
Abstract
The Toll-like receptor 3 (TLR3) agonist poly(I:C) is a promising adjuvant for cancer vaccines due to its induction of potent antitumor responses occurring primarily through the activation of dendritic cells (DCs) and natural killer (NK) cells. However, little is known about the role of TLR3 sensing of endogenous ligands in innate tumor immunosurveillance. Here, we investigated whether TLR3 could modulate immune responses and facilitate tumor control without administration of an agonist. We observed only limited impact of TLR3 deficiency on spontaneous carcinogenesis and primary growth of B16F10, E0771 or MC38 tumors when injected subcutaneously to mice. Nevertheless, TLR3 was observed to limit experimental B16F10 lung metastasis, an immunologic constraint dependent on both IFNγ secretion and NK cells. Interestingly, we observed that NK cells derived from Tlr3 null (Tlr3−/−) mice were hyporesponsive to cytokine stimulation. Indeed, compared with NK cells with intact TLR3, Tlr3−/− NK cells produced significantly reduced pro-inflammatory cytokines, including IFNγ, when incubated in the presence of different combinations of IL-12, IL-18 and IL-15. Bone-marrow chimera experiments established that competent NK cell responses required TLR3 sensing on radio-sensitive immune cells. Intriguingly, although CD8α DCs robustly express high levels of TLR3, we found that those cells were not necessary for efficient IFNγ production by NK cells. Moreover, the defective NK cell phenotype of Tlr3−/− mice appeared to be independent of the gut microbiota. Altogether, our data demonstrate a pivotal role of endogenous TLR3 stimulation for the acquisition of full NK cell functions and immune protection against experimental metastasis.
Keywords:
Abbreviations:
- Abs, antibodies
- Batf3, basic leucine zipper transcription factor ATF-like 3
- DC, dendritic cell
- dsRNA, double stranded ribonucleic acid
- IFN, interferon
- IL, interleukin
- IRF, interferon regulatory factor
- MCA, methylcholanthrene
- MDA5, melanoma differentiation associated protein 5
- MHC, major histocompatibility complex
- NF-κB, nuclear factor kappa-light chain enhancer of activated B cells
- NK, natural killer
- PMA, phorbol 12-myristate 13-acetate
- Poly(I:C), polyriboinosinic-polyribocytoidylic acid
- Rae-1, ribonucleic acid export 1
- TICAM, toll-interleukin I receptor domain containing molecule1
- TIR, Toll-interleukin I receptor
- TLR, Toll-like receptor
- TRIF, TIR domain-containing adaptor inducing IFNβ
Introduction
Toll-like receptors (TLRs) initiate immune responses upon recognition of pathogen- and damage-associated molecular patterns. Therefore, members of the TLR family play a crucial role in the protection against microbial infections and the maintenance of host homeostasis.Citation1 Among these, TLR3 is an endosomal receptor that senses viral dsRNA.Citation2 TLR3 is expressed by various immune or non-immune cells, including dendritic cells (DCs),Citation3 macrophages,Citation4 natural killer (NK) cells,Citation5,6 fibroblastsCitation7 and epithelial cells.Citation8 Sensing of viral dsRNA by TLR3 leads to the secretion of type I interferon (IFN) and other pro-inflammatory cytokines.Citation9 TLR3 interacts with the adaptor protein TIR domain-containing adapter-inducing IFNβ (TRIF), also known as Toll-interleukin I receptor domain containing molecule 1 (TICAM), to induce the activation of interferon regulatory factor 3 (IRF-3) and nuclear factor κB (NF-κB) transcription factors.Citation10 Interestingly, a TRIF-independent pathway of TLR3 signaling has recently been described.Citation11 This pathway involves the non-receptor tyrosine kinase c-Src and regulates cell migration.
Well known for its role in viral infections,Citation12,13 TLR3 can also protect against cancer. Indeed, the TLR3 agonist polyriboinosinic-polyribocytoidylic acid (poly I:C) efficiently suppresses tumor growth in miceCitation14,15 and TLR3 agonists are in Phase I/II trials as adjuvants for therapeutic vaccination against melanoma and breast cancer.Citation16 The anti-cancer properties of TLR3 agonists have been attributed to the direct apoptosis of TLR3-expressing tumor cellsCitation17,18 together with the induction of immune responses that overcome tumor tolerance.Citation19-21 However, the contribution of TLR3 to the antitumor properties of poly(I:C) remains unclear since this dsRNA analog can also be recognized by the cytosolic sensor melanoma differentiation associated protein 5 (MDA5).Citation22,23 In addition, little is known about TLR3 function in the absence of administration of its agonist. Interestingly, a study reported that transplanted mouse prostate adenocarcinoma grew faster in Tlr3−/− mice compared with WT mice, supporting a protective role for endogenous triggering of TLR3.Citation20 In humans, high levels of TLR3 expression have been associated either with goodCitation24,25 or poorCitation26 prognosis, depending on the malignancies. Thus, the exact role of TLR3 in tumor immunosurveillance remains to be characterized.
Among the different cellular mediators of the poly(I:C) induced-response, NK cells represent a major antitumor effector.Citation20,21 NK cells are innate lymphocytes that recognize and directly kill transformed cells.Citation27 In addition, activated NK cells release a myriad of pro-inflammatory factors, including interferon γ (IFNγ), tumor necrosis factor α (TNFα), colony stimulating factor 2 (CSF2, also known as GM-CSF), and the chemokines MIP1-α (CCL3), MIP1-β (CCL4) and RANTES (CCL5).Citation28 NK cell responses are controlled by the integration of signals from germline-encoded activating and inhibitory receptors that recognize molecules expressed on the surface of the target cells. Yet, the acquisition of full effector functions by NK cells requires additional signals provided by cytokines such as interleukin (IL)-2, IL-12, IL-15, IL-18 and type I IFN or by direct contact with accessory cells, often DCs.Citation29 Poly(I:C) has been shown to induce efficient NK cells responses, either by the direct activation of TLR3 on NK cellsCitation5,30 or via the activation of accessory cells.Citation21-23
Here, we investigated the role of TLR3 in NK cell activation and cancer immunosurveillance in the absence of administration of exogenous dsRNA. We showed that TLR3 modulates NK cell responses by endowing them with the ability to release high amounts of IFNγ in response to cytokine stimulation. In addition, we established that the TLR3 signaling pathway controlled the growth of Rae-1β expressing RMAS tumors as well as the metastatic spread of experimental B16F10 melanoma, both of which are known to be tightly controlled on the basis of NK cell effector function. This study demonstrates that TLR3 expression on immune cells regulates IFNγ secretion by NK cells independently of the gut microbiota and is essential to control metastatic spread of cancer.
Results
NK cells from Tlr3−/− mice are hyporesponsive to cytokine stimulation
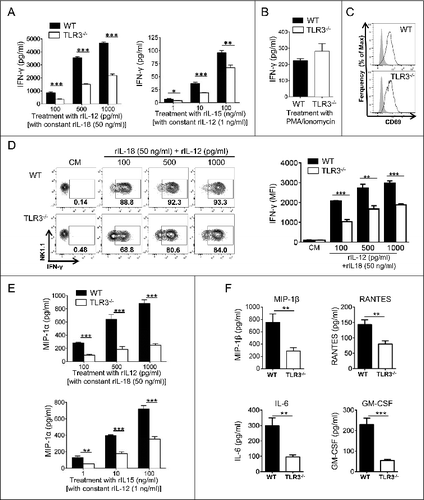
The ability of the TLR3 ligand poly (I:C) to activate NK cells is well established.Citation5,22 However, nothing is known about the influence of TLR3 on NK cell priming in the absence of administration of its agonist. To determine whether TLR3 signaling modulates NK cell ability to respond to cytokine stimulation, we purified NK cells from WT or Tlr3−/− mice (Sup. Fig. S1) and cultured them in the presence of different combinations of recombinant IL-12, IL-18 and IL-15. Interestingly, we observed that Tlr3−/− NK cells produced significantly less IFNγ than WT NK cells in response to cytokine stimulation (). By contrast, when cultured with phorbol 12-myristate 13-acetate (PMA)/ionomycin, no difference between Tlr3−/− and WT NK cells was observed in terms of IFNγ production (). Thus, the inherent ability of Tlr3−/− NK cells to produce IFNγ was not compromised. In addition, despite low levels of cytokine-induced IFNγ production, Tlr3−/− NK cells were efficiently activated upon IL-12/IL-18 stimulation, as assessed by their upregulation of CD69 (). Immunofluorescence staining and cytofluorimetric analysis confirmed that IL-12/IL-18 stimulated Tlr3−/− NK cells produced less IFNγ as compared with WT NK cells since both the percentage of IFNγ producing cells and the fluorescence intensity of the signal were reduced (). Finally, we detected lower levels of MIP-1α, MIP-1β, RANTES, IL-6 and GM-CSF in the supernatant of Tlr3−/− NK cells when cultured in the presence of IL-12/IL-18 or IL-12/IL-15 ( and F), indicating the signaling pathway controlling the production of all pro-inflammatory cytokines and chemokines by NK cells is defective in the absence of TLR3. Altogether, these data demonstrates that the presence of TLR3 regulates NK cell ability to produce of high levels of pro-inflammatory factors in response to cytokine stimulation.
Figure 1. NK cells from Tlr3−/− mice display defective cytokine production. NK cells were purified from the spleens of wild-type (WT) or Toll-like receptor 3 null (Tlr3−/−) mice and cultured together with variable concentration of recombinant (r) IL-12 with constant 50 ng/mL rIL-18 or with variable concentration of rIL-15 with constant concentration of 1 ng/mL rIL/12 (A, D, and E), with 500 pg/mL of rIL-12 combined with 50 pg/mL of rIL-18 (C, F) or with 10 ng/mL of PMA together with 1 μg/mL of ionomycin (B). After 16 to 20 h, supernatants were collected and IFNγ (A, B), MIP-1α (E) or MIP-β, RANTES, IL-6 and GM-CSF (F) production was assessed by CBA. Alternatively, cells were stained and NK cell activation (C) or IFNγ production (D) were analyzed by immunofluorescence staining and cytofluorimetric analysis. Data shown are representative of at least 2 independent experiments and presented as mean ± SD of 3 experimental replicates. The Mann-Whitney test was used to compare differences between WT and TLR3-/- NK cells; **P < 0.01; ***P < 0.001).
NK cell development and killing activity are not affected by TLR3 deficiency
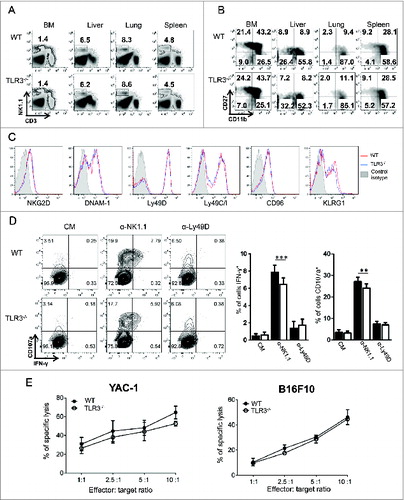
The decreased ability of NK cells from Tlr3−/− mice to produce cytokines could have been caused by a defect in NK cell development. To explore this possibility, NK cell numbers and maturation status were analyzed by flow cytometry. Tlr3−/− and WT mice showed similar percentages of NK cells in every organ analyzed, including bone marrow, liver, lung and spleen (). In addition, the absence of TLR3 did not affect NK cell maturation status, as determined by the combined expression of CD11b and CD27Citation31 (). Moreover, WT and Tlr3−/− NK cells expressed identical levels of NK cell activating (NKG2D, Ly49D, DNAM-1) or inhibitory (Ly49C/I, CD96, KLRG1) receptors (). We found that NK1.1 crosslinking induced IFNγ production and degranulation of Tlr3−/− NK cells, even though the response was slightly decreased compared to WT NK cells (). Very low degranulation was observed following anti-Ly49D stimulation, with no difference between WT and Tlr3−/− NK cells. Finally, we analyzed the ability of NK cells from Tlr3−/− mice to kill target cells in vitro. TLR3 deficiency did not affect NK cell cytotoxic activity against YAC-1 or B16F10 tumor cells (). Taken together, these results demonstrate that NK cells develop, mature and acquire killing capacities independently of TLR3. However, TLR3 is probably required at later stages to condition mature NK cells to release high levels of pro-inflammatory cytokines upon stimulation and to be fully responsive to NK1.1 ligation.
Figure 2. TLR3 is not required for NK cell development and acquisition of killing capacity. Bone marrow (BM), liver, lung and spleen from wild-type (WT) and Toll-like receptor 3 null (Tlr3−/−) mice were processed and the percentages (A) and maturation status (B) of natural killer (NK) cells (CD3− NK1.1+) were determined by flow cytometry. (C) Expression of activating and inhibitory receptors on splenic NK cells from WT (red) or Tlr3−/− mice (blue). Data shown are representative of 2 independent experiments each consisting of 3 mice per group. (D, E) Purified splenic NK cells from WT or Tlr3−/− mice were cultured for 3–5 d together with 10 ng/mL of IL-15/IL-15Rα. (D) NK cells were incubated in anti-NK1.1 or anti-Ly49D coated plates for 6 h and degranulation (measured by CD107a exposure) and IFNγ production were assessed by flow cytometry. Representative dot plots are depicted and graphs represent mean ± SD of pooled data from 2 (anti-Ly49D) to 3 (anti-NK1.1) experiments done in 3 to 5 experimental replicates. The Mann-Whitney test was used to compare difference between WT and TLR3-/- NK cells. Black bars: WT, White bars: TLR3-/-. (E) Cytotoxic activity against YAC-1 or B16F10 cells was determined in a standard 4 h 51Cr release assay. Graphs represent mean ± SD of triplicates from one experiment representative of 3 independent experiments.
NK cell production of high levels of IFNγ requires TLR3 expression on hematopoietic cells
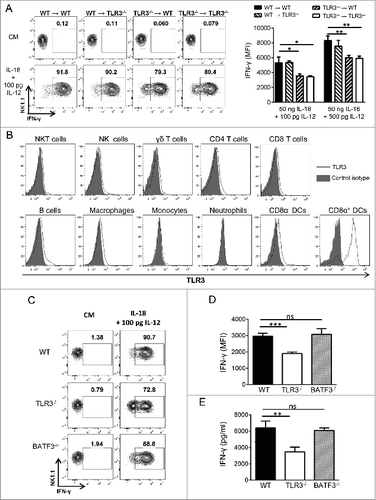
TLR3 is expressed on a wide range on immune cells but also on non-immune cells, including fibroblasts and epithelial cells.Citation2 To determine the contribution of TLR3 expression on non-hematopoietic cells to NK cell conditioning, we performed bone marrow chimera experiments in which lethally irradiated WT or Tlr3−/− mice were reconstituted with bone marrow cells from WT or Tlr3−/− mice. Eight weeks later, NK cells were purified from these mice and stimulated with IL-12/IL-18 (). As expected, NK cells obtained from Tlr3−/− mice reconstituted with Tlr3−/− bone marrow produced much less IFNγ than NK cells from WT mice reconstituted with WT bone marrow. Interestingly, NK cells from Tlr3−/− mice reconstituted with WT bone marrow were proficient in IFNγ production, whereas lower IFNγ levels were produced by NK cells from WT mice reconstituted with Tlr3−/− bone marrow. These data indicate that selective expression of TLR3 on immune cells is necessary and sufficient for the pre-conditioning of NK cells to cytokine stimulation.
Figure 3. TLR3 expression on immune cells other than CD8α DCs is required for the acquisition of full NK cell functions. (A) Lethally irradiated wild-type (WT) mice were reconstituted with bone marrow (BM) cells from WT or Toll-like receptor 3 null (Tlr3−/−) mice. Alternatively, lethally irradiated Tlr3−/− mice received BM cells from WT or Tlr3−/− mice. Eight weeks later, natural killer (NK) cells were isolated from the spleen, cultured for 16 to 20 h together with 50 ng/mL of rIL-18 and variable concentrations of rIL-12. IFNγ production was determined by immunofluorescence staining and flow cytometry. Graphs and dot plots showing mean ± SD of experimental triplicates from one representative experiment out of 2 are depicted. (B) TLR3 expression by splenic natural killer T (NKT) cells (TCRβ+ CD1d/α-GalCer tetramer+), NK cells (CD3− NK1.1+), γδ T cells (TCRγδ+ CD3+), CD4 T cells (TCRβ+ CD4+), CD8 T cells (TCRβ+ CD8+), B cells (CD19+ B220+), macrophages (CD11b+ F4/80+), monocytes (CD11b+ Ly6Chigh), neutrophils (CD11b+ Ly6G+), CD8α− DC (CD11c+ MHC-II+ CDα8−) and CD8α+ (CD11c+ MHC-II+ CD8α+) from WT mice was determined by intracellular staining. (C-E) NK cells were purified from the spleen of WT, TLR3−/− and Batf3−/− mice and cultured in the presence of 100 pg/mL of rIL-12 and 50 ng/mL of rIL-18 for 16 h. IFNγ production was determined by flow cytometry (C, D) and by Cytometric Bead Array (CBA) (E). Representative dot plots and graphs showing the mean ± SD of triplicates from one experiment representative of 3 independent experiments are depicted. The Mann-Whitney test was used to compare differences between mice in A, D and E; *P < 0.05; **P < 0.01; ***P < 0.001.
CD8α DCs are not required for TLR3-dependent NK cell conditioning
To get further insight on which immune cells were responsible for TLR3-dependent NK cell conditioning, we performed intracellular TLR3 staining on mouse splenocytes. We found that TLR3 was expressed at much higher levels on CD8α DCs than on other immune cells (). However, NK cells from Batf3−/− mice that lack CD8α DCsCitation32 produced IFNγ to similar levels to those produced by WT NK cells, indicating that CD8α DCs are not necessary for NK cell conditioning (). Taken as a whole, our results indicate that the acquisition of full effector functions by NK cells requires TLR3 expression on immune cells other than CD8α DCs.
Role of the gut-microbiota in TLR3-dependent NK cell conditioning
Although TLR3 is well known for recognizing virus dsRNA,Citation9 the factors driving TLR3 signaling in the absence of infection are unknown. Given that NK cell responses are compromised in germ-free miceCitation33 and that bacterial RNA can potentially activate TLR3,Citation34 we hypothesized that a defective sensing of the microbiota could account for the defective NK cells conditioning in Tlr3−/− mice. To explore this possibility, WT and Tlr3−/− mice were co-housed for at least 6 weeks before testing NK cell IFNγ production (Fig. S2A). NK cells from Tlr3−/− mice remained poor IFNγ producers compared with WT NK cells, even when the mice were co-housed. To confirm these data, we treated Tlr3−/− and WT mice with broad-spectrum antibiotics known to strongly reduce the gut microbiotaCitation35 for 3 to 4 weeks. IFNγ production by Tlr3−/− NK cells was still reduced compared with WT (Fig. S2B). These results strongly indicate that the acquisition of full effector functions by NK cells is independent of bacterial RNA sensing.
TRIF is not necessary for efficient NK cell response to cytokines
The adaptor protein TRIF interacts with the intracellular domain of TLR3Citation36 and poly(IC)-induced responses are abolished in Trif−/− mice.Citation10 However, a TRIF-independent pathway of TLR3 has recently been described.Citation11 Thus, we asked whether TRIF was involved in the TLR3-dependent sensitization of NK cells. For that, NK cells were purified from WT and Trif−/− mice and cultured together with IL-12/IL-18 or IL-15/IL-12. Interestingly, NK cells from both mice secreted comparable levels of IFNγ (Fig. S3A). Similar results were obtained regarding MIP-1α production (Fig. S3B). These data demonstrate that TLR3 signals through a TRIF-independent pathway to regulate NK cell responses to cytokine stimulation.
TLR3 is involved in NKG2D-mediated suppression of tumor progression
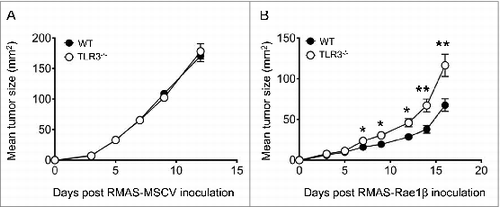
NK cells recognize various molecules upregulated on the surface of transformed cells.Citation28 Among them, ribonucleic acid export 1 (Rae-1) is a stress-induced molecule that triggers the NK cell activating receptor killer cell lectin-like receptor K1 (KLRK1, better known as NKG2D).Citation37 We asked whether TLR3 would be involved in the recognition and/or killing of Rae-1 expressing tumor cells by NK cells. To this aim, WT and Tlr3−/− mice were inoculated subcutaneously with RMAS tumor cells transfected with an empty vector (RMAS-MSCV) or with the Rae-1β molecule (RMAS- Rae-1β). Both mouse strains exhibited equal outgrowth of the RMAS-MSCV control cell line () whereas the growth of RMAS-Rae1β cells was significantly reduced in WT compared with Tlr3−/− mice (). These results demonstrate that TLR3 is also involved in NKG2D-dependent NK cell-mediated tumor suppression.
Figure 4. NKG2D-mediated suppression of tumor is attenuated in Tlr3−/− mice. Wild-type (WT) or Toll-like receptor 3 null (Tlr3−/−) mice were injected s.c. with (A) RMAS-MSCV or (B) RMAS-Rae1β tumor cells and tumor size was recorded. Data shown are representative of 2 independent experiments with 5–10 mice per group and presented as mean ± SEM. The Mann-Whitney test was used to compare differences between mice; **P < 0.01; *P < 0.05.
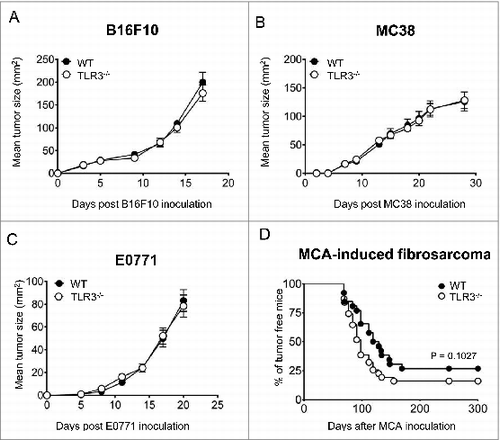
TLR3 is dispensable for the control of primary tumors or carcinogen-induced fibrosarcoma
To further investigate the role of TLR3 in tumor control, we used the B16F10 melanoma and MC38 colon adenocarcinoma syngeneic mouse models. We observed that each cell line had comparable growth rates upon transplantation in either WT or Tlr3−/− mice (). Next, we employed an orthotopic, syngeneic mouse tumor model established by injecting E0771 mammary carcinoma cells into the mammary fat pad. Here again, no difference in tumor growth was observed between WT and Tlr3−/− host mice (). We next sought to explore whether TLR3 signaling could participate in the immunosurveillance of de novo models of tumorigenesis. To this end, we took advantage of a carcinogen-induced fibrosarcoma model in which NK cell-mediated suppression of tumor initiation has been previously demonstrated.Citation38 WT and Tlr3−/− mice were injected with methylcholanthrene (MCA) and monitored for tumor development over 300 d (). Our results indicate a trend toward higher tumor incidence in Tlr3−/− mice but this difference did not reach significance. Overall, our data suggest that TLR3 signaling is not critical for the control of both carcinogenesis and primary tumor growth.
Figure 5. TLR3 is dispensable for the control of primary tumor growth and carcinogenesis. Wild-type (WT) and Toll-like receptor 3 null (Tlr3−/−) mice were injected s.c. with (A) B16F10 or (B) MC38 tumor cells (n = 5 mice per group). (C) Mice were orthotopically injected into the mammary fat pad with E0771 cells (n = 10 mice per group). Data shown are representative of 2 independent experiments and presented as mean ± SEM. (D) WT and Tlr3−/− mice were injected with 100 µg of MCA and subsequently monitored for tumor development over 300 d. Results are pooled from 2 independent experiments with a total of 26 WT and 31 Tlr3−/− mice. Statistical differences in tumor incidence were determined by Mantel Cox Log-rank test.
TLR3 protects against experimental metastasis through NK cells and IFNγ
Previous studies have established a fundamental role of NK cells in the control of B16F10 experimental metastasis.Citation39 This prompted us to investigate the role of TLR3 in this model. We found that Tlr3−/− mice displayed significantly higher numbers of lung metastases than WT mice following intravenous inoculation of B16F10 cells (). Thus, despite a negligible effect upon primary tumor growth, TLR3 controls the metastatic spread of B16F10 cells. By contrast, carcinogenesis, primary tumor growth and metastasis were unaffected in the Trif−/− mice (Sup. ). These experiments establish the importance of TRIF-independent signaling of TLR3 in the control of experimental B16F10 metastasis.
Figure 6. TLR3 participates in the control of metastasis in a NK cell- and IFNγ-dependent manner. (A-C) Wild-type (WT) and Toll-like receptor 3 null (Tlr3−/−) mice were challenged i.v. with B16F10 melanoma cells. For some experiments, mice were treated with natural killer (NK) cell-depleting anti-asialo-GM1 antibodies (B) or blocking antibodies against IFNγ (C). Symbols represent the number of lung metastases for individual mice 14 d following tumor inoculation. The Mann-Whitney test was used to compare differences between mice; **P < 0.01).
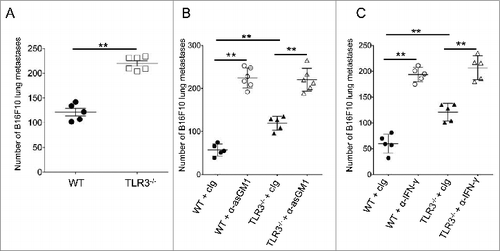
Finally, we aimed to investigate the mechanisms by which TLR3 controls metastasis. In particular, we wondered whether NK cells and the secretion of IFNγ were involved in this process. Mice injected intravenously with B16F10 cells were treated with anti-asialo GM1 antibodies (Abs) or anti-IFNγ Abs to deplete NK cells or neutralize IFNγ, respectively. Both treatments significantly increased metastatic burden in WT and Tlr3−/− mice (). These data confirm the role of NK cells and IFNγ in protecting against B16F10 metastasisCitation39,40 and indicate that these 2 components are still protective in the absence of TLR3 signaling. Importantly, no significant difference was observed between WT and Tlr3−/− mice when those mice were treated with anti-asialo GM1 or anti-IFNγ Ab, suggesting that the TLR3-mediated control of B16F10 metastasis requires both NK cells and IFNγ production. Altogether, these data demonstrate the importance of the TLR3 signaling pathway for the protection against experimental metastasis and establish that both NK cells and IFNγ secretion are necessary for such protection.
Discussion
The present study highlights the critical role played by TLR3 signaling in the immunosurveillance of cancers and metastases. Importantly we showed that in the absence of administration of any agonist, TLR3 considerably increased NK cell production of pro-inflammatory cytokines and protected against experimental metastasis.
NK cells are innate effector cells that were initially thought to recognize and eliminate their targets with fast kinetics, without prior sensitization. However, NK cell activity is tightly regulated because only NK cells that can engage self-MHC become fully competent, a processed called licensing.Citation41 In addition, similar to T cells, NK cells need to be primed to acquire full effector functions.Citation42 Here, we extended those concepts by demonstrating that constant triggering of TLR3 keeps mature NK cells in a “ready to go” state. This process takes place at steady-state and thus differs from NK cell-priming that occurs in a highly inflammatory context and from NK cell-licensing that ensures self-tolerance during NK cell development. Hence, we propose that TLR3 allows the conditioning of NK cells, an essential step to ensure immediate and potent NK cell responses to cytokine stimulation.
Data from the literature indicate that, unlike human NK cells,Citation5,6,30 purified mouse NK cells fail to respond to poly(I:C) in vitro.Citation23,44 Thus, even if our results did not formally exclude a direct role of TLR3 signaling on NK cells, it is probable that accessory cells are required for TLR3-dependent NK cell conditioning. Using BM-chimera mice, we demonstrated that the expression of TLR3 on hematopoietic cells is necessary and sufficient for the acquisition of full NK cell functions. These data are reminiscent of results showing that TLR3 signaling in BM-derived accessory cells, but not in stromal cells, drives NK cell activation.Citation22 Flow cytometry analyses established that TLR3 protein is preferentially expressed in CD8α DCs, a finding consistent with the high levels of TLR3 mRNA in these cells.Citation23,43 It has been previously described that CD8α DCs co-cultured with NK cells induce IFNγ production in response to poly(I:C).Citation23 However, the use of Baft3−/− mice (that lack CD8α DCs) excluded a predominant role for this DC subset in NK cell conditioning. It is noteworthy that such a negligible role of CD8α DCs has been previously reported following in vivo administration of poly(I:C).Citation22 Further work is needed to identify the cell type(s) necessary for TLR3-dependent conditioning of NK cells. Macrophages constitute a likely candidate since they have been found to induce NK cell IFNγ production when stimulated with poly(I:C).Citation44
IL-18 has been reported to regulate the ability of NK cells to produce IFNγ in response to cytokine stimulation.Citation45 An attractive hypothesis is that TLR3 signaling controls IL-18 production by accessory cells. However, we measured similar levels of IL-18 in the sera of naïve WT and Tlr3−/− mice (data not shown). In addition, we failed to detect any production of TNFα or IL-12 p70 in the sera of naïve mice. However, these data do not exclude the possibility that cytokine levels are regulated locally in a TLR3-dependent manner.
Our result that NK cells are hyporesponsive in Tlr3−/− mice suggests that repetitive stimulation by TLR3-ligand(s) controls NK cell activity. The ligands required for proper NK cell conditioning are still unknown. Although defective sensing of commensal bacterial mRNA could have explained NK cell hyporesponsiveness,Citation33 our data obtained in co-housed or antibiotic treated mice excluded a role of the gut microbiota. Interestingly, endogenous mRNA released from necrotic cells has the potential to activate TLR3.Citation46,47 Therefore, NK cell responsiveness could be modulated by TLR3-sensing of host-derived mRNAs. Alternatively, TLR3 could recognize nucleic acids from endogenous retrovirus.Citation48 Eventually, TLR3 might interact with other signaling pathways in a ligand-independent manner. Future studies should aim to define the nature of the ligand(s) recognized by TLR3, if any.
The absence of phenotype in Trif−/− mice despite defective NK cell responses and increased metastasis in Tlr3−/− mice was somehow surprising. Indeed, TLR3 was initially believed to signal exclusively through the adaptor protein TRIF.Citation10 However, an interesting study highlighted a new branch of TLR3 signaling that is TRIF-independent but does require the non-receptor tyrosine kinase Src.Citation11 TRIF-independent response to TLR3 stimulation affected the migration, adhesion and proliferation of multiple cell types and did not require gene-induction. Thus, although TRIF is essential for TLR3-mediated response to virus or poly(I:C) leading to IRF3 and NF-kB activation, another facet of TLR3-signaling may regulate a different kind of response in a TRIF-independent manner. It is tempting to speculate that distinct pathways of TLR3 signaling could be triggered in response to different signals (e.g., endogenous mRNA versus viral mRNA) or in different contexts (e.g., inflammation vs. homeostasis). Further investigation is warranted to determine whether the Src pathway or another yet unknown pathway is responsible for the TLR3-mediated control of NK cell reactivity.
Despite substantive data emphasizing the anticancer properties of poly (I:C),Citation16 it was previously unclear whether TLR3 could promote immunosurveillance without administration of its agonist. Interestingly, one study reported a role for endogenous TLR3 in restricting the primary growth of transplantable tumors.Citation20 The present work broadens TLR3 function to the control of Rae1β-expressing tumor cells and restriction of metastasis. Conversely, we found no significant contribution of TLR3 to carcinogenesis in the MCA-induced fibrosarcoma model. Moreover, the presence of TLR3 did not influence primary growth of subcutaneous tumors that did not express NKG2D-ligand. Hence, the tumor microenvironment probably determines the importance of TLR3 signaling for the induction of efficient antitumor immune responses. The availability of TLR3 ligands and the presence TLR3-expressing cells at the tumor site are likely to be determinant factors for TLR3 function.
Intriguingly, the tumor models controlled by TLR3 (e.g. RMAS-Rae1β tumor growth and B16F10 metastasis) have been previously shown to be suppressed by NK cells.Citation37,49,50 Despite normal in vitro NK cell cytotoxic activity and expression levels of NKG2D in Tlr3−/− mice, TLR3 deficiency may affect NK cell tumoricidal abilities in vivo. Alternatively, the defect in NK cells conditioning we observed might account for the higher sensitivity of Tlr3−/− mice to tumor challenge. We can hypothesize that cytokines present in the tumor microenvironment stimulate NK cell IFNγ production in WT mice, but hyporesponsive NK cells in Tlr3−/− mice would fail to secrete sufficient IFNγ. Our finding that protection against experimental B16F10 metastasis in WT mice requires both NK cells and IFNγ support this hypothesis. Nevertheless, the whole picture must be vastly more complex and probably includes several feedback loops and interactions between the various actors of the immune system. For instance, TLR3 signaling stimulates DCs or macrophages to prime NK cells.Citation22,44 In turn, NK cells regulate DC functions as well as T-cell responses.Citation29,51 Thus, TLR3 may control the priming of innate responses (of NK cells more particularly), further necessary to turn on adaptive immunity against cancer.
Overall, we provide evidence that TLR3 is involved in the NK cell-mediated suppression of experimental tumor metastasis. Notably, this study demonstrates a role for TLR3 in cancer immunosurveillance in the absence of exogenous application of its agonist. However, metastases development in TLR3 competent-hosts suggests that either TLR3 itself is insufficient to prevent metastatic spread or alternatively, that some mechanisms allow a proportion of malignant cells to escape TLR3-dependent immune responses. Indeed, one attractive possibility would be that tumors could modulate the availability of TLR3 ligands and/or TLR3 expression to escape the immune system. Our data provide clues to understand how the lack of TLR3 signaling down-regulates NK cell functions and leads to defective immune responses unable to constrain metastatic disease.
Materials and Methods
Mice
C57BL/6 WT mice were purchased from the Walter and Eliza Hall Institute of Medical Research (Victoria, Australia). C57BL/6 Tlr3−/−, C57BL/6 Trif−/− and C57BL/6 Batf3−/− mice were bred in house at the Peter MacCallum Cancer Center (Victoria, Australia) and then at the QIMR Berghofer Medical Research Institute (Queensland, Australia). Mice were used at 6-12 weeks of age and housed in specific pathogen-free conditions.
Antibodies and reagents
The flow cytometry antibodies anti-CD69 (H1.2F3), anti-CD107a (1D4B) and anti-NKp46 (29A1.4) were purchased from Biolegend. Anti-IFN-γ (XMG1.2), anti-NK1.1 (PK136), anti-CD3ξ (145–2C11) and anti-TCR-β (H57–597) antibodies were obtained from eBioscience. The BD Bioscience Cytofix/Cytoperm kit was used for intracellular staining. Recombinant IL-12p70 and IL-15 and IL-15/IL15Rα were purchased from eBioscience and recombinant IL-18 was obtained from R&D systems.
In vitro stimulation assays
NK cells were enriched using the NK cell isolation kit II mouse from Miltenyi Biotec and then purified using a FACS Aria II (BD Biosciences) and cultured with the indicated concentrations of cytokines or 10 ng/mL of PMA together with 1 μg/mL of ionomycin. After 16 h, cells were stained for flow cytometry analysis and/or supernatants were collected. Measurements of cytokine/chemokine production were performed using a Cytometric Bead Array (CBA) kit (BD Biosciences). Alternatively, NK cells were cultured for 4–5 d together with 10 ng/mL of IL-15/IL-15Rα (eBioscience). Then, NK cells were incubated in plates coated with anti-NK1.1 (clone PK136) or anti-Ly49D (clone 4E5) together with anti-CD107a antibodies and Golgi Plug and Golgi Stop (BD Bioscience). After 6 h, NK cells were stained for cytofluorimetric analysis.
In vitro cytotoxicity assay
Target cells were labeled with 100 µCi of 51Cr (Perkin Elmer) and co-cultured with IL15 stimulated NK cells at defined effector-to-target cell ratios. After 4 h incubation at 37°C, supernatants were harvested and the levels of 51Cr were quantified by gamma counter (Wallac Wizard). The percentage of specific killing was determined using the following formula: (Sample 51Cr release – spontaneous 51Cr release) / (Maximum 51Cr release – Spontaneous 51Cr release) × 100.
Organ processing
Lungs and livers were perfused with PBS to remove peripheral blood and incubated with 1 mg/mL collagenase type IV (Sigma-Aldrich), 0.1 mg/mL hyaluronidase (Sigma-Aldrich) and 0.02 mg/mL DNase I (Roche) for 45 min at 37°C. Cell suspensions obtained from spleen, BM, lungs and livers were treated with ammonium chloride potassium (ACK) lysis buffer to remove erythrocytes.
Bone marrow chimera
WT or Tlr3−/− mice received 2 irradiation doses of 552 cGy before being injected i.v. with 10 × 106 total BM cells from WT or Tlr3−/− mice. Mice were treated with neomycin for 2 weeks and used for experiments 8 weeks after reconstitution.
Antibiotic treatment
WT or Tlr3−/− mice were treated with 1g/L ampicillin, streptomycin and vancomycin, and 0.5 g/L metronidazol ad libidum for 3–4 weeks via the drinking water. Water containing antibiotic was exchanged every 3 d
Tumor models
For the MCA-induced fibrosarcoma model, mice were injected sc with 100 µL of corn oil containing 100 µg of MCA (Sigma-Aldrich) on the right hind flank. Development of fibrosarcoma was monitored weekly. For primary tumor growth 1 × 105 B16F10, 1 × 106 MC38, 1 × 106 RMAS-MSV or 1 × 106 RMAS-Rae1β cells were resuspended in 100 µL of PBS and injected s.c. on the right hind flank whereas 2 × 105 E0771 cells were injected into the 4th mammary fatpad. Tumor measurement was recorded as the product of 2 perpendicular diameters (mm2). For experimental metastasis, mice were injected i.v. with 2 × 105 B16F10 resupended in 200 µL of PBS and 14 d later lung metastases were counted using a dissection microscope. For some experiments, mice were treated with anti-asialo-GM1 (Wako Chemicals), anti-IFNγ (H22 from Robert Schreiber, Washington School of Medicine, St Louis, MO, USA) or IgG control antibodies (100–250 µg i.p.) on the day prior to tumor injection, the day of tumor inoculation, and then once a week thereafter.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
SUPPLEMENTAL MATERIAL
Supplemental data for this article can be accessed on the publisher's website.
Supplemental_Material.docx
Download MS Word (184.2 KB)Acknowledgments
The authors wish to thank Liam Town, Kate Elder and Joanne Sutton for breeding, genotyping and maintenance and care of the mice used in this study.
Funding
MJS was supported by a NH&MRC Australia Fellowship (628623).
References
- Janeway CA, Jr., Medzhitov R. Innate immune recognition. Annu Rev Immunol 2002; 20:197-216; PMID:11861602; http://dx.doi.org/10.1146/annurev.immunol.20.083001.084359.
- Matsumoto M, Seya T. TLR3: interferon induction by double-stranded RNA including poly(I:C). Adv Drug Deliv Rev 2008; 60:805-12; PMID:18262679; http://dx.doi.org/10.1016/j.addr.2007.11.005.
- Muzio M, Bosisio D, Polentarutti N, D'Amico G, Stoppacciaro A, Mancinelli R, van't Veer C, Penton-Rol G, Ruco LP, Allavena P, et al. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J Immunol 2000; 164:5998-6004; http://dx.doi.org/10.4049/jimmunol.164.11.5998.
- Heinz S, Haehnel V, Karaghiosoff M, Schwarzfischer L, Muller M, Krause SW, Rehli M. Species-specific regulation of Toll-like receptor 3 genes in men and mice. J Biol Chem 2003; 278:21502-9; PMID:12672806; http://dx.doi.org/10.1074/jbc.M301476200.
- Schmidt KN, Leung B, Kwong M, Zarember KA, Satyal S, Navas TA, Wang F, Godowski PJ. APC-independent activation of NK cells by the Toll-like receptor 3 agonist double-stranded RNA. J Immunol 2004; 172:138-43; http://dx.doi.org/10.4049/jimmunol.172.1.138.
- Girart MV, Fuertes MB, Domaica CI, Rossi LE, Zwirner NW. Engagement of TLR3, TLR7, and NKG2D regulate IFN-gamma secretion but not NKG2D-mediated cytotoxicity by human NK cells stimulated with suboptimal doses of IL-12. J Immunol 2007; 179:3472-9; http://dx.doi.org/10.4049/jimmunol.179.6.3472.
- Matsumoto M, Kikkawa S, Kohase M, Miyake K, Seya T. Establishment of a monoclonal antibody against human Toll-like receptor 3 that blocks double-stranded RNA-mediated signaling. Biochem Biophys Res Commun 2002; 293:1364-9; PMID:12054664; http://dx.doi.org/10.1016/S0006-291X(02)00380-7.
- Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun 2000; 68:7010-7; PMID:11083826; http://dx.doi.org/10.1128/IAI.68.12.7010-7017.2000.
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001; 413:732-8; PMID:11607032; http://dx.doi.org/10.1038/35099560.
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003; 301:640-3; PMID:12855817; http://dx.doi.org/10.1126/science.1087262.
- Yamashita M, Chattopadhyay S, Fensterl V, Zhang Y, Sen GC. A TRIF-independent branch of TLR3 signaling. J Immunol 2012; 188:2825-33; http://dx.doi.org/10.4049/jimmunol.1103220.
- Rudd BD, Smit JJ, Flavell RA, Alexopoulou L, Schaller MA, Gruber A, Berlin AA, Lukacs NW. Deletion of TLR3 alters the pulmonary immune environment and mucus production during respiratory syncytial virus infection. J Immunol 2006; 176:1937-42; http://dx.doi.org/10.4049/jimmunol.176.3.1937.
- Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci U S A 2004; 101:3516-21; PMID:14993594; http://dx.doi.org/10.1073/pnas.0400525101.
- Levy HB, Law LW, Rabson AS. Inhibition of tumor growth by polyinosinic-polycytidylic acid. Proc Natl Acad Sci U S A 1969; 62:357-61; PMID:4894326; http://dx.doi.org/10.1073/pnas.62.2.357.
- Forte G, Rega A, Morello S, Luciano A, Arra C, Pinto A, Sorrentino R. Polyinosinic-polycytidylic acid limits tumor outgrowth in a mouse model of metastatic lung cancer. J Immunol 2012; 188:5357-64; http://dx.doi.org/10.4049/jimmunol.1103811.
- Sharma S, Zhu L, Davoodi M, Harris-White M, Lee JM, St John M, Salgia R, Dubinett S. TLR3 agonists and proinflammatory antitumor activities. Expert Opin Ther Targets 2013; 17:481-3; PMID:23506058; http://dx.doi.org/10.1517/14728222.2013.781585.
- Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol 2006; 176:4894-901; http://dx.doi.org/10.4049/jimmunol.176.8.4894.
- Paone A, Starace D, Galli R, Padula F, De Cesaris P, Filippini A, Ziparo E, Riccioli A. Toll-like receptor 3 triggers apoptosis of human prostate cancer cells through a PKC-α-dependent mechanism. Carcinogenesis 2008; 29:1334-42; PMID:18566014; http://dx.doi.org/10.1093/carcin/bgn149.
- Shime H, Matsumoto M, Oshiumi H, Tanaka S, Nakane A, Iwakura Y, Tahara H, Inoue N, Seya T. Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proc Natl Acad Sci U S A 2012; 109:2066-71; PMID:22308357; http://dx.doi.org/10.1073/pnas.1113099109.
- Chin AI, Miyahira AK, Covarrubias A, Teague J, Guo B, Dempsey PW, Cheng G. Toll-like receptor 3-mediated suppression of TRAMP prostate cancer shows the critical role of type I interferons in tumor immune surveillance. Cancer Res 2010; 70:2595-603; PMID:20233880; http://dx.doi.org/10.1158/0008-5472.CAN-09-1162.
- Akazawa T, Ebihara T, Okuno M, Okuda Y, Shingai M, Tsujimura K, Takahashi T, Ikawa M, Okabe M, Inoue N, et al. Antitumor NK activation induced by the Toll-like receptor 3-TICAM-1 (TRIF) pathway in myeloid dendritic cells. Proc Natl Acad Sci U S A 2007; 104:252-7; PMID:17190817; http://dx.doi.org/10.1073/pnas.0605978104.
- McCartney S, Vermi W, Gilfillan S, Cella M, Murphy TL, Schreiber RD, Murphy KM, Colonna M. Distinct and complementary functions of MDA5 and TLR3 in poly(I:C)-mediated activation of mouse NK cells. J Exp Med 2009; 206:2967-76; PMID:19995959; http://dx.doi.org/10.1084/jem.20091181.
- Miyake T, Kumagai Y, Kato H, Guo Z, Matsushita K, Satoh T, Kawagoe T, Kumar H, Jang MH, Kawai T, et al. Poly I:C-induced activation of NK cells by CD8 α+ dendritic cells via the IPS-1 and TRIF-dependent pathways. J Immunol 2009; 183:2522-8; http://dx.doi.org/10.4049/jimmunol.0901500.
- Chew V, Tow C, Huang C, Bard-Chapeau E, Copeland NG, Jenkins NA, Weber A, Lim KH, Toh HC, Heikenwalder M, et al. Toll-like receptor 3 expressing tumor parenchyma and infiltrating natural killer cells in hepatocellular carcinoma patients. J Natl Cancer Inst 2012; 104:1796-807; PMID:23197495; http://dx.doi.org/10.1093/jnci/djs436.
- Hsu WM, Huang CC, Wu PY, Lee H, Huang MC, Tai MH, Chuang JH. Toll-like receptor 3 expression inhibits cell invasion and migration and predicts a favorable prognosis in neuroblastoma. Cancer Lett 2013; 336:338-46; PMID:23541683; http://dx.doi.org/10.1016/j.canlet.2013.03.024.
- Fernandez-Garcia B, Eiro N, Gonzalez-Reyes S, Gonzalez L, Aguirre A, Gonzalez LO, Del Casar JM, Garcia-Muniz JL, Vizoso FJ. Clinical significance of toll-like receptor 3, 4, and 9 in gastric cancer. J Immunother 2014; 37:77-83; PMID:24509170; http://dx.doi.org/10.1097/CJI.0000000000000016.
- Chan CJ, Andrews DM, Smyth MJ. Can NK cells be a therapeutic target in human cancer? Eur J Immunol 2008; 38:2964-8; PMID:18979512; http://dx.doi.org/10.1002/eji.200838764.
- Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science 2011; 331:44-9; PMID:21212348; http://dx.doi.org/10.1126/science.1198687.
- Degli-Esposti MA, Smyth MJ. Close encounters of different kinds: dendritic cells and NK cells take centre stage. Nat Rev Immunol 2005; 5:112-24; PMID:15688039; http://dx.doi.org/10.1038/nri1549.
- Sivori S, Falco M, Carlomagno S, Romeo E, Moretta L, Moretta A. Heterogeneity of TLR3 mRNA transcripts and responsiveness to poly (I:C) in human NK cells derived from different donors. Int Immunol 2007; 19:1341-8; PMID:17962643; http://dx.doi.org/10.1093/intimm/dxm105.
- Hayakawa Y, Smyth MJ. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J Immunol 2006; 176:1517-24; http://dx.doi.org/10.4049/jimmunol.176.3.1517.
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science 2008; 322:1097-100; PMID:19008445; http://dx.doi.org/10.1126/science.1164206.
- Ganal SC, Sanos SL, Kallfass C, Oberle K, Johner C, Kirschning C, Lienenklaus S, Weiss S, Staeheli P, Aichele P, et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity 2012; 37:171-86; PMID:22749822; http://dx.doi.org/10.1016/j.immuni.2012.05.020.
- Kariko K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005; 23:165-75; PMID:16111635; http://dx.doi.org/10.1016/j.immuni.2005.06.008.
- Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, Merad M. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 2014; 343:1249288; PMID:24625929; http://dx.doi.org/10.1126/science.1249288.
- Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-β induction. Nat Immunol 2003; 4:161-7; PMID:12539043; http://dx.doi.org/10.1038/ni886.
- Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci U S A 2001; 98:11521-6; PMID:11562472; http://dx.doi.org/10.1073/pnas.201238598.
- Smyth MJ, Crowe NY, Godfrey DI. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol 2001; 13:459-63; PMID:11282985; http://dx.doi.org/10.1093/intimm/13.4.459.
- Smyth MJ, Taniguchi M, Street SE. The anti-tumor activity of IL-12: mechanisms of innate immunity that are model and dose dependent. J Immunol 2000; 165:2665-70; http://dx.doi.org/10.4049/jimmunol.165.5.2665.
- Jiang Q, Wei H, Tian Z. IFN-producing killer dendritic cells contribute to the inhibitory effect of poly I:C on the progression of murine melanoma. J Immunother 2008; 31:555-62; PMID:18528299; http://dx.doi.org/10.1097/CJI.0b013e31817d8e75.
- Jonsson AH, Yokoyama WM. Natural killer cell tolerance licensing and other mechanisms. Adv Immunol 2009; 101:27-79; PMID:19231592; http://dx.doi.org/10.1016/S0065-2776(08)01002-X.
- Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 2007; 26:503-17; PMID:17398124; http://dx.doi.org/10.1016/j.immuni.2007.03.006.
- Edwards AD, Diebold SS, Slack EM, Tomizawa H, Hemmi H, Kaisho T, Akira S, Reis e Sousa C. Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 α+ DC correlates with unresponsiveness to imidazoquinolines. Eur J Immunol 2003; 33:827-33; PMID:12672047; http://dx.doi.org/10.1002/eji.200323797.
- Kasamatsu J, Azuma M, Oshiumi H, Morioka Y, Okabe M, Ebihara T, Matsumoto M, Seya T. INAM plays a critical role in IFN-gamma production by NK cells interacting with polyinosinic-polycytidylic acid-stimulated accessory cells. J Immunol 2014; 193:5199-207; http://dx.doi.org/10.4049/jimmunol.1400924.
- Chaix J, Tessmer MS, Hoebe K, Fuseri N, Ryffel B, Dalod M, Alexopoulou L, Beutler B, Brossay L, Vivier E, et al. Cutting edge: Priming of NK cells by IL-18. J Immunol 2008; 181:1627-31; http://dx.doi.org/10.4049/jimmunol.181.3.1627.
- Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem 2004; 279:12542-50; PMID:14729660; http://dx.doi.org/10.1074/jbc.M310175200.
- Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, Hogaboam CM, Kunkel SL. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med 2008; 205:2609-21; PMID:18838547; http://dx.doi.org/10.1084/jem.20081370.
- Yu P, Lubben W, Slomka H, Gebler J, Konert M, Cai C, Neubrandt L, Prazeres da Costa O, Paul S, Dehnert S, et al. Nucleic acid-sensing Toll-like receptors are essential for the control of endogenous retrovirus viremia and ERV-induced tumors. Immunity 2012; 37:867-79; PMID:23142781; http://dx.doi.org/10.1016/j.immuni.2012.07.018.
- Teng MW, Andrews DM, McLaughlin N, von Scheidt B, Ngiow SF, Moller A, Hill GR, Iwakura Y, Oft M, Smyth MJ. IL-23 suppresses innate immune response independently of IL-17A during carcinogenesis and metastasis. Proc Natl Acad Sci U S A 2010; 107:8328-33; PMID:20404142; http://dx.doi.org/10.1073/pnas.1003251107.
- Takeda K, Nakayama M, Sakaki M, Hayakawa Y, Imawari M, Ogasawara K, Okumura K, Smyth MJ. IFN-gamma production by lung NK cells is critical for the natural resistance to pulmonary metastasis of B16 melanoma in mice. J Leukoc Biol 2011; 90:777-85; PMID:21712396; http://dx.doi.org/10.1189/jlb.0411208.
- Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol 2008; 9:503-10; PMID:18425107; http://dx.doi.org/10.1038/ni1582.