
Abstract
Immunogenic cell death (ICD) offers interesting opportunities in cancer cell (CC) vaccine manufacture, as it increases the immunogenicity of the dead CC. Furthermore, fusion of CCs with dendritic cells (DCs) is considered a superior method for generating whole CC vaccines. Therefore, in this work, we determined in naive mice whether immunogenically killed CCs per se (CC vaccine) elicit an antitumoral immune response different from the response observed when immunogenically killed CCs are associated with DCs through fusion (fusion vaccine) or through co-incubation (co-incubation vaccine). After tumor inoculation, the type of immune response in the prophylactically vaccinated mice differed between the groups. In more detail, fusion vaccines elicited a humoral anticancer response, whereas the co-incubation and CC vaccine mainly induced a cellular response. Despite these differences, all three approaches offered a prophylactic protection against tumor development in the murine mammary carcinoma model.
In summary, it can be concluded that whole CC vaccines based on immunogenically killed CCs may not necessarily require association with DCs to elicit a protective anticancer immune response. If this finding can be endorsed in other cancer models, the manufacture of CC vaccines would greatly benefit from this new insight, as production of DC-based vaccines is laborious, time-consuming and expensive.
Abbreviations
| CC | = | cancer cell |
| CC-DC hybrid | = | cell formed through merger between a CC and a DC |
| CM | = | culture medium |
| CRT | = | calreticulin |
| CTL | = | cytotoxic T lymphocyte |
| DAMP | = | damage-associated molecular pattern |
| DC | = | dendritic cell |
| DC-CM | = | culture medium for dendritic cells |
| DMEM | = | Dulbecco's modified Eagle's medium |
| DPBS | = | Dulbecco's Phosphate-Buffered Solution |
| FACS | = | fluorescence-assisted cell sorting |
| HSP70 | = | heat shock protein 70 |
| ICD | = | immunogenic cell death |
| MHC | = | major histocompatibility complex |
| MTX | = | Mitoxantrone |
| PEG | = | polyethylene glycol |
| PRRs | = | pattern recognition receptors |
| s.c. | = | subcutaneous |
| SFM | = | serum-free medium |
| Th | = | T helper cell. |
Introduction
Cancer vaccines have emerged as a new therapeutic modality for cancer,Citation1 which aim to re-educate the immune system to recognize and eliminate CCs.Citation2 An additional aim of cancer vaccines is to create an immune memory in order to prevent cancer recurrence. As a single modality, cancer vaccines will in most cases be unable to eradicate established tumors.Citation3 However, they can reduce the risk of cancer recurrence,Citation4 increase survival timeCitation5,6 and induce regression of metastases.Citation7,8
One can divide cancer vaccines into (whole) cancer cell vaccines, genetic vaccines and peptide or protein vaccines.Citation9 There are some strong arguments to consider whole CC vaccines as superior to most other cancer vaccine types.Citation9 First of all, there is no need to identify tumor antigens or select immunodominant epitopes on the tumor antigens for vaccine generation. Furthermore, a great variety of antigens is offered by whole CCs which evoke a major histocompatibility complex (MHC)-independent broad-ranged anticancer response. Additionally, the CCs contain epitopes for parallel presentation to both CD8+ and CD4+ T cells, which greatly diminishes the risk of tumor escape. Autologous CCs have the added advantage of containing patient-specific unique mutated antigens. Furthermore, whole CC vaccines have shown a higher objective clinical response ratio than peptide, protein or genetic vaccines.Citation10-13 Despite the observed tumor eradication in experimental animal studies, limited success has been obtained so far in clinical trials for whole CC vaccines in humans.Citation13,14
Various forms of CC vaccines exist and those loaded ex vivo with DCs (DC-based vaccines) are considered superior to non-DC-based vaccines in stimulating anticancer immunity in vivoCitation15 as antigen presentation is facilitated in the former.Citation16 DC-based vaccines can be generated through various techniques. DCs can be loaded through co-incubation with different types of tumor antigens, namely, whole CCs, tumor lysate, apoptotic CCs, peptides, transfection of RNACitation10 or DNACitation17 or, alternatively, they can be fused with CCs.Citation18 DCs fused to CCs elicit levels of DC maturation and tumor-specific T cell activation that are superior to the levels obtained by DCs co-incubated or pulsed with CCs or tumor lysate, respectively.Citation8,18 In spite of these advantages, fusion vaccines are not common practice, since fusion is the most complicated method of loading DCs with CCs and has a low production efficiency.Citation8
The immunogenicity of whole CCs in vaccines can be increased through ICD.Citation19 In analogy, vaccines based on DCs pulsed with apoptotic cells are considered superior to those pulsed with whole cell lysate or necrotic cells.Citation18,20 ICD can be induced by chemical agents such as anthracyclines (e.g. Mitoxantrone – MTX) or by physical stimuli (e.g., UV light or γ-irradiation). The main characteristic of ICD-inducing stimuli is their ability to induce the expression or release of danger (or damage)-associated molecular patterns (DAMPs) from the killed cells. The DAMPs that are expressed or released by the CCs after ICD can interact with pattern recognition receptors (PRRs) present on immune cells. Binding of DAMPs to PRRs results in the release of immunostimulatory cytokines and chemokines by the immune cell. As a result, antigen presenting cells are stimulated to efficiently take up and process tumor antigens and cross-prime T cells.Citation19
In this article, we examined the in vivo effects of vaccines based on immunogenically killed CCs, induced by MTX, and whether the protective anticancer effects could be augmented through association with DCs (co-incubated or fused with these immunogenically killed CCs).
Results
In vitro optimization
Different MTX concentrations and incubation conditions were tested to induce ICD of the EO771 cells. The most optimal protocol for this purpose was 2 h of incubation in a 1 µM MTX-containing serum-free medium (SFM) followed by 22 h of incubation in SFM. This protocol yielded the highest expression of calreticulin (CRT) (35.39% ± 16.7%) and Heat Shock Protein 70 (HSP70) (50.64% ± 20.74%) on the surface of the immunogenically killed CCs. These MTX-treated cells also expressed significantly more CRT and HSP70 than the mildly stressed cells that were incubated in SFM (14.87% ± 9.63%, P = 0.01 and 23.44% ± 12.12%, P = 0.012, respectively) and the unstressed control cells that were incubated in culture medium (CM) (12.1% ± 3.2%, P = 0.003 and 17.49% ± 12.02%, P = 0.003, respectively).
MTX-treated EO771 cells are unable to induce tumors, since it was confirmed in vitro that EO771 cells treated with MTX do no longer multiply and die over a period of 3–4 d, whereas untreated EO771 cells continue to multiply.
We demonstrated that ICD has a positive influence on phagocytosis. We followed the phagocytosis of untreated and MTX-treated EO771 cells by DCs during 12 h. MTX-treated EO771 cells were much faster phagocytized by DCs than untreated EO771 cells. Depending on the time point, 2–2.5 times more cancer cell (CC)-DC hybrids were formed after co-incubation of DCs with MTX-treated EO771 cells than with untreated EO771 cells ().
Figure 1. Formation and characterization of CC-DC hybrids. (A) Phagocytosis of MTX-treated (dashed line) EO771 cells and untreated (solid line) EO771 cells by DCs during 12 h of co-incubation (n = 4, error bars ± 1 SD). (B) Hybrid formation between MTX-treated EO771 cells fused to DCs (dashed line) or co-incubated with DCs (solid line) (n = 4, error bars ±1 SD). (C) Influence of fusion or co-incubation of EO771 cells with DCs on functional DC maturation (IL-12 production) (n = 3, error bars ± 1 SD). (D) Influence of fusion or co-incubation of EO771 cells with DCs on phenotypical DC maturation (CD86 surface expression) (n = 3, error bars ±1 SD). *, P < 0.05 between groups.
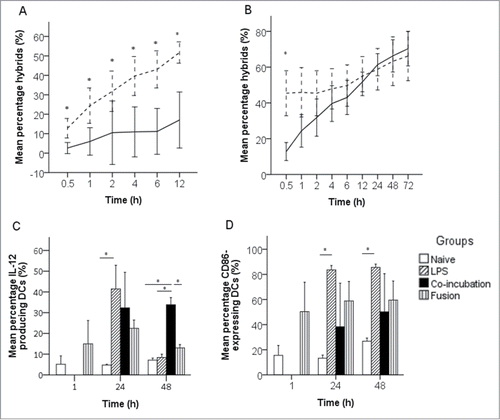
Subsequently, CC-DC hybrid formation via fusion or co-incubation of immunogenically killed CCs with DCs was compared and followed over 72 h. Thirty minutes after fusion or co-incubation, a significantly higher percentage of double-fluorescent (hybrids) cells was observed after fusion (P = 0.002). The instant formation of hybrids after fusion was confirmed by the observation that the generation of hybrids after co-incubation occurred much slower. Indeed, the percentage of hybrids in the co-incubation group gradually increased as a function of time and after 24 h the percentage of hybrids was the same as in the fusion group (). To ensure that real hybrids were measured and not merely aggregated cells, the formation of hybrids was confirmed through fluorescence microscope imaging (data not shown).
One can expect DCs to mature after fusion or co-incubation with immunogenically killed CCs.Citation21-24 However, maturation markers such as CD40 and CD86 can also be expressed by CCs.Citation25 Therefore, to unambiguously confirm DC maturation, the expression of CD40, CD86 and IL-12 by MTX-treated and untreated EO771 cells was measured. CD40 was highly expressed by MTX-treated and untreated EO771 cells, while CD86 and IL-12 were barely expressed by MTX-treated as well as untreated EO771 cells. Interestingly, MTX-treatment seemed to increase the expression of CD40, although statistical significance could not be reached (P = 0.053). In contrast, the expression of CD86 and production of IL-12 was not affected by MTX (P = 0.318 and 0.912, respectively). Since it was observed that the maturation marker CD40 was highly expressed on MTX-treated CCs, this marker was not optimal to evaluate maturation of CC-DC hybrids. Hence, CD86 and IL-12 were used as maturation markers. Twenty-four hours after fusion or co-incubation with immunogenically killed EO771 cells, CD86 expression as well as IL-12 production in DCs were not significantly higher than in naive DCs and did not reach the CD86 and IL-12 levels observed in LPS-treated DCs. After 48 h, the CD86 expression remained not significantly higher and, interestingly, the level of IL-12 expression dropped in DCs fused with immunogenically killed CCs or treated with LPS and even returned to the levels close to those of naive DCs. In contrast, the level of IL-12 production in DCs co-incubated with immunogenically killed CCs was significantly higher than in naive DCs and similar to the IL-12 level 24 h after co-incubation ().
In vivo effects of whole CC vaccines based on immunogenically killed CCs followed by tumor inoculation
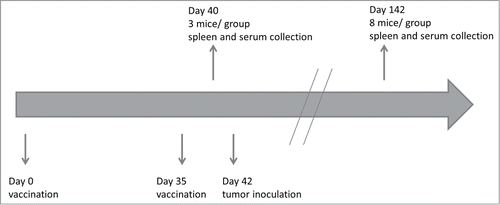
Three groups of 11 mice were vaccinated with either immunogenically killed EO771 cells (CC vaccine group), immunogenically killed EO771 cells co-incubated with DCs (co-incubation vaccine group) or fused with DCs (fusion vaccine group). A fourth group of 11 mice served as control and was treated with a saline solution (negative control group). The vaccination and sample collection schedule is shown in . The effect of immunogenically killed EO771 cells and co-incubation or fusion of immunogenically killed EO771 cells with DCs on the elicited immune response in mice was evaluated.
Figure 2. In vivo experimental setup. All mice received on days 0 and 35 in the flanks (left and right flank, respectively) a subcutaneous injection of phosphate buffered saline (control group) or 1 of the 3 vaccines (cancer cell vaccine, co-incubation vaccine or fusion vaccine). Three mice in each group were euthanized on day 40 and spleen and serum were collected to characterize the cellular and humoral response (tumor-specific cytotoxicity, activation of Th1 or Th2 effector cells, production of tumor-specific antibodies). Tumor inoculation was performed on day 42. Eight mice in each group were euthanized on day 142 (or earlier if the tumor would reach 1 cm2) and spleen and serum were collected for the characterization of the memory cellular and humoral response (tumor-specific cytotoxicity, activation of Th1 or Th2 memory cells, production of tumor-specific antibodies).
Five days after the second vaccination, cellular and humoral responses were evaluated in all groups. The percentages of activated tumor-specific IL-4+ CD4+ and CD8+ splenocytes were significantly higher in the co-incubation vaccine group than in the control group and the other vaccine groups. The latter groups did not generate more activated IL-4+ CD4+ and CD8+ splenocytes than the control group (). The percentages of activated tumor-specific IFNγ+ CD4+ and CD8+ splenocytes in the vaccinated groups were not significantly higher than in the control group (). The cellular response was also characterized by measuring the tumor-specific cytotoxicity (in vitro CC killing capacity of isolated splenocytes). Splenocytes from the CC vaccine group showed a significantly higher CC killing than splenocytes collected from the fusion vaccine or control group (), while those of the co-incubation vaccine group only differed significantly from the control group. Five days after the second vaccination, the humoral response (presence of antibodies against EO771 cells) was confirmed in all vaccinated groups, yet only the co-incubation and fusion vaccine group had significantly higher levels of EO771-specific IgG antibodies compared to the control group. The highest antibody levels were measured in the co-incubation vaccine group, which parallels the high percentage of activated IL-4+ (Th2) cells in this group ().
Figure 3. In vivo evaluation of vaccines based on immunogenically killed EO771 cells. (A) Activation of Th2 effector cells (IL-4+ CD4+ and CD8+ splenocytes) 5 d after the second vaccination (n = 3, ± 1 SD). (B) Activation of Th1 and CTL effector cells (IFNγ+ CD4+ and CD8+ splenocytes) 102 d after the second vaccination (n = 8, ± 1 SD). (C) The cellular response for tumor-specific cytotoxicity (in vitro cancer cell killing capacity of isolated splenocytes). In this cytotoxicity assay, the percentage of live EO771 cells inversely correlates with the efficacy of the vaccine/group (Pre n = 3, Post n = 8, ± 1 SD). (D) The humoral response (presence of antibodies against EO771 cells) (Pre n = 3, Post n = 8, ± 1 SD). Abbreviations: CCV, cancer cell vaccine group; CIV, co-incubation vaccine group; Control, control group; FV, fusion vaccine group; Post, 100 d post tumor inoculation or 102 d after the second vaccination; Pre, 2 d prior to tumor inoculation or 5 d after the second vaccination. *, P < 0.05 between groups.
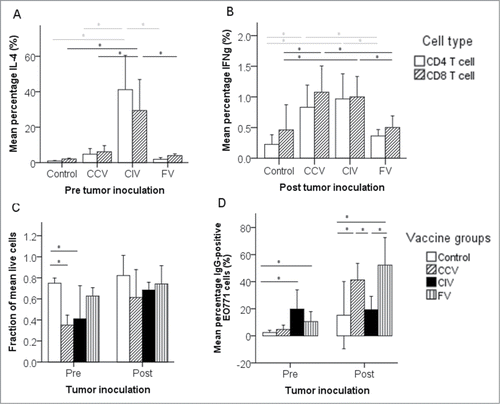
Table 1. Cellular and humoral responses in the different vaccine groups
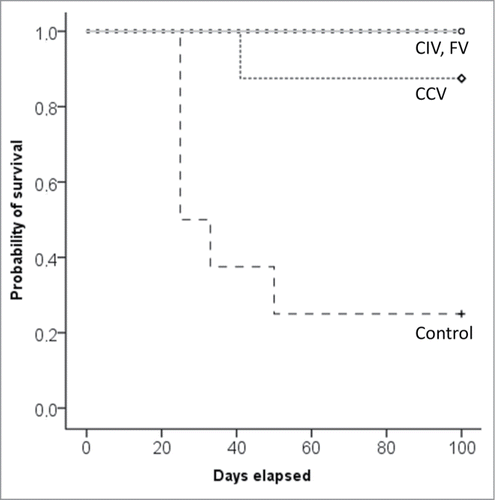
To assess the protective effect of the induced immune responses in the different vaccine groups, mice in all groups were challenged with the EO771 CCs. Six of the eight mice in the control group developed a tumor after tumor inoculation, which differed significantly from one in eight mice in the CC vaccine group (P = 0.041) and none in the co-incubation and fusion vaccine group (P = 0.015).
A 100 d after the tumor challenge, significant differences in the type of immune response were observed between the different treatment groups at the time of necropsy. The percentages of activated tumor-specific IL-4+ CD4+ and CD8+ splenocytes in any vaccine group were not significantly higher than in the control group (). In contrast, the percentages of activated tumor-specific IFNγ+ CD4+ and CD8+ splenocytes in mice of the CC and the co-incubation vaccine groups were significantly higher than in the control group (), indicating a clear memory T cell response. The tumor-specific cytotoxic activity of the isolated splenocytes was also higher in these groups than in the control group, but just did not reach the significance level (P = 0.064 and 0.085, respectively) (). On the other hand, no indications of a cellular memory could be found in the fusion vaccine group. A 100 d after the tumor challenge, the level of tumor-specific antibodies was, compared to the control group, significantly higher in the CC and fusion vaccine groups, but not in the co-incubation group (). Thus, a clear humoral memory was established in the CC and fusion vaccine group, but not in the co-incubation vaccine group.
Discussion
Association of CCs with DCs and ICD of CCs both improve the immunogenicity of whole CC vaccines. We examined whether the immunogenicity of immunogenically killed CCs could be further enhanced through association with DCs. The impact of these constructs was monitored by measuring cellular and humoral (memory) responses of all included mice groups ().
Although CCs used in whole CC vaccines are often irradiated prior to useCitation19,26,27 and irradiation is able to cause ICD,Citation28 we cannot assume the antitumoral effects of these vaccines arise from immunogenically killed CCs. Indeed, the question remains whether irradiation can specifically and efficiently induce ICD.Citation29 To unambiguously evaluate the effects of ICD, MTX, an agent well-known to trigger ICD, was used as the ICD-inducing stimulus.
None of the mice vaccinated with immunogenically killed CCs fused (fusion vaccine) or co-incubated (co-incubation vaccine) with DCs developed a tumor after tumor challenge. In the group that received only the immunogenically killed CCs one mouse developed a tumor after tumor challenge. In contrast, 75% of the mice of the control group developed a tumor after tumor challenge (). Therefore, it is sound to conclude that all tested vaccines induced a protective effect against cancer growth. Although all vaccinated groups were protected against tumor development, our experiments indicated that the immunological responses accountable for this protection were different between the different vaccine groups. As IL-4 is produced by activated T helper (Th) 2 cells and IFNγ is produced by activated Th1 cells and cytotoxic CD8+ T lymphocytes (CTL), these cytokines were chosen to indicate a Th2 and Th1 response, respectively.
Figure 4. Kaplan–Meier survival curve after tumor challenge of prophylactically vaccinated mice (n= 8/group). Abbreviations: CCV, cancer cell vaccine group; CIV, co-incubation vaccine group; Control, control group; FV, fusion vaccine group.
In the co-incubation vaccine group, tumor-specific cytotoxicity and humoral responses were present shortly after the second vaccination. Surprisingly, a 100 d later, a significant cellular memory response for CTL and Th1 activation was present, but only a trend toward increased tumor-specific cytotoxicity and humoral response was observed. This decline in cellular and humoral response in time could potentially be explained by the fact that the co-incubation vaccine induced such a powerful effector effect that the responding lymphocytes were driven into terminal differentiation and did not have the opportunity to develop a memory response.Citation30
In the fusion vaccine group, no cellular response was induced after vaccination and the observed humoral response appeared relatively weak. A 100 d after the tumor challenge, the humoral memory response demonstrated a 5-fold increase compared to the response before challenge, while a cellular response remained absent. The fact that none of the mice in this group developed a tumor suggests that a sole humoral response has protective effects against tumor development. The observed protection is potentially mediated through the Th2-cytokine IL-24.Citation31 Indeed, although Th2 cells have been associated in the past with a contribution to tumor growth,Citation32 large numbers of tumor-infiltrating Th2 cells are associated with significantly improved disease-free survival in human patients with classical Hodgkin lymphoma.Citation33
Vaccination with immunogenically killed CCs only induced a strong tumor-specific cytotoxicity response, but no humoral response. After the tumor challenge, there was a significant humoral and cellular memory response (for CTL and Th1 activation and a trend for tumor-specific cytotoxicity).
It is noteworthy that the initial cellular immune responses in the vaccinated groups seem to originate from CTLs with lytic activity that secrete no or low amounts of IFNγ, as we did not observe significantly higher amounts of IFNγ+ cells within 5 d after the second vaccination compared to the control group. Likewise, CTLs with lytic activity that do not secrete IFNγ have been reported by several other research groups.Citation34,35 Indeed, IFNγ and cytotoxicity are regulated independently in CD8+ T cells.Citation34
Many previous studies with CC-DC fusion vaccines mentioned an overnight incubation of the fusion mixture.Citation27,36-40 Yet our in vitro data revealed that during overnight incubation of the fusion mixture, CC-DC hybrids are also formed via phagocytosis of unfused CCs by unfused DCs. Therefore, it is very likely that in overnight incubated fusion mixtures, two types of CC-DC hybrids are present: hybrids that are formed via fusion as well as hybrids that occur after phagocytosis of CCs by DCs. In our study, the CC-DC hybrids were purified shortly after fusion and hence the formation of hybrids through phagocytosis was prevented. In the literature, humoral as well as cellular responses have been reported after vaccination with “unpurified” CC-DC fusion vaccines.Citation41 Our data documented that the fusion vaccine mainly operated through a humoral response, suggesting that the cellular response induced by “unpurified” fusion vaccines with overnight incubation most likely originates from CC-DC hybrids formed via phagocytosis and not via fusion. The lower immunogenicity of our fusion vaccine compared with the other fusion vaccines reported in the literature could be accredited to the use of pure CC-DC fusion hybrids. However, there are two other possible reasons that may explain why our fusion vaccine did not elicit a significant cellular response. First, it has been reported that in some CC-DC fusion hybrids based on mature DCs, like in this study, the anti-inflammatory cytokine IL-10 is produced, which tempers the cellular immune response and skews it toward a Th2 response.Citation42 Second, O'Connor et al.Citation43 described reversal of mature DCs to an immature state after fusionCitation43 that is less capable of homing toward the lymph node. Therefore, cellular responses might not be induced, as they are highly dose responsive,Citation44 whereas for antibody responses the minimal antigen threshold is relatively low.Citation45
The CC and the co-incubation vaccine group both induced cellular as well as humoral immune responses. It has been shown that primarily the lymph node residing CD169+ macrophages are essential for the cross-presentation of dead cell antigens.Citation46,47 However, a small contribution of migratory DCs, as present in the co-incubation group, cannot be ruled outCitation46 and might explain the differences in timing and intensity of the immune reactions observed between these two vaccine groups.
Although it has been reported that subcutaneous (s.c.) injection of EO771 cells near the fat pad of the fourth mammary gland into C57BL/6 mice resulted in tumor development in 97% of the inoculated mice,Citation48 gross tumor growth was only present in 75% of the non-vaccinated mice. Yet, the number of affected mice in the control group versus any of the vaccine groups was significantly different.
Since the fusion process can reverse the maturation state of DCs, preferably immature DCs should be harvested for vaccination with fusion-based vaccines.Citation43 DCs in this work were selected through their expression for CD11c in order to remove non-hybridized MTX-treated EO771 cells from the CC-DC mix. Since mature DCs express more CD11c than immature DCs,Citation49 mature DCs were more likely selected prior to fusion. Consequently, the obtained response in the fusion vaccine group is possibly lower than it could have been. A possible solution for fusion-based DC vaccines could be selection of DCs based on DC immaturity markers CCR1, CCR2 or CCR5Citation43 after negative selection of DCs. Alternatively, pre-conditioning of the vaccine site with a potent recall antigen (e.g. tetanus/diphtheria toxoid) could be used to improve the lymph node homing and efficacy of tumor-antigen-specific DCs.Citation50
In conclusion, based on our findings, we stipulate that vaccination with immunogenically killed CCs alone or with CC-DC hybrids obtained via co-incubation or fusion of immunogenically killed CCs with DCs, results in the protection of vaccinated mice against tumor development when challenged 5 d after the second vaccination. However, the immune responses responsible for this protection differ between the vaccine types. The immunogenically killed CCs without DCs protect the mice against tumor development via a cellular response only. The pre-inoculation protection in the fusion vaccine occurs mainly via a humoral response, whereas the co-incubation vaccine protects the mice via a cellular as well as a humoral immune response.
The current study indicates that vaccination with immunogenically killed CCs (with or without DCs) may be a useful prophylactic strategy for cancer growth. Based on the finding that a non-DC-based CC vaccine provided protection when using immunogenically killed CCs, the manufacture of CC vaccines can potentially be simplified, as the production of DC-based vaccines is laborious, time-consuming and expensive. Further research should be performed to confirm the advantages of ICD in CC vaccines. Additional cancer models should be tested to document whether other cancer types will reveal similar results.
Materials and Methods
Animals
Female C57BL/6 mice, aged 6–8 weeks, were purchased from Janvier Labs (Janvier Breeding Center, 2013-06-ENG-RM-20) and housed at the Laboratory for Gene Therapy at the University of Ghent. Experiments were conducted at age 16–52 weeks. This experiment was approved by the Ethical Committee (approval number EC2013/123).
Media and cell culture
Complete medium consisted of Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, 21041–033), supplemented with 100 IU/mL penicillin (Invitrogen, 15140-122) and 10% heat-inactivated fetal bovine serum (Thermoscientific, SV30160.03). EO771 cells, an aggressive estrogen receptor-positive poorly immunogenic mamma adenocarcinoma cell line (a kind gift from professor Jo Van Ginderachter), were grown in CM. Stable transduction of these cells with a luciferase gene was performed as described earlier by Van Impe.Citation51 Briefly, retrovirus was produced in HEK293T cells by calcium phosphate transfection, harvested after 48 and 72 h, filtered and concentrated by ultracentrifugation. Retroviral transduction was performed in the presence of 8 µg/mL polybrene and the efficiency was evaluated by bioluminescence. These cells were grown in DMEM, supplemented with 10% fetal bovine serum, 100 mg/mL streptomycin, 100 IU/mL penicillin and 1 mmol/mL L-glutamine (Invitrogen, 25030-081).
Bone-marrow-derived DCs were generated as described by Lutz.Citation52 Briefly, bone marrow cells were harvested from the femur and tibia of sacrificed mice and seeded at 2 × 106 cells per 100 mm dish in 10 mL DC-CM (i.e. CM with 20 ng/mL recombinant murine GM-CSF (Peprotech, 315-03) and 50 μM β-mercaptoethanol (Sigma-Aldrich, M3148-25ML). Five mL of DC-CM was added on day 3 and on days 6 and 8 collected, centrifuged after which the cell pellet was resuspended in 5 mL of fresh DC-CM and given back to the original plate. On day 10, non-adherent DCs were harvested by gentle pipetting.
In vitro evaluation of immunogenic cell death
Induction of ICD
EO771 cells were incubated for 2 h in SFM (DMEM, supplemented with 100 IU/mL penicillin) containing 1, 10 or 20 µM MTX (Sigma, M6545). Afterwards the MTX-treated EO771 cells were immediately analyzed for expression of the ICD markers CRT (Millipore, MABT145) and HSP70 (Abcam, ab45133) through anti-rabbit antibody (Invitrogen, A-11008), or further incubated during 22 h in the same MTX-containing medium or in SFM. As controls, EO771 cells were incubated either in SFM or in CM during 2 or 24 h (n = 4 for each condition tested). Expression of ICD markers on cells was analyzed by flow cytometry (Accuri C6, BD Biosciences). To generate the cell suspension for flow cytometry we compared forceful pipetting with trypsinization, and we found that cells harvested via trypsinization had a significantly lower expression of surface associated DAMPs than cells harvested via forceful pipetting (data not shown). Hence, all further cell harvesting was done through forceful pipetting.
Evaluation of cell death after Mitoxantrone treatment
Vaccines based on CCs should be free of multiplying CCs to eliminate the risk of inducing cancer after in vivo administration. For this purpose, cell death after MTX-treatment was evaluated in vitro. Luciferase-positive EO771 cells (1×104) were treated with 1 µM MTX in 100 μL CM or left untreated in 100 μL CM in a black 96-well plate. After 24, 48 and 72 h of incubation 10 μL of D-luciferin (Gold Biotechnology, LUCK-1G) was added per well and the plate was incubated at 37°C, 5% CO2 and 95% relative humidity for 15 min. The expression of luciferase (an indication of living EO771 cells) was measured with an IVIS Lumina II (Perkin-Elmer, Zaventem).
Preparation of vaccines
Cancer cell-DC fusion hybrids were prepared as follows. Bone-marrow-derived DCs and MTX-treated EO771 cells were mixed before fusion at DC:CC ratio (2:1) using 50% polyethylene glycol (PEG) (Sigma, P7306), as previously described.Citation53 Briefly, the DCs and MTX-treated EO771 cells were washed with DMEM and mixed before fusion at DC:CC ratio (2:1). The cell mix was spun for 5 min at 300 g after which the supernatant was removed. The pellet was broken by a 2 min pipetting in 1 mL of pre-warmed PEG (37˚C). Then, 1 mL of pre-warmed DMEM was slowly added over 1 min followed by slow addition of 3 mL of DMEM over a period of 3 min. This was followed by slow addition of 10 mL DMEM over a period of 1 min. The mixture was afterwards incubated for 5 min at 37˚C in a 15 mL final volume of DMEM, followed by centrifugation for 5 min at 350 g. The cell pellet of the fusion products was resuspended in 30 mL CM, unless otherwise specified, and plated in 3 100 mm Petri dishes for 1 h. After 1 h of incubation, (fused) DCs were magnetically separated from the MTX-treated EO771 cells in the CC-DC mix according to the manufacturer's instructions. Briefly, DCs were targeted with an antibody complex recognizing CD11c and dextran-coated magnetic particles (StemCell Technologies, Inc., 18780). Labeled cells were subsequently separated using an EasySep™ magnet (StemCell Technologies, Inc., 18000).
Cancer cell-DC hybrids were also generated via co-incubation of MTX-treated EO771 cells with DCs (co-incubation vaccine). In more detail, non-PEG-treated DCs and EO771 cells were co-cultured at the same DC:CC ratio (2:1). Unless otherwise stated, co-incubated cells were harvested 24 h after co-incubation. Like the fusion CC-DC hybrids the co-incubated CC-DC hybrids were magnetically separated from the CC-DC mix prior to further use.
Confirmation of hybrid formation
Before fusion or co-incubation the DCs and MTX-treated EO771 cells were fluorescently labeled using respectively the PKH67-Green Fluorescent Cell Linker Kit (Sigma, mini67 1 kit) and the CellVue® Claret Far Red Fluorescent Cell linker Kit (Sigma, MINCLARET-1KT). After labeling, the DCs and EO771 cells were mixed (DC:CC ratio 2:1) and were fused with 50% PEG or co-incubated as described above.
The fusion and phagocytosis efficiency (during co-incubation) was evaluated by assessing the percent of double-stained cells using flow cytometry and an inverted fluorescence microscope (Ti-S/L100). Prior to vaccine administration, the amount of hybrids was determined through flow cytometry. A gate was placed on the CD11c+-PE+-FL2+ cells and within this gate the number of MTX-treated FL4+ cells was determined (MTX is autofluorescent in the FL4 channel).
Influence of fusion or co-incubation on DC maturation
After staining with CD11c antibody (Biolegend, 117307), the phenotypical maturation markers CD40 (eBioscience, 11-0402) and CD86 (eBioscience, 11-0862-81) and the functional maturation marker IL-12 (eBioscience, 53-7123-80) were used to stain naive DCs, DCs co-incubated with LPS (Sigma, L4391-1MG) (100 ng/mL for 22 h), DCs co-incubated with MTX-treated EO771 cells and DCs fused to MTX-treated EO771 cells. In the latter two conditions the amount of maturation markers was determined after magnetic separation of the CC-DC hybrids. For IL-12 measurement, the cells were incubated during 4 h with Brefeldin A (1/1000 dilution) (Biolegend, 420601) to block secretion of cytokines and then stained with CD11c prior to fixation with a fix/perm buffer (eBioscience, 00-5123-43 and 00-5223). Afterwards the cells were permeabilized and stained with anti-IL-12 antibody in permeabilization buffer (eBioscience, 00-8333) for 30 min at 4°C, and then analyzed by flow cytometry.
In vivo experimental setup
In the vaccine groups, mice were s.c. vaccinated on day 0 in the left flank with 2 × 106 MTX-treated EO771 cells (CC vaccine group), with 2 × 106 MTX-treated EO771-DC hybrids obtained through co-incubation (co-incubation vaccine group), or with 2 × 106 MTX-treated EO771-DC hybrids obtained through fusion (fusion vaccine group). On day 35, a second vaccine was given in the right flank. The control group consisted of mice injected with a phosphate buffered saline solution (DPBS) (Invitrogen, 14190250) on days 0 and 35 (s.c.) in the flanks (left and right flank, respectively). The injections were performed with a hypodermic needle. Three mice in each group were euthanized on day 40 to characterize the cellular as well as the humoral response. Tumor inoculation was performed on day 42 with 5×105 EO771 cells in the right mammary fat pad of the fourth mammary gland with a hypodermic needle. Eight mice in each group were euthanized on day 142 (or earlier if the tumor would reach 1 cm2) for the memory cellular and humoral response. After euthanasia, spleen and serum were harvested ().
Cellular response: tumor-specific cytotoxicity
Splenocytes (2 × 105) from every mouse from every treatment group were co-incubated with luciferase-positive EO771 cells (1 × 104) in 100 μL CM in a black 96-well plate. After 24 h of co-incubation, 10 μL of D-luciferin (Gold Biotechnology, LUCK-1G) was added per well and incubated during 15 min at 37°C, 5% CO2 and 95% relative humidity. The expression of luciferase was measured with an IVIS Lumina II (Perkin-Elmer) and compared between the different treatment groups. The expression of luciferase is inversely correlated to CC death caused by tumor-specific cytotoxicity.
Cellular response: activation of Th1 or Th2 effector cells
The ability of splenocytes of vaccinated mice to produce IFNγ (marker for activated CTLs and Th1 cells) or IL-4 (marker for activated Th2 cells) when stimulated with EO771 whole cell lysate was evaluated. In detail, whole cell lysate of EO771 cells was developed as follows: cells were harvested with a scraper and resuspended in a DPBS solution at a final concentration of 8 × 107 cells/mL. A tablet cOmplete Mini (Roche, 5892970001) was added to the cells and the mixture was frozen quickly in a –154°C freezer for 10 min followed by rapid thawing in a 37°C water bath for 5 min. The quick freeze/thaw process was performed five times.
Subsequently, splenocytes were harvested and 1 × 107 splenocytes per mouse were incubated in 100 μL CM with 50 μL tumor lysate (the equivalent of 4 × 106 tumor cells) or without tumor lysate. After 18 h of incubation, Brefeldin A (1/1000 dilution) was added to block secretion of cytokines and the splenocytes were further incubated during 4 h. Afterwards, the splenocytes were harvested, washed and resuspended in fluorescence-assisted cell sorting (FACS) Buffer (BD Biosciences, 554656). Cell-surface staining with CD8+ antibody (Biolegend, 100705) or CD4+ antibody (Biolegend, 100509) was followed by fixation, permeabilization and intracellular staining with IL-4 antibody (Biolegend, 504103) and IFNγ antibody (Biolegend, 17-7311-81). The cells were analyzed with a flow cytometer and events in the lymphocyte gate were selected. Control for specificity of the cytokine response was evaluated by assessing non-stimulated splenocytes.
Humoral response
The presence of tumor-specific antibodies was determined as previously described.Citation54 Briefly, serum of each mouse was 1/8 diluted in FACS Buffer. Washed EO771 cells (1.25×105) were resuspended in 50 µL of this diluted serum and incubated for 1 h at 37°C, 5% CO2 and 95% relative humidity. Subsequently, the cells were washed and stained with anti-mouse IgG antibodies (Biolegend, 405308) and the number of positive cells was analyzed via flow cytometry. Control for specificity of staining included EO771 cells as such, EO771 cells that were not incubated with serum before being stained with the secondary antibodies and EO771 cells incubated with naive mouse serum before being stained with the secondary antibodies.
Statistics
All data were analyzed with the statistical software program SPSS (version 19.0). The parametric data were analyzed with one-way ANOVA, the non-parametric data with the Kruskall–Wallis test with Bonferroni correction. Proportion data were analyzed with Fisher's exact test. Statistical significance was determined at P ≤ 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by FWO grant G.0235.11N.
References
- Brown MP, Burdett N. Targeted therapies, aspects of pharmaceutical and oncological management. Cancer Forum 2013; 37:70-80
- Lasaro MO, Ertl HC. Targeting inhibitory pathways in cancer immunotherapy. CurrOpinImmunol 2010; 22:385-90
- Schlom J, Gulley JL, Arlen PM. Paradigm shifts in cancer vaccine therapy. Exp Biol Med(Maywood) 2008; 233:522-34
- Vermorken JB, Claessen AM, van TH, Gall HE, Ezinga R, Meijer S, Scheper RJ, Meijer CJ, Bloemena E, Ransom JH, et al. Active specific immunotherapy for stage II and stage III human colon cancer: a randomised trial. Lancet 1999; 353:345-50; PMID:9950438; http://dx.doi.org/10.1016/S0140-6736(98)07186-4
- Raez LE, Cassileth PA, Schlesselman JJ, Sridhar K, Padmanabhan S, Fisher EZ, Baldie PA, Podack ER. Allogeneic vaccination with a B7.1 HLA-A gene-modified adenocarcinoma cell line in patients with advanced non-small-cell lung cancer. J Clin Oncol 2004; 22:2800-7 ; http://dx.doi.org/10.1200/JCO.2004.10.197
- Finocchiaro LM, Fiszman GL, Karara AL, Glikin GC. Suicide gene and cytokines combined nonviral gene therapy for spontaneous canine melanoma. Cancer Gene Ther 2008; 15:165-72; PMID:18219342; http://dx.doi.org/10.1038/sj.cgt.7701096
- Hogge GS, Burkholder JK, Culp J, Albertini MR, Dubielzig RR, Keller ET, Yang NS, MacEwen EG. Development of human granulocyte-macrophage colony-stimulating factor-transfected tumor cell vaccines for the treatment of spontaneous canine cancer. HumGene Ther 1998; 9:1851-61
- Bergman PJ, McKnight J, Novosad A, Charney S, Farrelly J, Craft D, Wulderk M, Jeffers Y, Sadelain M, Hohenhaus AE, et al. Long-term survival of dogs with advanced malignant melanoma after DNA vaccination with xenogeneic human tyrosinase: a phase I trial. Clin Cancer Res 2003; 9:1284-90
- Serhal K, Baillou C, Ghinea N, Fontanges P, Dupuy FP, Lemoine FM, Lacave R. Characteristics of hybrid cells obtained by dendritic cell/tumour cell fusion in a T-47D breast cancer cell line model indicate their potential as anti-tumour vaccines. Int J Oncol 2007; 31:1357-65
- Chiang CL, Benencia F, Coukos G. Whole tumor antigen vaccines. Semin Immunol 2010; 22:132-43
- Chiang CL, Hagemann AR, Leskowitz R, Mick R, Garrabrant T, Czerniecki BJ, Kandalaft LE, Powell DJ, Jr., Coukos G. Day-4 myeloid dendritic cells pulsed with whole tumor lysate are highly immunogenic and elicit potent anti-tumor responses. PLoS One 2011; 6:e28732; http://dx.doi.org/10.1371/journal.pone.0028732
- Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011; 480:480-9; PMID:22193102; http://dx.doi.org/10.1038/nature10673
- Andersen BM, Ohlfest JR. Increasing the efficacy of tumor cell vaccines by enhancing cross priming. Cancer Lett 2012; 325:155-64; PMID:22809568; http://dx.doi.org/10.1016/j.canlet.2012.07.012
- Copier J, Dalgleish A. Overview of tumor cell-based vaccines. Int Rev Immunol 2006; 25:297-319; PMID:17169778; http://dx.doi.org/10.1080/08830180600992472
- Gong J, Koido S, Calderwood SK. Cell fusion: from hybridoma to dendritic cell-based vaccine. Expert Rev Vaccines 2008; 7:1055-68; PMID:18767954; http://dx.doi.org/10.1586/14760584.7.7.1055
- Gilboa E. DC-based cancer vaccines. J Clin Invest 2007; 117:1195-203; PMID:17476349
- Bagarazzi ML, Yan J, Morrow MP, Shen X, Parker RL, Lee JC, Giffear M, Pankhong P, Khan AS, Broderick KE, et al. Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci Transl Med 2012; 4:155ra38; http://dx.doi.org/10.1126/scitranslmed.3004414
- Fry TJ, Shand JL, Milliron M, Tasian SK, Mackall CL. Antigen loading of DCs with irradiated apoptotic tumor cells induces improved anti-tumor immunity compared to other approaches. Cancer Immunol Immunother 2009; 58:1257-64
- Kim TB, Park HK, Chang JH, Choi IH, Kim KH, Yoon SJ, Lee MS, Jung H, Kim CS. The establishment of dendritic cell-tumor fusion vaccines for hormone refractory prostate cancer cell. Korean J Urol 2010; 51:139-44
- Inzkirweli N, Guckel B, Sohn C, Wallwiener D, Bastert G, Lindner M. Antigen loading of dendritic cells with apoptotic tumor cell-preparations is superior to that using necrotic cells or tumor lysates. Anticancer Res 2007; 27:2121-9; PMID:17695495
- Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. NatRevCancer 2012; 12:860-75
- Kepp O, Tesniere A, Schlemmer F, Michaud M, Senovilla L, Zitvogel L, Kroemer G. Immunogenic cell death modalities and their impact on cancer treatment. Apoptosis 2009; 14:364-75; PMID:19145485; http://dx.doi.org/10.1007/s10495-008-0303-9
- Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, Kroemer G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ 2008; 15:3-12.
- Zitvogel L, Kroemer G. Introduction: the immune response against dying cells. Curr Opin Immunol 2008; 20:501-3; PMID:18657612
- Posner MR, Cavacini LA, Upton MP, Tillman KC, Gornstein ER, Norris CM, Jr. Surface membrane-expressed CD40 is present on tumor cells from squamous cell cancer of the head and neck in vitro and in vivo and regulates cell growth in tumor cell lines. Clin Cancer Res 1999; 5:2261-70; PMID:10473114
- Galea-Lauri J, Darling D, Mufti G, Harrison P, Farzaneh F. Eliciting cytotoxic T lymphocytes against acute myeloid leukemia-derived antigens: evaluation of dendritic cell-leukemia cell hybrids and other antigen-loading strategies for dendritic cell-based vaccination. Cancer Immunol Immunother 2002; 51:299-310; PMID:12111118
- Kao JY, Gong Y, Chen CM, Zheng QD, Chen JJ. Tumor-derived TGF-β reduces the efficacy of dendritic cell/tumor fusion vaccine. J Immunol 2003; 170:3806-11; PMID:12646647
- Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med 2005; 202:1691-701; PMID:16365148
- Golden EB, Pellicciotta I, Demaria S, Barcellos-Hoff MH, Formenti SC. The convergence of radiation and immunogenic cell death signaling pathways. Front Oncol 2012; 2:88; PMID:22891162
- Ribas A, Butterfield LH, Glaspy JA, Economou JS. Current developments in cancer vaccines and cellular immunotherapy. J Clin Oncol 2003; 21:2415-32; PMID:12805342
- Kreis S, Philippidou D, Margue C, Behrmann I. IL-24: a classic cytokine and/or a potential cure for cancer? J Cell Mol Med 2008; 12:2505-10; PMID:18505472
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet 2001; 357:539-45; PMID:11229684
- Schreck S, Friebel D, Buettner M, Distel L, Grabenbauer G, Young LS, Niedobitek G. Prognostic impact of tumour-infiltrating Th2 and regulatory T cells in classical Hodgkin lymphoma. Hematol Oncol 2009; 27:31-9; PMID:18924115
- Lim DG, Bieganowska BK, Freeman GJ, Hafler DA. Examination of CD8+ T cell function in humans using MHC class I tetramers: similar cytotoxicity but variable proliferation and cytokine production among different clonal CD8+ T cells specific to a single viral epitope. J Immunol 2000; 165:6214-20; PMID:11086055
- Bercovici N, Haicheur N, Massicard S, Vernel-Pauillac F, Adotevi O, Landais D, Gorin I, Robert C, Prince HM, Grob JJ, et al. Analysis and characterization of antitumor T-cell response after administration of dendritic cells loaded with allogeneic tumor lysate to metastatic melanoma patients. J Immunother 2008; 31:101-12; PMID:18157017
- Hayashi T, Tanaka H, Tanaka J, Wang R, Averbook BJ, Cohen PA, Shu S. Immunogenicity and therapeutic efficacy of dendritic-tumor hybrid cells generated by electrofusion. Clin Immunol 2002; 104:14-20; PMID:12139943
- Lee WT, Shimizu K, Kuriyama H, Tanaka H, Kjaergaard J, Shu S. Tumor-dendritic cell fusion as a basis for cancer immunotherapy. Otolaryngol Head Neck Surg 2005; 132:755-64; PMID:15886631
- Wei YC, Sticca RP, Li J, Holmes LM, Burgin KE, Jakubchak S, Bouton-Verville H, Williamson J, Meyer K, Evans L, et al. Combined treatment of dendritoma vaccine and low-dose interleukin-2 in stage IV renal cell carcinoma patients induced clinical response: A pilot study. Oncol Rep 2007; 18:665-71; PMID:17671717
- Tamai H, Watanabe S, Zheng R, Deguchi K, Cohen PA, Koski GK, Shu S. Effective treatment of spontaneous metastases derived from a poorly immunogenic murine mammary carcinoma by combined dendritic-tumor hybrid vaccination and adoptive transfer of sensitized T cells. Clin Immunol 2008; 127:66-77; PMID:18262845
- Cho EI, Tan C, Koski GK, Cohen PA, Shu S, Lee WT. Toll-like receptor agonists as third signals for dendritic cell-tumor fusion vaccines. Head Neck 2010; 32:700-7; PMID:19908319
- Rosenblatt J, Glotzbecker B, Mills H, Vasir B, Tzachanis D, Levine JD, Joyce RM, Wellenstein K, Keefe W, Schickler M, et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J Immunother 2011; 34:409-18; PMID:21577144
- Vasir B, Wu Z, Crawford K, Rosenblatt J, Zarwan C, Bissonnette A, Kufe D, Avigan D. Fusions of dendritic cells with breast carcinoma stimulate the expansion of regulatory T cells while concomitant exposure to IL-12, CpG oligodeoxynucleotides, and anti-CD3/CD28 promotes the expansion of activated tumor reactive cells. J Immunol 2008; 181:808-21; PMID:18566447
- Branham-O'Connor M, Li J, Kotturi HS, Yu X, Wagner TE, Wei Y. Fusion induced reversal of dendritic cell maturation: an altered expression of inflammatory chemokine and chemokine receptors in dendritomas. Oncol Rep 2010; 23:545-50; PMID:20043120
- Burchill MA, Tamburini BA, Pennock ND, White JT, Kurche JS, Kedl RM. T cell vaccinology: exploring the known unknowns. Vaccine 2013; 31:297-305; PMID:23137843; http://dx.doi.org/10.1016/j.vaccine.2012.10.096
- Hurn BA, Chantler SM. Production of reagent antibodies. Methods Enzymol 1980; 70:104-42; PMID:6999294
- Asano K, Nabeyama A, Miyake Y, Qiu CH, Kurita A, Tomura M, Kanagawa O, Fujii S, Tanaka M. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity 2011; 34:85-95; PMID:21194983; http://dx.doi.org/10.1016/j.immuni.2010.12.011
- Gerner MY, Torabi-Parizi P, Germain RN. Strategically localized dendritic cells promote rapid T cell responses to lymph-borne particulate antigens. Immunity 2015; 42:172-85; PMID:25607462; http://dx.doi.org/10.1016/j.immuni.2014.12.024
- Ewens A, Mihich E, Ehrke MJ. Distant metastasis from subcutaneously grown E0771 medullary breast adenocarcinoma. Anticancer Res 2005; 25:3905-15; PMID:16312045
- Xing F, Wang J, Hu M, Yu Y, Chen G, Liu J. Comparison of immature and mature bone marrow-derived dendritic cells by atomic force microscopy. Nanoscale Res Lett 2011; 6:455; PMID:21762525; http://dx.doi.org/10.1186/1556-276X-6-455
- Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, Congdon KL, Reap EA, Archer GE, Desjardins A, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015; 519:366-9; PMID:25762141; http://dx.doi.org/10.1038/nature14320.
- Van IK, Bethuyne J, Cool S, Impens F, Ruano-Gallego D, De WO, Vanloo B, Van TM, Lambein K, Boucherie C, et al. A nanobody targeting the F-actin capping protein CapG restrains breast cancer metastasis. Breast Cancer Res 2013; 15:R116; PMID:24330716; http://dx.doi.org/10.1186/bcr3585.
- Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods 1999; 223:77-92; PMID:10037236; http://dx.doi.org/10.1016/S0022-1759(98)00204-X
- Chien-Chun Pai T-FK, Simon J.T. Maoc Tien-Fu Chuanga, Chen-Si Lina, Rea-Min Chu. Immunopathogenic behaviors of canine transmissible venereal tumor in dogs following an immunotherapy using dendritic/tumor cell hybrid. Vet Immunol Immunopathol 2011; 139:187-99; PMID:21051091; http://dx.doi.org/10.1016/j.vetimm.2010.10.013
- Denies S, Cicchelero L, Van Audenhove I, Sanders NN. Combination of interleukin-12 gene therapy, metronomic cyclophosphamide and DNA cancer vaccination directs all arms of the immune system towards tumor eradication. J Control Release 2014; 187:175-82; PMID:24887014; http://dx.doi.org/10.1016/j.jconrel.2014.05.045