ-secretase is integral to adipocyte-initiated inflammatory signalling cascades. We first analysed stored whole adipose from the previously characterized cohort of animals that had adipocyte-specific blockade of γ-secretase activity, with concomitant altered adipocyte insulin sensitivity [Citation19]. Samples from the cohort were re-examined for alterations in inflammatory gene expression. However, analysis of our initial studies was complicated by the complex cellular makeup, including not only adipocytes, but the resident lymphocytes and other components of the stromal vascular fraction (SVF), of whole adipose tissue. Therefore, while γ-secretase has been implicated in immune signalling cascades and adipocytes separately, its specific role in adipocyte-initiated inflammatory signalling was further studied in vitro in 3T3-L1 adipocytes, limiting the noise from gene transcription and translation from the SVF. We determined whether γ-secretase inhibition is able to decrease resident adipose tissue macrophages and/or alter their activation. We further studied adipocyte-specific alterations and mechanisms of known adipokines’ secretion, exploring an initial pathway in which γ-secretase functions in regulation of adipose tissue inflammation.
Materials and methods
Experimental animals
Results described were obtained from stored samples from a previously described cohort [Citation19], wherein all procedures had been approved by the Columbia University Institutional Animal Care and Utilization Committee. In short, adiponectin-cre [Citation29] animals were crossed with Nicastrinflox/flox [Citation15] mice, all on the C57/BL6 background, to generate adiponectin(cre); Nicastrinflox/flox (A-Nicastrin) animals. Animals were weaned to either standard chow (Purina Mills 5053) or high-fat diet (18.4% calories/carbohydrates, 21.3% calories/protein and 60.3% calories/fat derived from lard; Harlan Laboratories, TD.06414).
Cell culture and adipocyte transfection
3T3-L1 preadipocytes (ATCC) were maintained in DMEM, high glucose GlutaMAX-I media (Gibco) with 10% Calf serum (Gibco) and differentiated according to normal specifications. RAW 264.7 macrophages (ATCC) were maintained under normal tissue culture procedures. Where specified, cells were treated with 200 nM N-[(1 S)-2-[[(7 S)-6,7-Dihydro-5-methyl-6-oxo-5 H-dibenz[b,d]azepin-7-yl]amino]-1-methyl-2-oxoethyl]-3,5-difluorobenzeneacetamid (DBZ, Tocris), cycloheximide (Sigma), and/or lipopolysaccharide (LPS, Sigma) added to concentrations noted.
Quantitative real time-PCR (qRT-PCR)
RNA was isolated from whole adipose tissue via the RNeasy Lipid mini-kit (Qiagen); RNA from whole cell lysates was obtained with the RNAeasy mini-kit (Qiagen). cDNA was generated from mouse adipose RNA extract as described [Citation19]; cDNA from 3T3L1 adipocyte RNA extracts was generated using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative real-time PCR was performed on a CFK96 Real-Time PCR detection system (Bio-Rad) with the Power SYBR Green PCR Master Mix (Applied Biosystems). Primers for all genes are available on request. Gene transcription was quantified using the ΔΔCt method using TATA Binding Protein (TBP) as a control to determine relative gene expression. Rapid analysis of inflammatory cascade gene expression was performed using a PrimePCR Custom 96 well plate (Bio-Rad) according to manufacturer’s specifications. Genes screened for included Adam17, Il4ra, Mrc1, Fos, Il1b, IL12a, Il6, Il10, Hes1, Jak1, Jun, Map2k1, Mapk1, Mapk3, Nfkb1, Nfkb2, Nr3c1, Ptpn11, Tnf, Ccl2, Ccl3, Nos2, Socs3, Stat3, Tbp, Gapdh, and Hprt.
ELISA and immunohistochemistry (IHC)
Cells were lysed in Whole Cell Lysis Buffer (20 mM Tris, pH 7.4 150 mM NaCl, 10% glycerol, 2% Nonidet P-40, 1 mM EDTA, pH 8.0, 0.1% SDS, 0.5% sodium deoxycholate) supplemented immediately prior to use with HALT Protease & Phosphatase Inhibitor Single-Use Cocktail (Roche). ELISAs for mouse MCP1 and IL-6, both secreted into media or from WCL as above, were performed according to manufacturer’s specifications (R&D Systems). For IHC whole adipose tissue was fixed in Z-fix (Anatech), paraffin embedded, sectioned and stained with H&E and F4/80 through the Advanced Tissue Pathology and Imaging Core in the Diabetes Research Centre at Columbia University Irving Medical Centre.
Macrophage migration
Media from 3T3L1 adipocytes pre-treated with ± DBZ overnight and then ± LPS for 6 hours was collected, filter sterilized, and utilized in a QCM 24-Well Colorimetric Cell Migration Assay (Millipore) with RAW 264.7 macrophages; at the end of the assay the insert was secondarily stained with DAPI and cells were manually counted.
Statistical analysis
All results are reported as mean ± SEM. Gene expression levels were compared using Students t-test. Experiments utilizing CHX/LPS or DBZ/LPS treatments were analysed by 2-way ANOVA with post-hoc Tukey HSD test. P values of <0.05 were considered significant.
Results
Decreased γ-secretase activity alters adipose tissue macrophages
We previously found that adipocyte-specific knockout of γ-secretase activity, by ablation of the non-redundant Nicastrin subunit (A-Nicastrin mice) decreased adipose insulin sensitivity [Citation19]. However, those initial studies did not examine the local changes to the SVF within the adipose tissue. We, therefore, examined several markers of the SVF from the epididymal fat pad (eWAT). Emr1 (F4/80), a well-known marker of adipose tissue macrophages, which increases in response to HFD ()), was lower in A-Nicastrin eWAT adipose tissue compared to that from cre-negative littermates independent of diet ()). GSI was able to similarly inhibit EMR1 in WT animals ()). Thus, inhibition of γ-secretase chemically or by disruption of the complex reduces macrophage markers in adipose tissue.
Figure 1. Adipocyte-specific γ-secretase blockade alters adipose inflammation. (a) Relative gene transcription of F4/80 (Emr1) was examined from eWAT of C67/Bl6 mice maintained on chow or HFD. (b) F4/80 transcription from eWAT from A-Nicastrin mice and their cre (-) littermate controls maintained on chow or HFD. (c) F4/80 expression was measured from eWAT adipose depots from wild type chow-fed animals treated with vehicle or 2 mg/kg GSI for 5 days (d) Relative transcription of markers of macrophage activation were compared between HFD-fed A-Nicastrin mice and their cre (-) littermate controls. (e) Representative images from adipose stained for macrophages via anti-F4/80 IHC in HFD-fed A-Nicastrin mice and their cre (-) littermate controls (f) Relative expression of total T-cell and T-reg cell markers, (g) MHC-II associated proteins, and (h) inflammatory cytokines from HFD-fed A-Nicastrin animals and their cre (-) littermates are shown (n = 7/condition, error bars are ± SEM, * = p < 0.05)
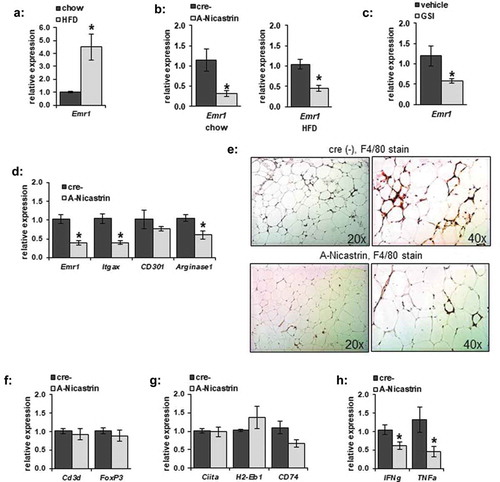
We next determined what immunological cell lines might be disrupted at the transcriptional level in adipose tissue from A-Nicastrin mice. In eWAT from HFD-fed animals, transcription of markers of M1 and M2a polarization, Itgax (Cd11 c) and Arginase1 respectively, were decreased in a similar fashion to F4/80 ()); CD301, another M2 marker, was not significantly affected. Histologically, F4/80(+) cells and crown-like structures in HFD-fed A-Nicastrin adipose were reduced compared to WT controls ()). These observed macrophage changes were not accompanied by a decrease in expression of a broad marker of T cells (Cd3d) or T regulatory cells (FoxP3) ()), as noted by expression of Cd3d and FoxP3 respectively. Transcription of MHCII markers Ciita as well as H2-Eb1 and CD74 were unaffected in A-Nicastrin mice ()). IFNg and TNFa transcription were decreased in A-Nicastrin mice, likely reflective of a decrease in local pro-inflammatory cytokine production.
γ-secretase blockade alters basal adipocyte inflammatory cytokine expression
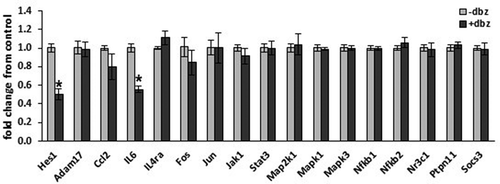
Due to the complex makeup of adipose tissue, we specifically focused on the adipocyte to determine changes in inflammatory gene expression caused by GSI in vitro. 3T3-L1 adipocytes were incubated overnight in the absence or presence of 200 nM DBZ, a potent γ-secretase inhibitor. Total RNA was purified and analysed for a broad set of genes involved in inflammatory signalling by qRT-PCR (). Consistent with inhibition of Notch signalling known to occur with GSI, Hes1 expression was significantly down-regulated. Basal IL6 transcription was significantly decreased by γ-secretase inhibition, whereas basal levels of ccl2 and several other genes within inflammatory signalling cascades were unaffected. These data indicate that γ-secretase inhibition does not broadly change basal adipocyte inflammatory cytokines or transcription factors but can specifically suppress basal IL6 transcription.
Figure 2. Adipocyte inflammatory gene expression is altered in response to γ-secretase blockade. Fully differentiated 3T3-L1 adipocytes were incubated in the absence or presence of γ-secretase-inhibitor DBZ (200 nM) for 18 hours. Total RNA was purified and gene expression analysed by the ΔΔCt method on a PrimePCR Custom 96 well plate (n = 5–6/condition, error bars are ± SEM, * = p < 0.05)
γ-secretase blockade can inhibit LPS-induced IL-6 and MCP1 transcription and alters basal and stimulated IL-6 secretion
LPS is known to induce a variety of inflammatory signals within adipocytes. As basal transcription levels of the cytokine IL-6 were affected by GSI, we sought to determine whether γ-secretase could alter LPS-stimulated transcription of IL-6 or MCP1. We incubated 3T3-L1 adipocytes in the absence or presence of DBZ overnight, followed by stimulation with 100 ng/mL. IL6 and MCP1 transcription were significantly increased by LPS stimulation, and partially blocked by GSI (,b)). To determine if this had an effect beyond the transcriptional level, cytokine secretion was next examined. Stimulated secretion of cytokines such as IL-6 may be regulated at the transcriptional level as well as at via secretory machinery [Citation30]. As expected, cycloheximide pre-treatment inhibited LPS-induced secretion of both IL-6 and MCP1 from adipocytes (,d)). We wondered if the full effect of GSI was due purely to blockade of transcription and translation of IL6 or MCP1, or if there was also a further blocking effect on stored cytokine secretion through changes in secretory machinery. Pre-treatment with the γ-secretase inhibitor DBZ did not alter the cycloheximide effect even though a small amount of IL-6, but not MCP1, was still secreted (,f)). A small pool of IL-6 can, therefore, be secreted even without active gene translation, and this pool appears resistant to the effects of GSI. Taken together, these data indicate that γ-secretase inhibition prevents full induction of IL-6 and MCP1 gene transcription, and that secretion of IL-6 and MCP1 are dependent on gene translation in adipocytes.
Figure 3. LPS-stimulated adipocyte IL-6 and MCP1 secretion is decreased by γ-secretase inhibition. (a) Fully differentiated 3T3-L1 adipocytes were exposed to LPS at 100 ng/mL for 6 hours in the absence or presence of overnight exposure to 200 nM DBZ. Total RNA was purified from the adipocytes and gene expression for IL-6 (a) or MCP1 (ccl2, b) was analysed by the ΔΔCt method, comparing to unstimulated adipocytes (n = 6/condition, error bars are ± SEM, * = p < 0.05). IL-6 (c) or MCP1 (d) secretion was measured by ELISA from conditioned media from LPS-treated 3T3-L1 adipocytes in the absence or presence of a 2 hour pre-incubation with cycloheximide (CHX) (n = 6/condition, fold change from untreated samples, error bars are ± SEM, * = p < 0.05 from untreated media). Media was similarly analysed for IL-6 (e) or MCP1 (f) secretion from LPS-stimulated 3T3-L1 adipocytes treated with CHX that had been previously treated overnight in the absence or presence of 200 nM DBZ. Increasing amounts of LPS were used to stimulate 3T3-L1 adipocytes following the absence or presence of DBZ (200 nM, overnight exposure) and IL-6 (g) or MCP1 (h) levels were assess by ELISA (n = 6/condition, error bars are ± SEM, * = p < 0.05 from the untreated samples.)
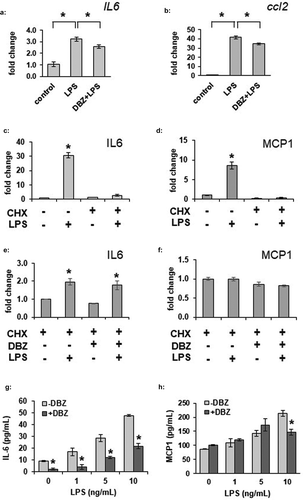
To determine whether LPS-induced IL-6 secretion could be affected by γ-secretase inhibition, we incubated adipocytes overnight in the absence or presence of DBZ, then stimulated them with increasing amounts of LPS. Pre-treatment of adipocytes with DBZ significantly attenuated adipocyte IL-6 secretion both in the basal and LPS-stimulated states ()). In stark contrast to the limited effects on adipocyte IL-6 secretion, and despite the significant change in transcription, LPS-induced MCP1 secretion was not drastically affected by GSI ()), suggesting stored MCP1 might not be dependent on rapid changes in expression in adipocytes.
LPS-stimulation of IL-6 intracellular expression can persistently be inhibited by GSI
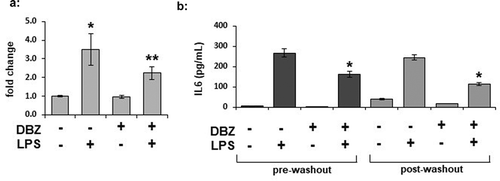
To determine if γ-secretase blockade decreased IL-6 intracellular protein levels in both the basal and LPS-stimulated state, we measured IL-6 levels from 3T3L1 whole cell lysates by ELISA ()). The LPS-stimulated increase in IL-6 accumulation within the cell was blocked by pre-treatment with DBZ. To determine whether or not the effects of γ-secretase blockade on IL-6 secretion can persist after removal of the LPS stimulus, we incubated adipocytes in the absence or presence of DBZ overnight, then stimulated for 6 hours with LPS. We then washed the cells extensively and incubated them for an additional 18 hours in regular adipocyte maintenance media. LPS levels were decreased following the wash (data not shown). Despite the washout of the LPS and DBZ, the decrease in IL-6 secretion was maintained ()).
Figure 4. IL-6 expression is regulated by γ-secretase in adipocytes, and secretion can be persistently inhibited by γ-secretase inhibition. Intracellular expression of IL-6 was measured by ELISA from whole cell lysates from 3T3L1 adipocytes pretreated with 200 nM DBZ overnight and then treated with LPS for 6 hours. (n = 6/condition, error bars are ± SEM, * = p < 0.05 from the untreated sample). (b) The persistence of alteration in IL-6 secretion was determined in 3T3-L1 adipocytes treated with DBZ and LPS as previously examined (pre-washout), with the cells then washed 3x with PBS and placed in adipocyte maintenance medium for an additional 18 hours (post-washout); media collected at the 2 time points were analysed by ELISA for IL-6 (n = 6/condition, error bars are ± SEM, * = p < 0.05 from the untreated samples.)
γ-secretase blockade does not alter L1-induced macrophage migration in vitro, but alters IL-6-induced activation
To determine whether GSI-treated adipocytes could alter macrophage migration, we pre-treated adipocytes overnight in serum-free media with DBZ followed by LPS stimulation to produce conditioned media. The conditioned media was then used media to analysed for its ability to induce macrophage translocation. Media collected from adipocytes following LPS stimulation induced macrophage translocation across a Boyden chamber ()). Conditioned media from GSI-treated adipocytes stimulated macrophage migration in a similar manner.
Figure 5. γ-secretase inhibition does not significantly alter adipocyte LPS-stimulated macrophage attraction or soluble IL6 receptor concentration but does alter adipocyte-mediated IL-6 activation of macrophages. (a) RAW 264.7 macrophage translocation was determined in a Boyden chamber after 6 hours of exposure to serum-free (SF) or media from 3T3L1 adipocytes pre-treated with DBZ and/or LPS (n = 6 per condition, * = p < 0.05 from SF control media). (b) Secretion of soluble IL-6 R was measured by ELISA from media from 3T3L1 adipocytes pretreated with 200 nM DBZ overnight and then treated with LPS for 6 hours (n = 6 per condition) (c) Macrophage IL-10 secretion was measured from RAW 264.7 treated with preconditioned media from 3T3-L1 adipocytes pre-treated in the absence (L1-) or presence (L1+) of 200 nM DBZ. Preconditioned media also received further pre-treatment (tx): L1- samples were also incubated with or without IL-6 blocking antibody andL1+ samples were incubated with and without exogenous IL-6. (* = p < 0.05 from – DBZ IgG control or +DBZ samples, respectively.) (d) Fold change of VEGFa transcription from RAW 264.7 macrophages following 6 hours of exposure to media from 3T3-L1 adipocytes pre-treated with DBZ or LPS (* = p < 0.05 from control sample, ** = p < 0.05 from +LPS sample)
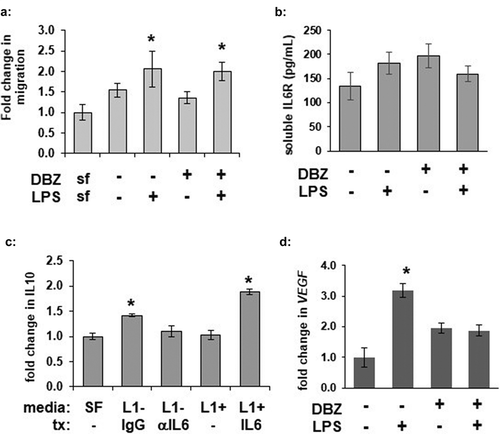
IL-6 has previously been suggested to mediate adipocyte inflammatory signalling, and macrophages can secrete IL10 in response to IL-6 [Citation31]. Furthermore, this could be a result of adipose IL-6 trans-signalling, wherein soluble IL-6-receptor (sIL6 R) can bind local IL-6 and activate inflammatory signalling in nearby cells expressing gp130 [Citation32]. Soluble IL6 R secretion was not affected by GSI ()). As IL-6 secretion even in the basal state appeared to be decreased by γ-secretase inhibition, and to avoid complications from residual LPS affecting macrophage function, media from unstimulated adipocytes was used to activate macrophages in vitro. Conditioned media from cells pre-treated with a GSI induced significantly less macrophage IL10 secretion, which could be rescued with addition of exogenous IL-6 ()). Similarly, GSI reduced another IL-6 target, VEGFa transcription in alternatively polarized M2d macrophages [Citation33], ()). These data are consistent with a requirement for γ-secretase function in adipocyte-secreted IL-6 activation of macrophages, likely primarily through alterations of IL-6 expression and secretion.
Discussion
Adipose inflammation has long been associated with insulin resistance and Type 2 diabetes [Citation34]. Our data further support the hypothesis that signalling cascades within adipocytes participate in regulation of the local inflammatory state. Previous work has shown that γ-secretase blockade in adipocytes leads to decreased insulin sensitivity, in a Notch-independent manner [Citation19]. Our current work suggests that adipocyte γ-secretase blockade simultaneously reduces adipocyte-mediated inflammation, likely by interfering with IL-6 transcription, expression, and secretion, to alter macrophage activation.
When subjecting samples from a previous study to further scrutiny, we found that either genetic or pharmacologic GSI led to a decrease in macrophage markers in adipose tissue. While pharmacologic treatment could lead to non-specific effects of GSI on macrophages, the genetic blockade was specific to adipocytes. Classic markers of macrophage polarization were affected as well, though several other cell lines were not affected when examined at the transcriptional level. Taken together, these data indicated that adipocyte-specific GSI could alter the local inflammatory state of adipose tissue in vivo.
IL-6 has been identified as a significant mediator of adipose inflammation [Citation35]. Macrophage IL-6 expression is known to be down-regulated by γ-secretase inhibition [Citation36], and we have observed a similar effect both at the transcriptional and translational levels in adipocytes. This suggests that paracrine inflammatory signalling can be initiated purely by the adipocyte, via a similar mechanism to macrophage regulation, leading to broader adipose inflammation alterations as seen in the A-Nicastrin mouse. The transcriptional regulation likely accounts for at least a part of its persistent effect, as downstream IL-6 function can be regulated through expression of both the cytokine and its endosome machinery. These disparate effects, on both IL-6 expression itself, as well as the secretory machinery, might explain why short-term blockade of γ-secretase leads to a decrease in basal secretion of IL-6, but does not alter its intracellular concentration. Finally, as expected, adipocyte IL-6 can affect macrophage activation, with altered IL-10 secretion and VEGFa expression from macrophages exposed to preconditioned media from 3T3L1 adipocytes. These changes were inhibited by prior γ-secretase blockade of the adipocytes, and could be rescued with exogenous IL-6. Taken together, these data suggest that adipocyte γ-secretase blockade can decrease IL-6-mediated activation of macrophages. This correlates with the suggested role of IL-6 in limiting obesity-related insulin resistance in mice [Citation31]. Local alterations in IL-6 paracrine signalling originating in adipocytes might then explain at least a portion of the localized insulin resistance of the A-Nicastrin mice. Most intriguingly, our data is consistent with work on adipose IL-6 trans-signalling, wherein soluble IL-6-receptor (sIL6 R) can bind IL-6 and activate inflammatory signalling in nearby cells expressing gp130 [Citation32]. Blockade of IL-6 trans-signalling in adipose decreased macrophage recruitment, but did not alter insulin resistance, similar to our in vitro findings. While sIL6 R secretion was not altered in this specific adipocyte cell line, decreased downstream signalling through gp130 could occur via the decrease in IL-6 itself. This would correlate with our observed in vivo decrease in macrophage recruitment. It is possible that this might occur via altered signalling within CD4 + T-cells, which aid in regulation of obesity-induced inflammation [Citation37], and which can express gp130. Further work into alterations of γ-secretase signalling in vivo will be needed to determine if local γ-secretase changes in IL-6 signalling via gp130 are affected in both Notch-dependent (i.e. transcriptional regulation) and Notch-independent (i.e. IL-6-trans signalling) manners.
Our data are broadly consistent with the theory of a ‘healthy’ adipose inflammatory state [Citation38,Citation39]. Adipose tissue macrophages (ATMs) have been shown to play different roles in adipose regulation, including activation of pathways increasing lysosomal function, thereby buffering increased local lipid concentrations [Citation40]. Worsening insulin sensitivity and resulting lipolysis are directly associated with alterations in immune response through a variety of pathways [Citation41,Citation42]. We hypothesize that with the worsening insulin sensitivity in adipose tissue following GSI (i.e. with the previously observed increased in NEFA [Citation19],) and these newly observed changes in IL-6 secretion, that GSI can lead to a significant degree of localized adipose insulin resistance.
Our study has some inconsistencies. We initially observed that adipocyte-specific γ-secretase deficiency leads to altered adipose inflammation, implying that adipocytes may regulate both macrophage infiltration and/or activity. However, due to the complexity of the whole tissue, interpretation of the transcriptional studies was difficult. For example, adipocytes are already known to regulate macrophage function via a variety of inflammatory cascades beyond IL-6 [Citation32]. Adipocytes may present antigens via MHC-II molecules, which can also activate CD4 + T-cells and in turn promote ATM M1 polarization and inflammatory cytokine secretion in HFD-induced obesity [Citation43]. We did not observe a change in whole-tissue expression of MHCII-associated genes, despite the observed decrease in ATMs. This might be explained by the possibility of up-regulated MHCII expression on other cells within the adipose tissue SVF. Another possible contradiction involves MCP1. MCP1 has been well characterized for its role in macrophage recruitment and adipose inflammation. However, in our system, MCP1 was only mildly affected in its LPS-stimulated transcription and secretion. Interestingly, MCP1 secretion has been suggested to primarily be from adipocyte progenitors in vivo, not adipocytes themselves [Citation44]. Therefore, while MCP1 is a known potent macrophage attractant, alterations in MCP1 (or lack thereof) likely did not lead to the decrease in adipose macrophage presence in the A-Nicastrin animals, which would have normal preadipocytes. Future work examining other cell line and cytokine changes will be needed to determine the root cause of the decrease in local macrophages, i.e. through alterations in IL-6 trans signalling.
Therefore, our current work is consistent adipocyte IL-6 expression, secretion, and function in adipose inflammation being regulated in part via pathways utilizing γ-secretase. Further exploration into the specific mechanisms of how adipocytes can alter the activation of ATMs, which serve a variety of roles in both the lean and obese states [Citation9], is warranted as γ-secretase inhibition appears to profoundly affect local adipose inflammation.
Acknowledgments
The authors would like to acknowledge April Teague for her excellent technical support at the University of Oklahoma Health Sciences Center.
The authors have no conflicts to disclose.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Walker CG, Zariwala M, Holness M, et al. Diet, obesity and diabetes: a current update. Clin Sci (Lond). 2007;112(2):93–111. DOI:10.1042/CS20060150
- Kahn SE, Haffner SM, Heise MA, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355(23):2427–2443. DOI:10.1056/NEJMoa066224
- Group TS. A clinical trial to maintain glycemic control in youth with type 2 diabetes. N Engl J Med. 2012;366(24):2247–2256.
- McCrindle BW. Cardiovascular consequences of childhood obesity. Can J Cardiol. 2015;31(2):124–130.
- Pollock NK. Childhood obesity, bone development, and cardiometabolic risk factors. Mol Cell Endocrinol. 2015;410:52–63.
- Zimmermann MB, Aeberli I. Dietary determinants of subclinical inflammation, dyslipidemia and components of the metabolic syndrome in overweight children: a review. Int J Obes (Lond). 2008;32(Suppl 6):S11–8.
- Balistreri CR, Caruso C, Candore G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediators Inflamm. 2010;2010:802078.
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121(6):2111–2117.
- Morris DL, Singer K, Lumeng CN. Adipose tissue macrophages: phenotypic plasticity and diversity in lean and obese states. Curr Opin Clin Nutr Metab Care. 2011;14(4):341–346.
- Steiner H, Fluhrer R, Haass C. Intramembrane proteolysis by gamma-secretase. J Biol Chem. 2008;283(44):29627–29631.
- Fluhrer R, Multhaup G, Schlicksupp A, et al. Identification of a beta-secretase activity, which truncates amyloid beta-peptide after its presenilin-dependent generation. J Biol Chem. 2003;278(8):5531–5538. DOI:10.1074/jbc.M211485200
- Okochi M, Steiner H, Fukumori A, et al. Presenilins mediate a dual intramembranous gamma-secretase cleavage of Notch-1. Embo J. 2002;21(20):5408–5416. DOI:10.1093/emboj/cdf541
- Wunderlich P, Glebov K, Kemmerling N, et al. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and gamma-secretase-dependent intramembranous cleavage. J Biol Chem. 2013;288(46):33027–33036. DOI:10.1074/jbc.M113.517540
- Swiatek PJ, Lindsell CE, Del Amo FF, et al. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994;8(6):707–719. DOI:10.1101/gad.8.6.707
- Pajvani UB, Shawber CJ, Samuel VT, et al. Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat Med. 2011;17(8):961–967. DOI:10.1038/nm.2378
- Weinmaster G, Kopan R. A garden of Notch-ly delights. Development. 2006;133(17):3277–3282.
- Ba K, Yang X, Wu L, et al. Jagged-1-mediated activation of notch signalling induces adipogenesis of adipose-derived stem cells. Cell Prolif. 2012;45(6):538–544. DOI:10.1111/j.1365-2184.2012.00850.x
- Huang Y, Yang X, Wu Y, et al. gamma-secretase inhibitor induces adipogenesis of adipose-derived stem cells by regulation of Notch and PPAR-gamma. Cell Prolif. 2010;43(2):147–156. DOI:10.1111/j.1365-2184.2009.00661.x
- Sparling DP, Yu J, Kim K, et al. Adipocyte-specific blockade of gamma-secretase, but not inhibition of Notch activity, reduces adipose insulin sensitivity. Mol Metab. 2016;5(2):113–121. DOI:10.1016/j.molmet.2015.11.006
- Bi P, Shan T, Liu W, et al. Inhibition of Notch signaling promotes browning of white adipose tissue and ameliorates obesity. Nat Med. 2014;20(8):911–918. DOI:10.1038/nm.3615
- Yuan JS, Kousis PC, Suliman S, et al. Functions of notch signaling in the immune system: consensus and controversies. Annu Rev Immunol. 2010;28(1):343–365. DOI:10.1146/annurev.immunol.021908.132719
- Singh CR, Bakhru P, Khan A, et al. Cutting edge: nicastrin and related components of γ-secretase generate a peptide epitope facilitating immune recognition of intracellular mycobacteria, through MHC Class II-dependent priming of T cells. J Immunol. 2011;187(11):5495–5499. DOI:10.4049/jimmunol.1100521
- Stephens JM, Lee J, Pilch PF. Tumor necrosis factor-alpha-induced insulin resistance in 3T3-L1 adipocytes is accompanied by a loss of insulin receptor substrate-1 and GLUT4 expression without a loss of insulin receptor-mediated signal transduction. J Biol Chem. 1997;272(2):971–976.
- Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol. 2005;25(10):2062–2068.
- Vieira-Potter VJ. Inflammation and macrophage modulation in adipose tissues. Cell Microbiol. 2014;16(10):1484–1492.
- Murray RZ, Stow JL. Cytokine secretion in macrophages: SNAREs, Rabs, and membrane trafficking. Front Immunol. 2014;5:538.
- Schaffler A, Scholmerich J. Innate immunity and adipose tissue biology. Trends Immunol. 2010;31(6):228–235.
- Yamashita A, Soga Y, Iwamoto Y, et al. DNA microarray analyses of genes expressed differentially in 3T3-L1 adipocytes co-cultured with murine macrophage cell line RAW264.7 in the presence of the toll-like receptor 4 ligand bacterial endotoxin. Int J Obes (Lond). 2008;32(11):1725–1729. DOI:10.1038/ijo.2008.153
- Eguchi J, Wang X, Yu S, et al. Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 2011;13(3):249–259. DOI:10.1016/j.cmet.2011.02.005
- Manderson AP, Kay JG, Hammond LA, et al. Subcompartments of the macrophage recycling endosome direct the differential secretion of IL-6 and TNFalpha. J Cell Biol. 2007;178(1):57–69. DOI:10.1083/jcb.200612131
- Mauer J, Chaurasia B, Goldau J, et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol. 2014;15(5):423–430. DOI:10.1038/ni.2865
- Kraakman MJ, Kammoun H, Allen T, et al. Blocking IL-6 trans-signaling prevents high-fat diet-induced adipose tissue macrophage recruitment but does not improve insulin resistance. Cell Metab. 2015;21(3):403–416. DOI:10.1016/j.cmet.2015.02.006
- Roszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm. 2015;2015:816460.
- Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72(1):219–246.
- Eder K, Baffy N, Falus A, et al. The major inflammatory mediator interleukin-6 and obesity. Inflamm Res. 2009;58(11):727–736. DOI:10.1007/s00011-009-0060-4
- Wongchana W, Palaga T. Direct regulation of interleukin-6 expression by Notch signaling in macrophages. Cell Mol Immunol. 2012;9(2):155–162.
- Cho KW, Morris D, DelProposto J, et al. An MHC II-dependent activation loop between adipose tissue macrophages and CD4+ T cells controls obesity-induced inflammation. Cell Rep. 2014;9(2):605–617. DOI:10.1016/j.celrep.2014.09.004
- Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184.
- Wernstedt Asterholm I, Tao C, Morley T, et al. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014;20(1):103–118. DOI:10.1016/j.cmet.2014.05.005
- Lumeng CN, DeYoung SM, Bodzin JL, et al. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007;56(1):16–23. DOI:10.2337/db06-1076
- Hu X, Cifarelli V, Sun S, et al. Major role of adipocyte prostaglandin E2 in lipolysis-induced macrophage recruitment. J Lipid Res. 2016;57(4):663–673. DOI:10.1194/jlr.M066530
- Kosteli A, Sugaru E, Haemmerle G, et al. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest. 2010;120(10):3466–3479. DOI:10.1172/JCI42845
- Deng T, Lyon CJ, Minze LJ, et al. Class II major histocompatibility complex plays an essential role in obesity-induced adipose inflammation. Cell Metab. 2013;17(3):411–422. DOI:10.1016/j.cmet.2013.02.009
- Kaplan JL, Marshall MA, McSkimming C, et al. Adipocyte progenitor cells initiate monocyte chemoattractant protein-1-mediated macrophage accumulation in visceral adipose tissue. Mol Metab. 2015;4(11):779–794. DOI:10.1016/j.molmet.2015.07.010