
ABSTRACT
As a part of cellular pathogen defense, IFNγ triggers induction of NADPH oxidase NOX2, which produces superoxide into phagosomes of immune cells. Recent data show that a similar mechanism can also operate in IFNγ-mediated anticancer control. IFNγ is capable of inducing expression of constitutively active NADPH oxidase NOX4 in tumor cells leading to generation of reactive oxygen species (ROS) damaging DNA, activation of DNA damage response and cell cycle arrest/premature cellular senescence.
As one of immune system mediators, interferon gamma (IFNγ) orchestrates the function of various immune cells in organismal protection against pathogens and also links the innate and adaptive arms of immunity. Besides its function in antiviral and antibacterial immunity, the crucial role of IFNγ in tumor immunosurveillance/anticancer immunity, as well as in cancer immunoediting, is well documented.Citation1 In simple terms, the anticancer effects of IFNγ are bimodal. Firstly, IFNγ can affect cancerous cells directly by inhibition of their proliferation and sensitization to cell death. Additionally, IFNγ modulates cancer development indirectly, for example by suppression of tumor-associated angiogenesis and/or activation of various components of immune system-mediated antitumor defense, such as via upregulating expression of MHC class I and II molecules on tumor cells or enhancing antitumor cytotoxicity of NK cells. Like type I interferons, IFNγ has been found to induce, besides cell death (apoptosis and necrosis), also premature cellular senescence in human cells in vitro via DNA damaging activity mediated by ROS (references see inCitation2). This senescence-inducing antitumour effect of IFNγ was also demonstrated in vivo using murine model of pancreatic β-cell cancer.Citation3 The latter study showed that IFNγ produced together with TNFα by CD4+TH1 lymphocytes limited the growth of β-cancer cells via induction of p16INK4a/pRb-dependent senescence, which required also intact STAT1 and TNFR1 signaling. Nevertheless, it is noteworthy that the direct effects of IFNγ on tumor cells can be cell and tumor type-specific and even opposite cell responses (e.g., proliferation induction) can be observed.
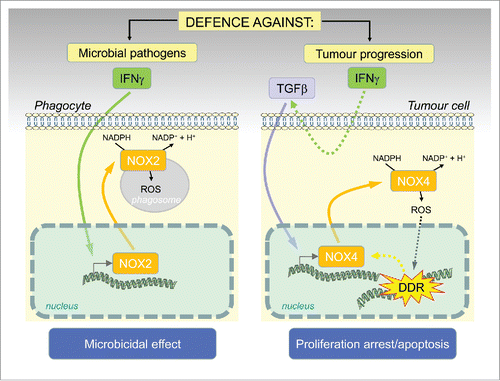
The antimicrobial activities of IFNγ are also linked to IFNγ-mediated induction of superoxide generation during respiratory burst of phagocytes (neutrophils and macrophages) via activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs). NOXs are membrane-associated multicomponent enzymes that catalyze one electron transfer from NAD(P)H to O2. The reaction product, superoxide anion, is a potent ROS undergoing further chemical and enzymatic exchanges leading to generation of other ROS such as hydrogen peroxide, hydroxyl or nitrogen radicals (arising after reaction of superoxide with nitric oxide to form peroxynitrite). The seven mammalian enzymes NOX1-5 and DUOX1/2 differ in subunit composition, subcellular localization, mechanism of activation and function. Besides the host defense mediated by microbicidal effects of ROS carried by both immune-system and non-immune cells such as intestinal epithelia, NOXs are involved in diverse physiological and pathophysiological processes in mammals. Importantly, NOX1 and NOX4 are causally involved in ROS-induced DNA damage during development of various forms of senescence.Citation4-6 In our recent study,Citation2 we elucidated the mechanism of NOX-mediated ROS generation in senescence induced by IFNγ. Previously, IFNγ has been reported as a potent inducer of NOX1 and NOX2 and it was suggested that their transcriptional activation is mediated via transcription factors STAT1 and IRF1, as both NOX1/2 genes contain corresponding DNA binding elements (reviewed in ref.Citation7). We showed that while IFNγ induces both NOX1 and NOX4 during development of senescence in human HeLa cells, NOX4 alone is the factor responsible for inducing the DNA damage (note NOX4 is constitutively active, i.e., it does not require coactivator regulatory subunits, in contrast to most other NOXs). Our findings point out that IFNγ can trigger NOX-mediated oxidative burst not only in immune cells but also in non-immune (cancerous) cells () and this mechanism might serve as an effective tool for control of malignancy by the immune system.Citation3 Unexpectedly, the effect of IFNγ on NOX4 expression in HeLa cells was indirect and mediated via TGFβ signaling activated downstream of IFNγ pathway. This discovery links IFNγ/STAT and TGFβ/SMAD signaling modules into one machinery operating to restrain cancer cell proliferation by the immune system. It can be envisaged that abrogation of signaling components of either IFNγ/STAT or TGFβ/SMAD signaling modules can modify the antiproliferative response of cancer cells to IFNγ. Indeed, this was supported by our additional observation that TC-1 tumor cells harbouring an intact JAK/STAT signaling pathway but unresponsive to IFNγ in terms of senescence development, lacked induction of NOX4, DNA damage response and activation of cell cycle checkpoints.Citation2 It should be further explored whether other tumor cells resistant to IFNγ-mediated antiproliferative effects share such lack of NADPH oxidase expression.
Figure 1. Schematic representation of the novel role of IFNgamma/NOX4 induced ROS production as a barrier against tumorigenesis: similarly as in phagocytes, where activation of ROS by IFNgamma/NOX2 results in a ‘respiratory burst’ and pathogen killing, generation of ROS in tumor cells caused by IFNgamma-induced TGFbeta activation of NOX4 results in DNA damage and proliferation arrest/cellular senescence or apoptosis.
From a perspective of disease pathogenesis, some recent studies provided striking observations about a complex interplay among cytokine signaling, regulation of expression of NADPH oxidases and DNA damage response.Citation8-10 Weyemi et al., showed that ROS generated by NOX4 and NOX5 in human primary fibroblasts exposed to ionizing radiation contribute significantly to radiation-induced DNA damage, as the extent of post-irradiation DNA damage can be suppressed by inhibition of either NOX4 or NOX5. Intriguingly, as shown in another study,Citation6 DUOX1 is specifically and persistently upregulated in irradiated thyrocytes in a radiation dose-dependent manner, via p38MAPK/IL13 signaling, thereby contributing (via H2O2 production) to persistent DNA damage and senescence-like growth arrest. Notably, pretreatment of thyrocytes with catalase, a scavenger of H2O2, led to decreased expression of DUOX1, whereas H2O2 had opposite effect, indicating the existence of a self-amplification mechanism. Even more puzzling but underscoring these two studies are the findings of Fandy et al.,Citation10 which might add the missing piece of the puzzle to mutual interplay between NOXs and DNA damage signaling. The authors showed that 5-aza-2′-deoxycytidine (DAC) induced expression of several NOX isoforms in leukemia cells resulting in ROS generation, cell cycle arrest and apoptosis. Surprisingly, inhibition of ATM, the crucial kinase orchestrating DNA damage response, diminished DAC-induced NOX4 upregulation suggesting that NOX4 levels are controlled by DNA damage signaling. Although the reasons for existence of such self-amplified DNA damage promoting mechanism are presently unclear, it can be foreseen as a component of antimicrobial/antiviral/anticancer immunity poised to eliminate infected or malignant cells.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol 2006; 6:836-48; PMID:17063185; http://dx.doi.org/10.1038/nri1961
- Hubackova S, Kucerova A, Michlits G, Kyjacova L, Reinis M, Korolov O, Bartek J, Hodny Z. IFN[gamma] induces oxidative stress, DNA damage and tumor cell senescence via TGF[beta]/SMAD signaling-dependent induction of Nox4 and suppression of ANT2. Oncogene 2015; [Epub ahead of print]; PMID:25982278; http://dx.doi.org/10.1038/onc.2015.162
- Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013; 494:361-5; PMID:23376950; http://dx.doi.org/10.1038/nature11824
- Weyemi U, Lagente-Chevallier O, Boufraqech M, Prenois F, Courtin F, Caillou B, Talbot M, Dardalhon M, Al Ghuzlan A, Bidart JM et al. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 2012; 31:1117-29; PMID:21841825; http://dx.doi.org/10.1038/onc.2011.327
- Hubackova S, Krejcikova K, Bartek J, Hodny Z. IL1- and TGFbeta-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘Bystander senescence’. Aging (Albany NY) 2012; 4:932-51; PMID:23385065
- Kodama R, Kato M, Furuta S, Ueno S, Zhang Y, Matsuno K, Yabe-Nishimura C, Tanaka E, Kamata T. ROS-generating oxidases Nox1 and Nox4 contribute to oncogenic Ras-induced premature senescence. Genes Cells 2013; 18:32-41; PMID:23216904; http://dx.doi.org/10.1111/gtc.12015
- Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radical Biol Med 2007; 43:319-31; PMID:17602947; http://dx.doi.org/10.1016/j.freeradbiomed.2007.03.028
- Weyemi U, Redon CE, Aziz T, Choudhuri R, Maeda D, Parekh PR, Bonner MY, Arbiser JL, Bonner WM. Inactivation of NADPH oxidases NOX4 and NOX5 protects human primary fibroblasts from ionizing radiation-induced DNA damage. Radiat Res 2015; 183:262-70; PMID:25706776; http://dx.doi.org/10.1667/RR13799.1
- Ameziane-El-Hassani R, Talbot M, de Souza Dos Santos MC, Al Ghuzlan A, Hartl D, Bidart JM, De Deken X, Miot F, Diallo I, de Vathaire F et al. NADPH oxidase DUOX1 promotes long-term persistence of oxidative stress after an exposure to irradiation. Proc Natl Acad Sci U S A 2015; 112:5051-6; PMID:25848056; http://dx.doi.org/10.1073/pnas.1420707112
- Fandy TE, Jiemjit A, Thakar M, Rhoden P, Suarez L, Gore SD. Decitabine induces delayed reactive oxygen species (ROS) accumulation in leukemia cells and induces the expression of ROS generating enzymes. Clin Cancer Res 2014; 20:1249-58; PMID:24423613; http://dx.doi.org/10.1158/1078-0432.CCR-13-1453