
ABSTRACT
Genetically engineered T cells that express chimeric antigen receptors (CAR+) are heterogeneous and thus, understanding the immunotherapeutic efficacy remains a challenge in adoptive cell therapy. We developed a high-throughput single-cell methodology, Timelapse Imaging Microscopy In Nanowell Grids (TIMING) to monitor interactions between immune cells and tumor cells in vitro. Using TIMING we demonstrated that CD4+ CAR+ T cells participate in multi-killing and benefit from improved resistance to activation induced cell death in comparison to CD8+ CAR+ T cells. For both subsets of cells, effector cell fate at the single-cell level was dependent on functional activation through multiple tumor cells.
Adoptive immunotherapy of chimeric antigen receptor T cells (CAR+ T cells) represents one of the most significant treatments against leukemia and lymphoma where it has been shown to be successful in mediating superior and sustained remission in previously relapsed and refractory cancer patients.Citation1-3 Because they express this hybrid immunoreceptors, CAR+ T cells directly recognize tumor associated antigen (TAA) on the surface of the cancer cells, bypassing conventional T-cell recognition that necessitates presentation by the human leukocyte antigen (HLA) molecules. New generations of CAR+ T cells constructs include one or two additional co-stimulatory molecules which further bestow activation to the T cells. Despite the success of these therapies however, the definition of the potency of the inoculum remains unclear. For instance, both pre-clinical and clinical studies in adoptive immunotherapy have shown improved benefits of infusing CD4+ and CD8+ T cells together for mediating antitumor response compared to infusing CD8+ T cells only.Citation4,5 The direct comparison of potency and kinetics of conjugation of these different T cell subsets with tumor cells, at the single-cell level in high-throughput, are lacking. Flow cytometry can identify phenotypic characteristics of immune cells at the single-cell level and in high throughput, but it cannot be used to study the real-time interactional behavior of immune cells. Multi-photon microscopy is a powerful tool used in tracking and studying immune cells in vivo, but it is limited with respect to the number of events observed per unit of time, thus making statistical decision challenging. There is a need for novel technologies to study the potency of individual immune cells both pre-infusion and post-infusion, and this single-cell technology ideally needs to be adaptive, reliable, scalable, high-throughput, and fast. Given the nature of adoptive immunotherapy in which infusions and treatments may be time-sensitive, rapid screening of potency of the inoculums may be paramount in identifying and generating suitable treatments personalized for each patient. We developed a high-throughput in vitro Time-lapse Imaging Microscopy in Nanowell Grids (TIMING) platform which allows the interrogation of thousands of single-cell level interactions between T cells and tumor cells in confined nanowells.Citation6-8 Specifically, we compared the potency and kinetics of CD19-specific CD8+ CAR+ T cells (CAR8) and CD4+ CAR+ T cells (CAR4) in their interactional behavior with CD19+ tumor cells.Citation7
At E:T of 1: 2–5, we found that both CAR8 and CAR4 cells can participate in multi-killing through conjugation to multiple tumor cells, although CAR4 cells experience a significant delay in kinetics of killing the tumor cells ().Citation7 Interestingly, the duration of conjugation leading to killing of the first, second, and third tumor cells were not significantly different, demonstrating no change in the efficiency of killing. In order to provide a mechanistic basis for the differential kinetics, we tested relative expression of the cytotoxic protease, granzyme B (GzB). Consistent with the kinetic results, we found that CAR8 cells had significantly higher expression of GzB compared with individual CAR4 cells. We further confirmed that GzB-mediated cytotoxicity is the major mechanism of killing mediated by these CAR+ T cells by co-incubating them with tumor cell lines in the presence/absence of EGTA calcium chelator.
Figure 1. TIMING methodology provides insight into function and fate of CAR+ T cells at the single-cell level. (A) Comparative schematic of CAR8 cells and CAR4 cells with respect to cytolytic efficiency and resistance to AICD. Both CAR8 (left) and CAR4 (right) cells participate in simultaneous conjugation with multiple tumor cells to engage in multi-killing, but CAR8 cells do so at a faster rate than CAR4 cells most likely due to higher GzB content. In addition, CAR4 cells benefits from improved AICD resistance compared to CAR8 cells upon killing the tumor cells. (B) Resistance to AICD was more pronounced in both CAR8 cells and CAR4 cells that participate in multi-killing compared to mono-killing, providing insights into the observation that CAR+ T cell fate at the single-cell level is dependent on functional antigenic activation.
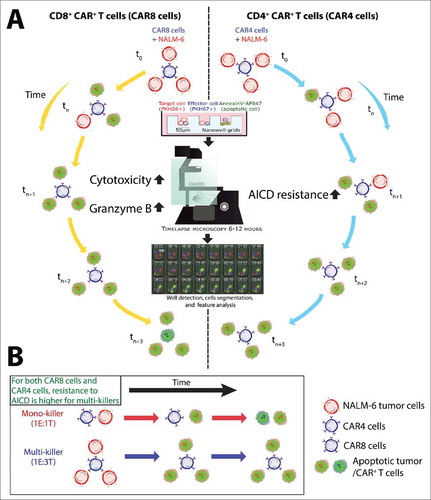
The ability of T cells to mediate antitumor response and to persist in vivo is one of the hallmarks of therapeutic success.Citation9 Since T cells can undergo apoptosis upon functional activation, we investigated the frequency and kinetics of them undergoing apoptosis. Remarkably, the frequency and kinetics of activation-induced cell death (AICD) was dependent on functional activation through multiple tumor cell lysis events. Although it is known that T cell populations undergo activation, expansion, and subsequent contraction, our results provide direct linkage to functional activation at the single-cell level modulating T-cell fate, in vitro (). Comparisons of the CAR8 cells and CAR4 cells demonstrated that CAR8 cells underwent AICD faster than CAR4 cells, upon antigen limitation. Together, these data are consistent with observations made in the clinic, where CAR+ T cells regulate their numbers based on the tumor bioburden i.e. they proliferate to greater numbers after infusion to respond to a large number of tumor cells, and then decreased in numbers as tumor cells were killed by the CAR+ T cells.Citation2,10 Overall, our short TIMING data (12 h) suggest that multi-killing, GzB content, and resistance to AICD may serve as critical components in determining the functional attributes of the inoculum that contribute to overall therapeutic success.
New immunobiology is needed to help define a T-cell product suitable for infusion. The large number of tumor-associated antigens, designs of immunoreceptors, T-cell subset(s) to be modified is but a few of the variables that aggregate to define the therapeutic potential of genetically modified T cells. Mouse models of comparative oncology are not amenable to scale up, and do not always predict efficacy in human trials.
We propose that the a priori identification of genetically modified T cells capable of mediating antitumor effects in humans can only be solved using high-throughput approaches supported by robust computational algorithms. Thus, our TIMING platform may enable the identification of this potency and thus help the design and manufacture of future generation of cellular infusion products.
Disclosure of potential conflicts of interest
LJN Cooper reports receiving commercial research grant from Immatics; has received speakers bureau honoraria from Miltenyi Biotec; has ownership interest (including patents) in Ziopharm, Intrexon, Sangamo Biosciences, and Targazyme; and is a consultant/advisory board member for Ferring Pharmaceuticals. No potential conflicts of interest were disclosed by the other authors.
References
- Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 2013; 5:177ra38; PMID:23515080; https://doi.org/10.1126/scitranslmed.3005930
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. New Engl J Med 2013; 368:1509-18; PMID:23527958; https://doi.org/10.1056/NEJMoa1215134
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl J Med 2014; 371:1507-17; PMID:25317870; https://doi.org/10.1056/NEJMoa1407222
- Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, Palmer DC, Chan CC, Klebanoff CA, Overwijk WW, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol 2005; 174:2591-601; PMID:15728465; https://doi.org/10.4049/jimmunol.174.5.2591
- Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003; 421:852-6; PMID:12594515; https://doi.org/10.1038/nature01441
- Liadi I, Roszik J, Romain G, Cooper LJ, Varadarajan N. Quantitative high-throughput single-cell cytotoxicity assay for T cells. J Visualized Exp 2013; e50058; PMID:23407457; https://doi.org/10.3791/50058
- Liadi I, Singh H, Romain G, Rey-Villamizar N, Merouane A, Adolacion JR, Kebriaei P, Huls H, Qiu P, Roysam B, et al. Individual motile CD4+ T cells can participate in efficient multikilling through conjugation to multiple tumor cells. Cancer Immunol Res 2015; 3:473-82; PMID:25711538; https://doi.org/10.1158/2326-6066.CIR-14-0195
- Romain G, Senyukov V, Rey-Villamizar N, Merouane A, Kelton W, Liadi I, Mahendra A, Charab W, Georgiou G, Roysam B, et al. Antibody Fc engineering improves frequency and promotes kinetic boosting of serial killing mediated by NK cells. Blood 2014; 124:3241-9; PMID:25232058; https://doi.org/10.1182/blood-2014-04-569061
- Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 2011; 3:95ra73; PMID:21832238; https://doi.org/10.1126/scitranslmed.3002842
- Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012; 119:2709-20; PMID:22160384; https://doi.org/10.1182/blood-2011-10-384388