
ABSTRACT
Th22 cells traffic to and retain in the colon cancer microenvironment, and target core stem cell genes and promote colon cancer stemness via STAT3 and H3K79me2 signaling pathway and contribute to colon carcinogenesis. However, whether Th22 cells affect colon cancer cell proliferation and apoptosis remains unknown. We studied the interaction between Th22 cells and colon cancer cells in the colon cancer microenvironment. Colon cancer proliferation was examined by flow cytometry analysis and H3 thymidine incorporation. Cell cycle related genes were quantified by real-time PCR and Western blotting. We transfected colon cancer cells with lentiviral vector encoding specific gene shRNAs and used chromatin immunoprecipitation (ChIP) assay to determine the genetic signaling involved in interleukin (IL)-22-mediated colon cancer cell proliferation. We showed that Th22 cells released IL-22 and stimulated colon cancer proliferation. Mechanistically, IL-22 activated STAT3, and subsequently STAT3 bound to the promoter areas of the Polycomb Repression complex 2 (PRC2) components SUZ12 and EED, and stimulated the expression of PRC2. Consequently, the activated PRC2 catalyzed the promoters of the cell cycle check-point genes p16 and p21, and inhibited their expression through H3K27me3-mediated histone methylation, and ultimately caused colon cancer cell proliferation. Bioinformatics analysis revealed that the levels of IL-22 expression positively correlated with the levels of genes controlling cancer proliferation and cell cycling in colon cancer. In addition to controlling colon cancer stemness, Th22 cells support colon carcinogenesis via affecting colon cancer cell proliferation through a distinct histone modification.
KEYWORDS:
Abbreviations
| CCNA1 | = | cyclin A1 |
| CCND1 | = | cyclin D1 |
| CCND3 | = | cyclin D3 |
| CCNE1 | = | cyclin E1 |
| CCNE2 | = | cyclin E2 |
| ChIP | = | Chromatin immunoprecipitation |
| DOT1L | = | disruptor of telomeric silencing1-like |
| EED | = | embryonic ectoderm development |
| EZH2 | = | zeste homolog 2 |
| FACS | = | Flow cytometry analyzer |
| FDR | = | false discovery rate |
| GSEA | = | Gene Set Enrichment Analysis |
| IL-22 | = | interleukin-22 |
| PRC2 | = | Polycomb Repression complex 2 |
| RT-PCR | = | reverse-transcriptase polymerase chain reaction |
| STAT3 | = | signal transducers and activators of transcription protein 3 |
| SUZ12 | = | suppressor of zeste 12 |
| TCGA | = | the Cancer Genomic Atlas project. |
Introduction
IL-22 is mainly produced by innate lymphoid cell (ILC22) and IL-22+CD4+ T (Th22) cells.Citation1–4 It has been reported that IL-22 is predominantly expressed by CD4+ T cells in the human colorectal cancer microenvironment.Citation5–8 As its receptor is only expressed on epithelial cells,Citation9 it is reasonable that IL-22 protects epithelial mucosa from bacterial infection and inflammation damage in mouse models.Citation10–12 We have recently reported that human Th22 cells are recruited into the colon cancer microenvironment and promote colon cancer stemness through STAT3-dependent pathway.Citation5 However, it is unknown whether Th22 cells and/or Th22 cell-derived IL-22 can target colon cancer cell proliferation and apoptosis.
Histone modification plays an important role in cancer development and progression. Trimethylation of histone H3 lysine 27 (H3K27me3), catalyzed by the enhancer of zeste homolog 2 (EZH2), is mainly related to gene repression and oncogenic activation in several types of cancer.Citation13-16 This catalyzation requires the presence of two additional proteins, embryonic ectoderm development (EED) and suppressor of zeste 12 (SUZ12). These proteins constitute the PRC 2Citation14-17 and contributes to tumorigenesis.Citation14-16 Disruptor of telomeric silencing1-like (DOT1L)-mediated H3K79me2 is associated with gene activation.Citation13 Th22 cell-derived IL-22 can activate DOT1L and promote colon cancer stemness via H3K79me2 targeted core stem cell genes.Citation5 However, it is unknown if the PRC2 components or the DOT1L and H3K79me2 signaling pathway is involved in the control of colon cancer cell proliferation and apoptosis.
In the current work, we have studied the interaction between Th22 cells and colon cancer cells in the human colon cancer microenvironment. We found that Th22 cell-derived IL-22 target the PRC2 components and stimulate colon cancer cell proliferation.
Results
Th22 cell-derived IL-22 induces colon cancer proliferation
We have recently, demonstrated that Th22 cells traffic to and retain in the colon cancer microenvironment; and Th22 cell-derived IL-22 targets core stem cell genes and promotes colon cancer stemness and contributes to colon carcinogenesis.Citation5 However, whether Th22 cells and IL-22 affect colon cancer cell proliferation and apoptosis remains unknown.
To address this question, we performed Gene Set Enrichment Analysis (GSEA) using high throughput RNA-sequencing data of the GC cohort of the Cancer Genomic Atlas project (TCGA). GSEA is designed to detect coordinated differences in expression of predefined sets of functionally related genes.Citation18 We found that the most significantly enriched functional categories upon IL-22 positive profile were associated with multiple processes involved in cell proliferation (). The analysis supports the hypothesis that IL-22 may be a critical regulator of colon cancer cell proliferation.
Figure 1. Th22 cell-derived IL-22 stimulates colon cancer cell proliferation. (A) GSEA analysis in the association between IL-22 and cell proliferation pathways in the TCGA colon cancer dataset. n = 224, nominal p < 0.05, false discovery rate [FDR] q < 0.25, red bar: positively correlated genes, blue bar: negatively correlated genes. (B) Effect of endogenous IL-22 on primary colon cancer cell proliferation. Single cells including colon cancer cells and immune cells were isolated from fresh colon cancer tissue and cultured with or without anti-IL-22 antibody for 24 h. Cell proliferation was tested by H3 Thymidine Incorporation. Results are expressed as the mean of CPM ± SD. One of three patients with triplicates is shown. *p < 0.05. (C) Effect of endogenous Th-22-derived IL-22 on the established primary colon cancer cell proliferation. Freshly sorted colon cancer associated CD4+ T cells were stimulated with anti-CD3 and anti-CD28 for 3 d. Established primary colon cancer cells (C1) were cultured with these T cell supernatants with or without anti-IL-22 antibody for 24 h. Tumor cell proliferation was evaluated with thymidine incorporation. Results are expressed as the mean of CPM ± SD. One of three patients with triplicates is shown. *p <0.05. (D–F) Effect of exogenous IL-22 on colon cancer cell proliferation. DLD-1 (D-F), HT29 (F), C1 and C2 primary colon cancer cells (F) were treated with or without recombinant IL-22 for 24 h. Cell proliferation was evaluated with Ki67 expression by FACS (D) and thymidine incorporation (E). The absolute cell numbers were counted and relative cell numbers were shown (F). One of four repeats is shown. *p <0.05.
![Figure 1. Th22 cell-derived IL-22 stimulates colon cancer cell proliferation. (A) GSEA analysis in the association between IL-22 and cell proliferation pathways in the TCGA colon cancer dataset. n = 224, nominal p < 0.05, false discovery rate [FDR] q < 0.25, red bar: positively correlated genes, blue bar: negatively correlated genes. (B) Effect of endogenous IL-22 on primary colon cancer cell proliferation. Single cells including colon cancer cells and immune cells were isolated from fresh colon cancer tissue and cultured with or without anti-IL-22 antibody for 24 h. Cell proliferation was tested by H3 Thymidine Incorporation. Results are expressed as the mean of CPM ± SD. One of three patients with triplicates is shown. *p < 0.05. (C) Effect of endogenous Th-22-derived IL-22 on the established primary colon cancer cell proliferation. Freshly sorted colon cancer associated CD4+ T cells were stimulated with anti-CD3 and anti-CD28 for 3 d. Established primary colon cancer cells (C1) were cultured with these T cell supernatants with or without anti-IL-22 antibody for 24 h. Tumor cell proliferation was evaluated with thymidine incorporation. Results are expressed as the mean of CPM ± SD. One of three patients with triplicates is shown. *p <0.05. (D–F) Effect of exogenous IL-22 on colon cancer cell proliferation. DLD-1 (D-F), HT29 (F), C1 and C2 primary colon cancer cells (F) were treated with or without recombinant IL-22 for 24 h. Cell proliferation was evaluated with Ki67 expression by FACS (D) and thymidine incorporation (E). The absolute cell numbers were counted and relative cell numbers were shown (F). One of four repeats is shown. *p <0.05.](/cms/asset/e250fec0-b193-4fed-bb49-42041edcd2b9/koni_a_1082704_f0001_oc.gif)
To test this hypothesis, we freshly prepared single cells from colon cancer tissues, which contained tumor cells and infiltrating T cells, and cultured these single cells with monoclonal anti-IL-22 antibody. We observed that anti-IL-22 reduced primary colon cancer cell proliferation (). We established primary colon cancer cells from colon cancer patients. Then, we cultured primary colon cancer cells with the supernatants of primary T cells isolated from colon cancer tissues with or without anti-IL-22. Colon cancer associated T cells stimulated primary colon cancer cell proliferation, and anti-IL-22 blocked this effect (). Recombinant IL-22 also stimulated colon cell proliferation, as shown by an increased Ki67 expression (), H3 thymidine incorporation () and the cell numbers of DLD-1, HT-29 and two primary colon cancer cells (C1, C2) (). Furthermore, IL-22 had no effects on Annexin V+ colon cancer cells in the regular culture (Fig. S1A). Five-Fluorouracil (5-Fu) treatment induced colon cancer cell apoptosis. IL-22 didn't affect 5-Fu-mediated colon cancer apoptosis (Fig. S1B – S1C). IL-22 is primary from Th22 cells.Citation5 Thus, the data indicate that Th22-derived IL-22 stimulates colon cancer cell proliferation and has no effect on their apoptosis.
IL-22 targets cyclin genes and controls colon cancer cell proliferation
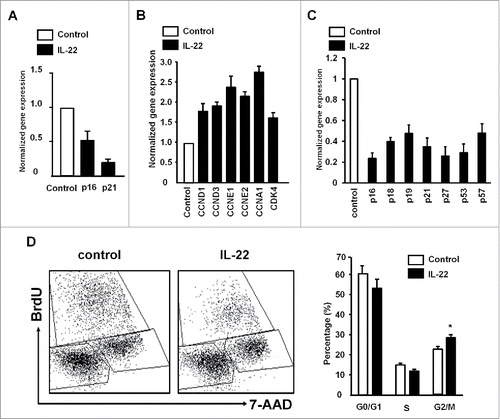
We next examined the potential gene targets of IL-22 in colon cancer cell proliferation. Real-time PCR revealed that IL-22 reduced the expression of several cyclin-dependent kinase inhibitors including p16 (CDKN2A), p21 (CDKN1A) (), p18 (CDKN2C), p19 (CDKN2D), p27 (CDKN1B), p53 and p57 (CDKN1C) (Fig. S2). In line with this observation, IL-22 increased the mRNA levels of several proliferation positive regulators including cyclin A1 (CCNA1), cyclin D1 (CCND1), cyclin D3 (CCND3), cyclin E1 (CCNE1), cyclin E2 (CCNE2) and CDK4 (). The effects were similar in primary colon cancer cells we established in the laboratory (). FACS revealed that IL-22 increased colon cancer cells in G2/M phase (). Collectively, the results indicate that IL-22 targets cyclin genes, promotes cell cycle progress and controls colon cancer cell proliferation.
Figure 2. IL-22 targets cyclin genes and controls colon cancer cell proliferation. (A–C) Effect of IL-22 on the expression of cell proliferation genes in colon cancer cells. Colon cancer cells were cultured with IL-22 for 12 h. The levels of cyclin-dependent kinase inhibitors and cyclins were quantified by real-time PCR in DLD-1 (A, B) and primary colon cancer cells (C). Results are expressed as the relative values (mean ± SD). One of three experiments with triplicates is shown. p < 0.05. (D) Effect of IL-22 on colon cancer cell cycles. DLD-1 cells were cultured with IL-22 for 24 h. Expression of BrdU and 7-AAD was analyzed by FACS. Results are shown as the percent of cells in G0/G1, S, G2/M. n = 4, p <0.05.
IL-22 induced colon cancer proliferation depends on H3K27me3, not H3K79me2
We have recently shown that IL-22 induced colon cancer stemness via DOT1L-mediated H3K79me2.Citation5 To test if IL-22-induced colon cancer cell proliferation depends on DOTL1, we treated colon cancer cells with EPZ004777, a specific DOT1L inhibitor.Citation19 EPZ004777 had no effect on colon cancer cells (Fig. S3A) proliferation induced by IL-22. We genetically knocked down DOT1L with shDOT1L (Fig. S3B). shDOT1L specifically reduced H3K79me2 (Fig. S3B), but had no effect on cell proliferation stimulated by IL-22 (Fig. S3C). The results indicate that IL-22-induced colon cancer proliferation is not DOT1L dependent.
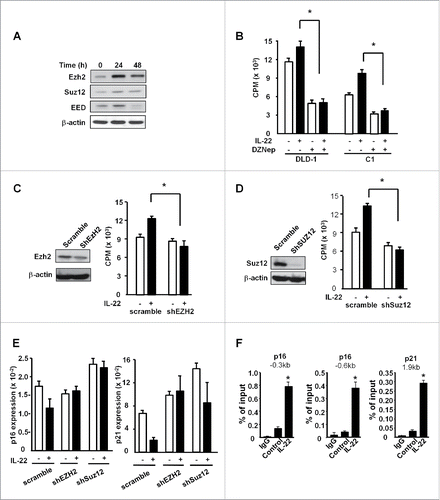
It has been reported that the PRC2 complex including EZH2, EED and SUZ12 is involved in the regulation of cancer cell proliferation.Citation14-16 In support of this possibility, IL-22 significantly increased the expression levels of EZH2, SUZ12 and EED (). We treated colon cancer cells with the PRC2 inhibitor, DZNep. DZNep significantly blocked DLD-1 and primary colon cancer cell proliferation induced by IL-22 (). Genetic silencing of EZH2 () and SUZ12 () with shRNAs slowed down colon cancer cell proliferation induced by IL-22. Thus, PRC2 controls IL-22-induced colon cancer cell proliferation.
Figure 3. IL-22 induced colon cancer proliferation depends on H3K27me3, not H3K79me2. (A) Effect of IL-22 on PCR2 component expression in colon cancer cells. DLD-1 cells were cultured with IL-22 (20ng/mL) for 24 or 48 h. Expression of EZH2, EED, SUZ12 was detected by Western blotting. One of three experiments is shown. (B) Effect of DZNep on IL-22-induced colon cancer cell proliferation. DLD-1 and C1, primary colon cancer cells were treated with IL-22 and EZH2 inhibitor DZNep for 24 h. Cell proliferation was detected by H3 thymidine incorporation. n = 4, p < 0.05. (C, D) Effect of shEZH2 and shSUZ12 on DLD-1 cell proliferation. DLD-1 cells were transfected with control vector (scramble), shEZH2 (C) and shSUZ12 (D), and stimulated with IL-22 for 24 h. EZH2 expression was detected by Western blotting (left). Cell proliferation was detected by H3 thymidine incorporation (right). n = 4, * p < 0.05. (E) Effect of shEZH2 and shSUZ12 on the expression of p16 and p21 in DLD-1 cells. DLD-1 cells were transfected with control vector (scramble), shEZH2 and shSUZ12, and stimulated with IL-22 for 24 h. P16 and p21 expression was quantified by real-time PCR. n = 3, p < 0.05. (F) Effect of IL-22 on the occupancies of H3K27me3 in the promoters of p16 and p21 in DLD-1 cells. DLD-1 cells were cultured with IL-22 for 24 h. The occupancies of H3K27me3 in the promoters of p16 and p21 were determined by ChIP assays. n = 3, * p < 0.05.
We next studied whether PRC2 targets cycling inhibitor genes in colon cancer cells. Real-time PCR demonstrated that shEZH2 or shSUZ12 increased the expression of p16 and p21 (). Furthermore, ChIP assay showed high-level of H3K27me3 occupancies in the promoters of p16 and p21, which were further increased with IL-22 stimulation (). Taken together, the data suggest that IL-22-induced colon cancer proliferation is mediated by PRC2 and H3K27me3, but not DOT1L and H3K79me2.
IL-22 promoted STAT3 binding to PRC2 promoters
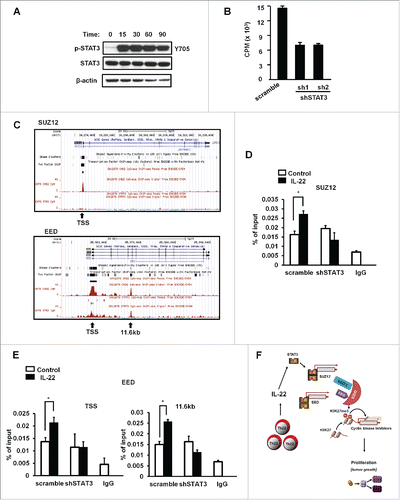
We have recently demonstrated that STAT3 is essential for IL-22-induced colon cancer stemness.Citation5 In line with this, we confirmed that IL-22 strongly activated STAT3 in colon cancer cells (). To examine whether the effect of IL-22 on colon cancer proliferation was STAT3 dependent, we used lentiviral vectors encoding two shRNAs for STAT3 to knock down the expression of STAT3 (Fig. S4A). shSTAT3 efficiently reduced IL-22-mediated STAT3 activation (Fig. S4A) and colon cancer cell proliferation (). shSTAT3 reversed the effects of IL-22 on the expression of p16 and p18, the cyclin-dependent kinase inhibitor genes (Fig. S4B). IL-22 stimulated the expression of PRC2 components (). We hypothesized that IL-22 activated STAT3, STAT3 bound to the promoters of PRC2 and controlled colon cancer proliferation via H3K27me3. In support of this possibility, bioinformatics analysis revealed that STAT3 has binding sites at the promoters of SUZ12 and EED () (http://www.sabiosciences.com/chipqpcrsearch.php). We performed ChIP assay with different pairs of primers in the proximal promoters of SUZ12 and EED, covering the putative STAT3 binding sites. We found that IL-22 significantly increased the binding of STAT3 to the promoters of SUZ12 () and EED (). The effects were abrogated in shSTAT3 expressing colon cancer cells (). As a positive control, we showed that IL-22 enhanced STAT3 binding to c-fos promoter, which was reduced in shSTAT3 expressing colon cancer cells (Fig. S4C). Knockdown of SUZ12 didn't change the phosphorylation level of STAT3 in colon cancer cells stimulated with IL-22 (Fig. S4D). Thus, IL-22 activates and promotes STAT3 binding to PRC2 promoters, and stimulates colon cancer cell proliferation by targeting cyclin repressors via H3K27me3 ().
Figure 4. IL-22 promoted STAT3 binding to the PRC2 promoters. (A) Activation of STAT3 by IL-22. Colon cancer cells were treated with IL-22 for different time points. Phosphorylated STAT3 and STAT3 proteins were detected by immunoblotting. One of three experiments is shown. (B) Effect of shSTAT3 on IL-22-induced DLD-1 cell proliferation. DLD-1 cells were transfected with control vector (scramble) or two vectors encoding shSTAT3, and stimulated with IL-22 for 24 h. Cell proliferation was detected by H3 thymidine incorporation. n = 3, p < 0.05. (C) Based on the ENCODE STAT3-ChIP-Seq data base, STAT3 occupancy on the promoter areas of SUZ12 and EED is shown. (D, E) Effect of IL-22 on the occupancy of STAT3 in the promoters of PRC2 components in DLD-1 cells. DLD-1 cells expressing control vector or shSTAT3 were cultured with IL-22 for 0.5 h. The occupancy of STAT3 in the promoters of SUZ12 (D) and EED (E) was determined by ChIP assays. n = 3, p < 0.05. (F) Scheme of the mode of action of Th22 cells in colon cancer cell proliferation. Th22 cells release IL-22, activate STAT3, and STAT3 binds to the promoters of PRC2 components causing tumor cell proliferation via H3K27me3-mediated p16 and p21 repression.
Discussion
In this study, we have observed that Th22 cell-derived IL-22 targets PRC2 complex and controls colon cancer cell proliferation.
In support of our observation, a procarcinogenic role for IL-22 has been observed in several mouse models including colitis-associated cancer,Citation20,21 hepatocellular carcinoma,Citation22 non-small cell lung cancer and breast cancer.Citation23,24 However, the mode of action of IL-22 is poorly understood in these tumor bearing mouse models. Furthermore, there is limited information on IL-22 oncogenic activity in patients with colon cancer.Citation5,25 Th22 cells may be expanded in the colon cancer environment in response to inflammation and over-absorption of luminal microbial products.Citation26 We have recently demonstrated that IL-22 is basically derived from Th22 cells in the human colon cancer microenvironment, and Th22 cell-derived IL-22 promotes colon cancer stemness via inducing DOT1L-mediated H3K79me2 in the core stem cell genes. In the current work, we have shown that Th22 cell-derived IL-22 activates STAT3, STAT3 binds to PRC2 complex, removes H3K27me3 histone modification in the promoters of p16 and p21, and consequently causes colon cancer cell proliferation. Therefore, Th22 cells contribute to colon carcinogenesis through two distinct histone modification-associated mechanisms.
High-levels of PCR2 components, EZH2, EED and SUZ12, are linked to active oncogenic activities in several types of human cancer.Citation14–16 However, it is poorly understood how PRC2 components are regulated in the tumor microenvironment. Genomic loss of microRNA101 and microRNA26a may be associated with enhanced expression of EZH2.Citation27 Interestingly, we have found that IL-22 strongly activate STAT3 in colon cancer cells and stimulates the expression of PRC2 complex. Given that IL-22 is derived from Th22 cells, the interaction between Th22 cells and cancer cells activates oncogenic PRC2 gene expression and consequently endows their oncogenic activities. Thus, our observation has revealed a novel tumor immune evasion mechanism whereby tumor cells recruit Th22 cells to form a favorable niche for tumor stemness and proliferation.
We have previously shown that STAT3 regulates colon cancer invasion by EZH2 mediated-histone modification of Vitamin D receptor.Citation28 Now we have analyzed the ChIP sequencing data and found that STAT3 has several binding sites in the promoters of SUZ12 and EED. In support of this, STAT3 can directly bind to the promoter of SUZ12 and EED, regulating their expression. Thus, we provide a new insight into the interplay between PRC2 and STAT3 in the context of Th22 (IL-22) biology.
In summary, we have demonstrated that Th22 cell-derived IL-22 activates STAT3, targets PRC2 to induce human colon cancer cell proliferation via the H3K27me3 of cell cycle check-point genes p16 and p21, and stimulates DOT1L to promote human colon cancer cell stemness via H3K79me2 of core stem cell genes.Citation5
Materials and methods
Cell culture
We prepared single cell suspension and established primary colon cancer cell lines (C1, C2, C3) from fresh colon cancer tissues.Citation5 CD4+ T cells were enriched and subsequently sorted from fresh colon cancer environmental single cell suspension. DLD-1 cells were obtained from ATCC one month before starting experiments. We didn't do authentication. Primary colon cancer cells and DLD-1 were cultured with the colon cancer environmental CD4+ T cells or with recombinant IL-22 (R&D) 20 ng/mL for different time points. Neutralizing anti-human IL-22 was added in the culture as indicated. The PRC2 inhibitor DZNep (5 µM) or DOT1L inhibitor EPZ004777 (10 µM) was added in the culture as indicated.
Lentiviral transduction
We used lentiviral vector encoding gene specific shRNAs (STAT3, EZH2, SUZ12) and scramble particles (Puromycin resistant) to transduce colon cancer cells and established stable cell lines. The transduction efficiency was confirmed by vector GFP expression and Western blotting.
Real-Time reverse-transcriptase polymerase chain reaction (RT-PCR)
The mRNAs were quantified by real-time RT-PCR. Specific primers were included in the supplementary information (Table S1). SYBR Green Master Mix was used to detect fluorescence. Relative expression was calculated according to the Ct value with normalization to GAPDH.
Western blot
Western Blot assay was performed with specific antibodies against human STAT3, phosphorylate STAT3 (Cell Signaling), EED, SUZ12(Santa Cruz biotechnology), EZH2 (Abcam), H3K27me3 and H3K79me2 (Millipore).
Chromatin immunoprecipitation (ChIP)
ChIP assay was performed according to the protocol (Upstate, Millipore) as previously described.Citation5,29 Crosslinking was performed with 1% formaldehyde or 1% paraformaldehyde for 10 min. To enhance cell lysis, we ran the lysate through a 27 g needle three times and flash froze it in -80°C. Sonication was then performed with the Misonix 4,000 water bath sonication unit at 15% amplitude for 10 min. Protein/DNA complex was precipitated by specific antibodies against H3K27me3 (Millipore), STAT3 (Cell Signaling) and IgG control (Millipore). Then DNA was purified using DNA Purification Kit (Qiagen). ChIP-enriched chromatin was used for Real-Time PCR, relative expression level is normalized to Input. Specific primers are listed in supplementary information (Table S2).
Statistical analyses
Mann–Whitney U test or Student's t test was used in the statistical analysis. p values less than 0.05 were considered significant. Statistical analyses were done with SAS 9.3 software. Data were shown as mean ± SEM or mean ± SD.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
Danfeng Sun, Yanwei Lin, Nisha Nagarsheth, Dongjun Peng and Shuang Wei performed the experiments, analyzed data, and wrote the paper. Jie Hong, Haoyan Chen and Jingyuan Fang provided Gene Set Enrichment Analysis. Emina Huang provided clinical specimens and intellectual support. Ilona Kryczek and Weiping Zou designed the experiments and wrote the paper. All authors read and approved the manuscript.
1082704_supplemental_files.pdf
Download PDF (521.8 KB)Acknowledgments
We thank Deborah Postiff, Michelle Vinco and Jackline Barikdar in the Tissue Procurement Core for their technical assistance.
Funding
This work is supported (in part) by the National Institutes of Health grants (CA123088, CA099985, CA156685 and CA171306 for WZ; CA142808 and CA157663 for EH), and (in part) by the grants from National Natural Science Foundation of China (81320108024).
References
- Deng J, Liu Y, Lee H, Herrmann A, Zhang W, Zhang C, Shen S, Priceman SJ, Kujawski M, Pal SK et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell 2012; 21:642-54; PMID:22624714; http://dx.doi.org/10.1016/j.ccr.2012.03.039
- Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol 2011; 12:21-7; PMID:21113163; http://dx.doi.org/10.1038/ni.1962
- Basu R, O'Quinn DB, Silberger DJ, Schoeb TR, Fouser L, Ouyang WJ, Hatton RD, Weaver CT. Th22 Cells Are an Important Source of IL-22 for Host Protection against Enteropathogenic Bacteria. Immunity 2012; 37:1061-75; PMID:23200827; http://dx.doi.org/10.1016/j.immuni.2012.08.024
- Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012; 485:599-604; PMID:22660318; http://dx.doi.org/10.1038/nature11139
- Kryczek I, Lin Y, Nagarsheth N, Peng D, Zhao L, Zhao E, Vatan L, Szeliga W, Dou Y, Owens S et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 2014; 40:772-84; PMID:24816405; http://dx.doi.org/10.1016/j.immuni.2014.03.010
- Yu LZ, Wang HY, Yang SP, Yuan ZP, Xu FY, Sun C, Shi RH. Expression of interleukin-22/STAT3 signaling pathway in ulcerative colitis and related carcinogenesis. World J Gastroenterol 2013; 19:2638-49; PMID:23674871; http://dx.doi.org/10.3748/wjg.v19.i17.2638
- Jiang R, Wang H, Deng L, Hou J, Shi R, Yao M, Gao Y, Yao A, Wang X, Yu L et al. IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer 2013; 13:59; PMID:23379788; http://dx.doi.org/10.1186/1471-2407-13-59
- Andoh A, Zhang Z, Inatomi O, Fujino S, Deguchi Y, Araki Y, Tsujikawa T, Kitoh K, Kim-Mitsuyama S, Takayanagi A et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology 2005; 129:969-84; PMID:16143135; http://dx.doi.org/10.1053/j.gastro.2005.06.071
- Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity 2004; 21:241-54; PMID:15308104; http://dx.doi.org/10.1016/j.immuni.2004.07.007
- Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med 2008; 14:275-81; PMID:18264110; http://dx.doi.org/10.1038/nm1710
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 2008; 14:282-9; PMID:18264109; http://dx.doi.org/10.1038/nm1720
- Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, Lehr HA, Hirth S, Weigmann B, Wirtz S et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med 2009; 206:1465-72; PMID:19564350; http://dx.doi.org/10.1084/jem.20082683
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129:823-37; PMID:17512414; http://dx.doi.org/10.1016/j.cell.2007.05.009.
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002; 419:624-9; PMID:12374981; http://dx.doi.org/10.1038/nature01075
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A 2003; 100:11606-11; PMID:14500907; http://dx.doi.org/10.1073/pnas.1933744100
- Mimori K, Ogawa K, Okamoto M, Sudo T, Inoue H, Mori M. Clinical significance of enhancer of zeste homolog 2 expression in colorectal cancer cases. Eur J Surg Oncol 2005; 31:376-80; PMID:15837043; http://dx.doi.org/10.1016/j.ejso.2004.11.001
- Cao R, Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev 2004; 14:155-64; PMID:15196462; http://dx.doi.org/10.1016/j.gde.2004.02.001
- Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics 2007; 23:3251-3; PMID:17644558; http://dx.doi.org/10.1093/bioinformatics/btm369
- Yu W, Chory EJ, Wernimont AK, Tempel W, Scopton A, Federation A, Marineau JJ, Qi J, Barsyte-Lovejoy D, Yi J et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun 2012; 3:1288; PMID:23250418; http://dx.doi.org/10.1038/ncomms2304
- Kamanaka M, Huber S, Zenewicz LA, Gagliani N, Rathinam C, O'Connor W, Jr., Wan YY, Nakae S, Iwakura Y, Hao L et al. Memory/effector (CD45RB(lo)) CD4 T cells are controlled directly by IL-10 and cause IL-22-dependent intestinal pathology. J Exp Med 2011; 208:1027-40; PMID:21518800; http://dx.doi.org/10.1084/jem.20102149
- Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, Hedl M, Zhang W, O'Connor W, Jr., Murphy AJ et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012; 491:259-63; PMID:23075849; http://dx.doi.org/10.1038/nature11535
- Jiang R, Tan Z, Deng L, Chen Y, Xia Y, Gao Y, Wang X, Sun B. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology 2011; 54:900-9; PMID:21674558; http://dx.doi.org/10.1002/hep.24486
- Zhang W, Chen Y, Wei H, Zheng C, Sun R, Zhang J, Tian Z. Antiapoptotic activity of autocrine interleukin-22 and therapeutic effects of interleukin-22-small interfering RNA on human lung cancer xenografts. Clin Cancer Res 2008; 14:6432-9; PMID:18927282; http://dx.doi.org/10.1158/1078-0432.CCR-07-4401
- Kim K, Kim G, Kim JY, Yun HJ, Lim SC, Choi HS. Interleukin-22 promotes epithelial cell transformation and breast tumorigenesis via MAP3K8 activation. Carcinogenesis 2014; 35:1352-61; PMID:24517997; http://dx.doi.org/10.1093/carcin/bgu044
- Huang YH, Cao YF, Jiang ZY, Zhang S, Gao F. Th22 cell accumulation is associated with colorectal cancer development. World J Gastroenterol 2015; 21:4216-24; PMID:25892871; http://dx.doi.org/10.3748/wjg.v21.i14.4216
- Kim CJ, Nazli A, Rojas OL, Chege D, Alidina Z, Huibner S, Mujib S, Benko E, Kovacs C, Shin LY et al. A role for mucosal IL-22 production and Th22 cells in HIV-associated mucosal immunopathogenesis. Mucosal Immunol 2012; 5:670-80; PMID:22854709; http://dx.doi.org/10.1038/mi.2012.72
- Nishikawa S, Dewi DL, Ishii H, Konno M, Haraguchi N, Kano Y, Fukusumi T, Ohta K, Noguchi Y, Ozaki M et al. Transcriptomic study of dormant gastrointestinal cancer stem cells. Int J Oncol 2012; 41:979-84; PMID:22735680.
- Lin YW, Ren LL, Xiong H, Du W, Yu YN, Sun TT, Weng YR, Wang ZH, Wang JL, Wang YC et al. Role of STAT3 and vitamin D receptor in EZH2-mediated invasion of human colorectal cancer. J Pathol 2013; 230:277-90; PMID:23424038; http://dx.doi.org/10.1002/path.4179
- Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, Vatan L, Szeliga W, Mao Y, Thomas DG et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 2013; 39:611-21; PMID:24012420; http://dx.doi.org/10.1016/j.immuni.2013.08.025