
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Human telomerase reverse transcriptase (hTERT) is overexpressed in more than 85% of human cancers regardless of their cellular origin. As immunological tolerance to hTERT can be overcome not only spontaneously but also by vaccination, it represents a relevant universal tumor associated antigen (TAA). Indeed, hTERT specific cytotoxic T lymphocyte (CTL) precursors are present within the peripheral T-cell repertoire. Consequently, hTERT vaccine represents an attractive candidate for antitumor immunotherapy. Here, an optimized DNA plasmid encoding an inactivated form of hTERT, named INVAC-1, was designed in order to trigger cellular immunity against tumors. Intradermal injection of INVAC-1 followed by electrogene transfer (EGT) in a variety of mouse models elicited broad hTERT specific cellular immune responses including high CD4+ Th1 effector and memory CD8+ T‑cells. Furthermore, therapeutic INVAC‑1 immunization in a HLA-A2 spontaneous and aggressive mouse sarcoma model slows tumor growth and increases survival rate of 50% of tumor-bearing mice. These results emphasize that INVAC-1 based immunotherapy represents a relevant cancer vaccine candidate.
Introduction
Among a wide range of tumor antigens,Citation1 hTERT is a very attractive and important target for anticancer immunotherapeutic approaches. Human TERT is the rate-limiting catalytic subunit of the telomerase complex, a ribonucleoprotein enzyme which synthesizes telomeric DNA at chromosome ends.Citation2 It prevents apoptosis through the telomere‑dependent pathway promoting cell immortality effectively allowing cells to grow indefinitely. These properties have been described in malignant cell transformation as some of the hallmarks in cancer genesis.Citation3 Human TERT is overexpressed in the vast majority (80–90%) of cancer cells regardless of origin,Citation4 and is associated with poor prognosis.Citation5 This overexpression leading to presentation of hTERT peptides at the surface of tumor cells enables recognition and destruction of these tumor cells by a natural anti‑hTERT immune response. This response has been highlighted in some cancer patients revealing natural break of self‑tolerance.Citation6 Given its widespread expression in tumor cells, hTERT is considered to be a near universal TAA.Citation7 The crucial role of hTERT in oncogenesis justifies its use in clinical immunotherapy as a treatment for cancer.
To date, different clinical approaches have been explored based on MHC class I or II restricted hTERT peptides, autologous antigen-presenting cells (APCs; dendritic cells or B lymphocytes) loaded with hTERT peptides or transduced with hTERT mRNA.Citation8 Unfortunately, these strategies suffer from some limitations and disadvantagesCitation9 and have had limited clinical impact for most patientsCitation7 despite the induction of specific immune responses.
DNA vaccines offer numerous advantages such as the flexibility to incorporate easily multiple genes which encode either full‑length or partial tumor antigens and/or immunostimulatory molecules.Citation10 In addition, several add-on features have been explored such as the use of strong viral promoters, codon‑optimized genes, the addition of immunoglobulin sequences or the enrichment of CpG motifs in the plasmid backbone.Citation11 On top of this DNA is safe, stable and easy to manufacture. Despite these traits initial DNA vaccination studies showed that DNA was generally poorly immunogenic,Citation12 especially when self‑antigens were studied. Recent improvements in EGT, or DNA electroporation, such as the use of a sequence of high volt-low volt pulses have greatly increased the efficiency of DNA vaccination.Citation13 There are now many ongoing clinical trials using DNA vaccination for cancer immunotherapy or against infectious diseases.Citation14
We report the development of INVAC-1, a DNA plasmid for cancer immunotherapy. It encodes a modified full-length hTERT protein devoid of telomerase catalytic activity fused to human ubiquitin. INVAC-1 triggered broad and strong hTERT specific cytotoxic and Th1 immune responses in different mouse strains immunized through the intradermal route (ID) combined with an optimized EGT protocol. It shows an antitumor activity against a spontaneous and aggressive mouse sarcoma. These pharmacological data support the use of INVAC-1 in human clinical trials for the treatment of various types of cancers.
Results
Expression, subcellular localization and catalytic inactivity of INVAC‑1 hTERT protein
INVAC‑1 DNA construct encodes a nine bp deletion coding for three critical amino acids (867‑869, Valine-Aspartic Acid-Aspartic Acid or VDD) in the catalytic site. They are highly conserved across polymerases and mutation of any of these residues resulted in catalytic inactivation.Citation15 The nucleolar localization sequence (NoLS) at the N-terminal region (residues 1–47) was deleted and replaced by a complete 76 residue human ubiquitin (Ubi, MW = 8.4 kDa) moiety. Ubiquitin-fused proteins enter into a rapid proteasome-dependent degradation pathway leading to enhanced MHC class I peptide presentation and improved specific immune responses to a variety of antigens.Citation16 Telomerase expression in HEK293T cells was assessed by protein gel blotting (). INVAC‑1 construct expressed 2 distinct hTERT specific proteins, a weak upper band corresponding to the Ubi-hTERT fusion protein at the predicted size of 127.4 kDa and a major lower band corresponding probably to INVAC‑1 hTERT protein lacking the ubiquitin sequence (119 kDa). By contrast, wild‑type hTERT protein was detected at 124.5 kDa as expected. The INVAC‑1 hTERT protein signal disappeared over time indicating that it was rapidly degraded, likely through the ubiquitin‑dependent proteasome pathway, in contrast to wild-type hTERT protein which was stable out to 96 h ().
Figure 1. In vitro characterization of INVAC-1 hTERT protein. (A) Schematic maps and alignments of wild‑type hTERT and INVAC‑1 hTERT proteins. VDD: deletion in catalytic site at position AAs 867‑869. NoLS: Nucleolar Localization Signal. Ubi: ubiquitin, F/R/G: Amino acids flanking the NoLS to Ubi substitution. (B) Expression kinetics of wild‑type hTERT and INVAC‑1 hTERT protein monitored from18 to 96h post‑transfection into HEK293T cells. hTERT protein detection was performed using an anti‑hTERT rabbit monoclonal antibody. NTC: Empty vector backbone as negative control. β‑actin protein detection was used as a loading control assessment. (C) Intracellular localization of wild‑type hTERT and INVAC‑1 hTERT proteins in transfected QT6 cells visualized 24h post‑transfection with an anti‑hTERT rabbit monoclonal antibody and a goat anti‑rabbit secondary antibody‑Alexa Fluor 488® conjugate (green fluorescence). NTC: Empty vector backbone as negative control. NT: Non‑transfected cells. The nuclei were stained with DAPI (blue). The cells were analyzed for both fluorescence wavelengths (merged) upon fluorescence microscopy (x63). (D) Neutralization of INVAC‑1 telomerase catalytic activity. Total cell proteins were extracted from wild‑type hTERT and INVAC‑1 transfected CrFK cells and telomerase activity was assessed by TRAP assay. RTA (sample/positive control ratio) of INVAC‑1 compared to wild‑type hTERT and non-treated (NT) CrFK cells are displayed (n = 3 for 2.1 μg of total protein samples) using absorbance measurements values (OD450/690 nm). Mann–Whitney non-parametric test against non‑treated CrFK cells, **p < 0.01.
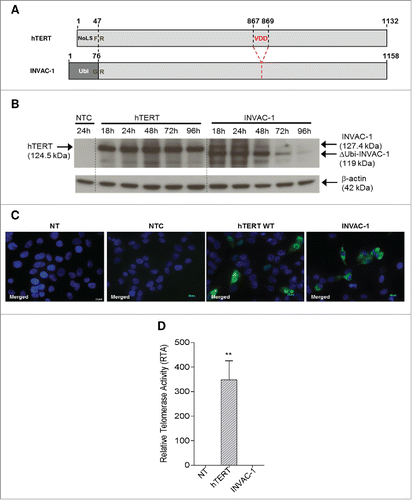
The impact of the NoLS deletion on the cellular localization was assessed in transfected QT6 cells, which do not express cross-reacting telomerase antigens, the avian and human enzymes sharing ∼50% amino acid homology. INVAC‑1 hTERT protein was localized in the cytoplasm, both at 24 h () and 48 h (data not shown), while wild‑type hTERT was mainly detected in the nucleus and nucleolus showing that the deletion of the nucleolar localization signal drastically altered the subcellular distribution.
In order to confirm the lack of INVAC‑1 hTERT activity, a critical requirement for any human trials, telomerase activity was determined in transfected CrFK cells using a telomeric repeat amplification protocol (TRAP) assay. Relative telomerase activity (RTA) data showed that INVAC‑1 hTERT protein was completely devoid of any telomerase activity in comparison to wild‑type hTERT control (). Taken together, these results demonstrate that INVAC‑1 hTERT protein displayed the characteristics and properties expected given the modifications engineered into the INVAC‑1 plasmid construct.
INVAC‑1 induces broad and strong hTERT specific T‑cell responses
CD8+ T-cells are known to be essential effectors involved in the elimination of tumor cells while CD4+ play a major role in orchestrating the global antitumor response.Citation17,18 To evaluate whether DNA vaccination with INVAC‑1 could trigger specific CD8+/CD4+ T‑cell-mediated immune responses, C57BL/6 (H2‑b) and BALB/c (H2‑d) mice and Tg mice (HLA‑B7 and HLA‑A2/DR1) were immunized with INVAC‑1. Fourteen days after, specific immune responses in spleen were monitored via an IFNγ ELISpot assay using specific hTERT peptides. As shown in , INVAC‑1 immunized mice generated a significantly higher hTERT specific CD8+ T‑cell response compared to any strain of control mouse (p < 0.01). In the same way, a significant CD4+ T‑cell response restricted to HLA‑DRB1 was detected in HLA‑A2/DR1 immunized mice in comparison to controls (p < 0.01).
Figure 2. INVAC‑1 induces hTERT specific T‑cell responses in mice. (A) C57BL/6 mice (6 mice per group), BALB/c mice (6‑7 mice per group), HLA‑B7 mice (4‑6 mice per group) and HLA‑A2/DR1 mice (5-6 mice per group) were immunized once. Fourteen days later, an IFNγ ELISpot assay was performed with splenocytes stimulated with a pool of hTERT restricted peptides according to mouse MHC. IFNγ hTERT specific CD8+ or CD4+ T‑cells/200,000 splenocytes are represented as mean ± SD. Mann–Whitney non-parametric test against mice control, **p < 0.01. (B) HLA‑A2/DR1 mice (3‑5 mice per group) were immunized twice (prime-boost) with INVAC‑1 (D0 and D21). At D31, splenocytes were Ficoll purified and stimulated with a pool of 3 hTERT specific peptides restricted to HLA‑DRB1. Supernatants from stimulated cells were recovered and tested in a cytokine binding assay in order to evaluate the concentration of Th1, Th2 and Th17 cytokines secreted by hTERT specific CD4+ T‑cells. Cytokine concentrations in pg/mL are represented as mean ± SD. Mann–Whitney non-parametric test against mice control, *p < 0.05.
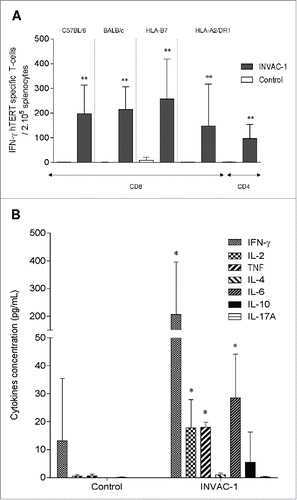
CD4+ T helper 1 (Th1) cells secreting several cytokines such as IFNγ, TNF and IL‑2 are particularly important for the induction of efficient cell‑mediated immunity against tumors. Consequently, the polarization profile of hTERT specific CD4+ T‑cells induced in HLA‑A2/DR1 after INVAC‑1 vaccination was investigated using a cytokine binding assay. Results showed not only that significant concentrations of IL‑2, TNF and IFNγ, Th1 cytokines (), but also IL‑6 were detected in supernatants from immunized mice in comparison with control mice (p < 0.05). Thus, INVAC‑1 vaccination is able to promote expansion of hTERT specific CD8+ T‑cells and specific CD4+ T‑cells with a predominant Th1 profile.
Prime‑boost vaccination enhanced, shortened and broadened hTERT specific responses
Most vaccination protocols are based on a prime‑boost regimen in order to improve the frequency of vaccine specific immune responses. Consequently, the impact of prime‑boost vaccination on the generation of hTERT specific CD8+ T‑cell response was evaluated in C57BL/6 immunized four times with INVAC‑1. The hTERT specific CD8+ T‑cell response was monitored in PBMCs over time by an IFNγ ELISpot assay using H2‑Kb/Db restricted hTERT peptides (). hTERT specific CD8+ T‑cell responses were observed 14 d post‑priming (mean # spots: 38, p <0.001). The second vaccination (first boost) shortened the time to generate hTERT specific immune responses (10 d after the boost) although this response was not higher (mean # spots: 40, p <0.001). Interestingly, this immune response was long lasting since it was still detected at D118 (mean # spots: 9) suggesting the establishment of a memory response. As is often observed following CD8+ T‑cell activation and expansion, a contraction phase occurs before the generation of a stable memory population.Citation19 To demonstrate this hypothesis, mice were vaccinated twice more at D123 and D144. As expected, responses were faster and higher (at D154, mean #spots: 96, p < 0.001) confirming the establishment of a hTERT specific CD8+ memory response.
Figure 3. Prime‑boost vaccination enhanced, shortened and broadened hTERT‑specific T‑cell responses. (A) Ten C57BL/6 mice were immunized four times with INVAC-1 (D0, D21, D123 and D144). Peripheral blood was collected before the first immunization D0, and at D7, D14, D21, D33, D40, D118, D132, D139, D154, D159 and D166. PBMCs were Ficoll purified and stimulated with a pool of 4 hTERT specific peptides restricted to the H2‑Kb/Db and analyzed by an IFNγ ELISPOT assay. Black arrows represent days of vaccination. IFNγ hTERT specific CD8+ T‑cells/200,000 PBMCs are represented as mean ± SD. Mann–Whitney non-parametric test against D0, *p < 0.05. (B) Seven to 13‑weeks‑old C57BL/6 mice (6 mice per group) were immunized twice (D0 and D21). At D31, splenocytes were Ficoll purified and stimulated with 13 pools of hTERT 15‑mer overlapping peptides (10 peptides/pool) and analyzed by an IFNγ ELISPOT assay. IFNγ hTERT specific T‑cells /200,000 splenocytes are represented as mean ± SD.
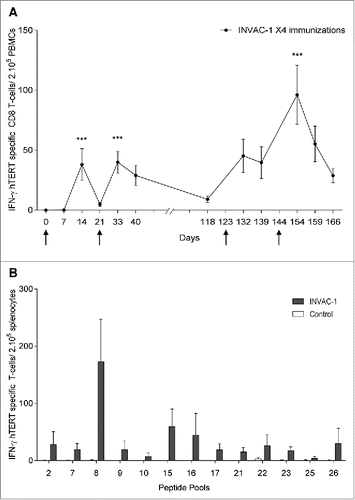
The breadth of the hTERT specific immune response was studied in a homologous prime‑boost approach in HLA‑B7 mice immunized with INVAC‑1 at D0 and D21. At D31, this immune response was analyzed by an IFNγ ELISpot assay using 13 pools of overlapping peptides spanning the entire hTERT sequence. As shown in , INVAC‑1 immunization induced a broad repertoire of T‑cells against numerous hTERT epitopes (at least 13 epitopes) as all peptide pools were able to stimulate T‑cells.
Generation of hTERT specific cytotoxic CD8+ T cell responses
Having established that the INVAC‑1 DNA vaccine was able to induce CD4+ and CD8+‑mediated immunity, it was essential to establish that CD8+ T‑cells displayed cytotoxic activity. Toward this end, we focused on granzyme B (GrB), a key mediator of target cell death secreted by T‑cells. The GrB secretion was evaluated using an ELISpot assay in HLA‑B7 Tg mice immunized twice. As shown in , a higher frequency of hTERT specific CD8+ T‑cells secreting GrB was detected in splenocytes from INVAC‑1 immunized mice as compared to controls (p < 0.01). In a second step, the in vivo cytotoxic activity was evaluated by flow cytometry using CFSE–labeled and peptide‑pulsed splenocytes in mice. To this end, C57BL/6 mice were immunized twice and the specific killing activity was evaluated 10 d post‑vaccination. There was a decrease of highly-labeled CFSE cells pulsed with the immunodominant peptide p660 in INVAC‑1 immunized mice as compared to controls. There was also a slight decrease of medium-labeled CFSE cells pulsed with the subdominant peptide p1034 (). Approximately 50% of p660 pulsed‑cells and 14% of p1034 pulsed‑cells were killed in INVAC‑1 immunized mice ().
Figure 4. INVAC‑1 induces hTERT specific cytotoxic T‑cells. (A) HLA‑B7 mice (4‑6 mice per group), were immunized twice (D0 and D21). At D31, an ELISpot granzyme B (GrB) assay was performed with splenocytes stimulated with a pool of hTERT HLA‑B7 restricted peptides. GrB hTERT specific CD8+ T‑cells/200,000 splenocytes are represented as mean ± SD. Mann–Whitney non-parametric test against mice control, **p < 0.01. (B) C57BL/6 mice (8 mice per group) were immunized twice (D0 and D21). At D31, syngeneic splenocytes, pulsed with individual hTERT peptides restricted to the H2‑Kb/Db (either p660 or p1034) were labeled with CFSE and injected IV to immunized mice. After 15‑18 h, the disappearance of peptide‑pulsed cells in spleens was analyzed by flow cytometry. (C) Percent of killing was presented as mean ± SD. (D) C57BL/6 mice (6 mice per group) were injected s.c. in the right flank with 2 × 105 TC‑1 cells. On day 5 (when tumors are palpable), mice were immunized with INVAC‑1 or left non-treated (NT). Fourteen days later, mice were sacrificed and splenocytes, tumor draining lymph nodes and tumor infiltrated lymphocytes were isolated. An ELISpot IFNγ was performed with H2‑Kb/Db restricted peptides (p429, p660, p1021 and p1034). IFNγ hTERT specific CD8+ T-cells/200,000 cells are represented as mean ± SD. Mann–Whitney non-parametric test **p < 0.01.
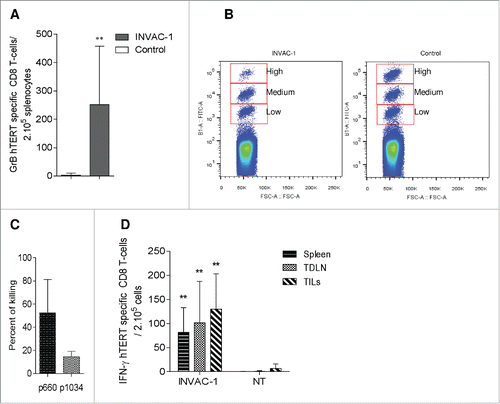
CTL infiltration in tumors is of paramount importance for the efficiency of cancer immunotherapy approaches. Therefore, the infiltration of hTERT specific CD8+ T‑cells in tumors was assessed. TC‑1 bearing mice were vaccinated at D5 (palpable tumors), sacrificed at D19 and splenocytes, tumor draining lymph nodes cells and tumor infiltrated lymphocytes were isolated and assessed with an IFNγ ELISpot assay using H2‑Kb/Db restricted peptides. As shown in , hTERT specific CD8+ T‑cells were significantly detected in all samples from INVAC‑1 immunized mice as compared to controls (p < 0.01) suggesting the capacity of these cells to circulate and migrate into the tumor. Hence, INVAC‑1 immunization generated hTERT specific CD8+ T cells that exhibited in vivo cytotoxic activity probably through the GrB mediated pathway and which can infiltrate the tumor.
hTERT specific immune response mediated by INVAC‑1 delays tumor growth
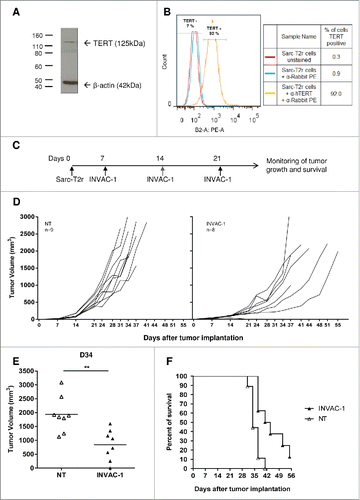
To evaluate whether the INVAC-1 mediated immune response can have an antitumor effect, the Sarc-T2r tumor model was used. This tumor cell line was characterized by Western blot () and flow cytometry () using specific anti-hTERT monoclonal antibodies. At least 92% of Sarc-T2r cells in culture expressed the mTERT protein at the predicted size of 125 kDa. HLA-A2/DR1 mice were inoculated with 2 × 105 Sarc-T2r cells via s.c. route on D0. On D7 (palpable tumors), D14 and D21 mice were immunized with INVAC‑1 or left non-treated (). Individual tumor growth is shown in . INVAC‑1 delayed the growth of Sarc-T2r tumors. At D34, when the majority of mice were still alive, tumor volumes of mice immunized with INVAC‑1 were smaller as compared to non-treated mice (p < 0.01) (). INVAC‑1 also induced longer survival (). Four out of eight INVAC‑1‑immunized mice were still alive at D41 while all mice in the non‑treated (NT) group were either dead or euthanized. In conclusion, vaccination with INVAC‑1 delayed tumor growth and improved survival rate of tumor‑bearing HLA‑A2/DR1 mice.
Figure 5. INVAC-1 induces antitumor immunity. (A) Murine TERT protein detection into Sarc-T2r cells was performed using an anti‑hTERT rabbit monoclonal antibody. β‑actin protein detection was used as a loading control assessment. (B) Staining of Sarc-T2r cells was achieved with the same anti-hTERT antibody followed by a secondary goat anti‑rabbit PE antibody (orange histogram). Sarc-T2r cells stained with secondary goat anti‑rabbit PE antibody (blue histogram) and unstained Sarc-T2r cells (red histogram) were used as controls. (C) HLA‑A2/DR1 mice (8–9 mice per group) were injected s.c. in the right flank with 2 × 105 Sarc-T2r cells. On days 7 (when tumors are palpable), D14 and D21, mice were immunized with INVAC‑1 or left non-treated (NT). Tumor sizes were measured twice a week. (D) Plots represent tumor volume growth kinetics of individual mice for both groups. (E) Tumor volumes of individual mice on day 34, when the majority of mice were still alive. Tumor volumes are represented as mean ± SD. Mann–Whitney non-parametric test **p < 0 .01. (F) Kaplan–Meier plot of overall survival.
Discussion
Human TERT is considered as a near universal tumor antigen for immunotherapy approaches and thus represents a relevant target for such a therapeutic strategies. Indeed, it has been demonstrated that hTERT specific T‑cell responses can be induced in vitro in HLA-A*0201 patients with prostate cancer as well as in healthy donors suggesting the existence of T‑cell precursors for hTERT which is the basis for breaking tolerance.Citation6 In addition, previous clinical vaccination strategies using the telomerase antigen as target, such as GV1001 peptide showed induction of a robust hTERT specific immune response in pancreatic and NSCLC cancer with clinical benefits for responding patients.Citation8 Many groups have demonstrated natural anti‑hTERT CTLs and CD4+ T‑cell responses as Godet et al.Citation20 These findings justified our choice to develop INVAC-1, a hTERT DNA vaccine for cancer immunotherapy.
INVAC‑1 incorporates a number of safety features. Deletion of the catalytic VDD triplet ensured a inactive form of the protein that was essential given that hTERT plays a critical role in tumorigenesis process. The NoLS deletion led to drastic alteration of its subcellular distribution. In addition, this modified protein was fused to ubiquitin taking into account the so‑called N‑end rule (destabilizing residue at N‑terminal position) to oriente its degradation through the ubiquitin-dependent proteasome pathway. Protein expression assay showed that INVAC-1 hTERT was rapidly degraded by the proteasome in contrast with wild‑type hTERT. Proteasome targeting systems such as ubiquitin fusion and N‑end rule have been shown to play a key role for efficient proteasomal degradation, and subsequently for antigen presentation through the MHC class I pathway.Citation21 Indeed, ubiquitin‑fused DNA vaccines have show a significant improvement of the antigen‑specific cellular immune response.Citation16
Several studies were carried out to optimize the immunization procedure for INVAC‑1 vaccine delivery (data not shown), especially the EGT process.Citation13 DNA vaccination through the ID route has been shown to be more efficient than intramuscular or subcutaneous routes of immunization.Citation22 Indeed, skin is readily accessible, presents a large surface for vaccination and constitutes a relevant immunogenic tissue due to a rich network of immunocompetent APCs such as Langerhans cells and dermal DCs.Citation23 To enhance the immune response elicited by INVAC-1 vaccination, we developed EGT using skin‑specific parameters to be used in humans with the CLINIPORATOR®2 (IGEA, Italy).Citation13
Recent improvements in EGT technology have reemphasized the interest in DNA vaccination.Citation10 Indeed, EGT was shown to induce a robust antigen-specific T-cell response in HPV-infected women.Citation24 This combination is a relevant alternative to peptide and viral vector-based immunotherapy vaccine strategies since EGT enhances DNA uptake and improves peptide presentation through MHC molecules by antigen presenting cells in vivo.Citation25 Furthermore, DNA is suitable for the expression of large proteins such as hTERT, for which peptides may be presented by various MHC molecules matching the majority of HLA haplotypes in the human population.Citation9
Numerous studies have demonstrated that CD4+ Th1 and CD8+ immune responses elicited by DNA vaccination are crucial to mediate efficient antitumor immune responses.Citation10 INVAC‑1 immunization induced high frequencies of hTERT specific CD4+ and CD8+ T‑cells producing IFNγ in different mouse strains. These results confirmed that hTERT antigen was well processed in vivo through both endogenous and exogenous pathways allowing peptide epitope presentation on numerous MHC class I and class II. We have also shown that hTERT specific CD4+ T‑cells elicited by INVAC‑1 presented a Th1 polarization profile. These results are consistent with other studies demonstrating that the antigen specific CD8+ T‑cell priming and expansion require the help of antigen specific CD4+ Th1 cells.Citation26 Similarly, Antony et al. demonstrated that the maintenance of antigen specific CD8+ T‑cells was also dependent on IL‑2-secreting CD4+ Th1 cells. Furthermore, the secretion of IL‑2 supports the establishment of memory response which has been shown to be important for improving the frequency of specific T‑cells.Citation27 In this study, we demonstrated that a homologous prime‑boost regimen amplified hTERT specific CD8+ T‑cells by generating a large number of secondary hTERT specific responses and by expanding rapidly the existing antigen specific memory T‑cells which encounter the same antigen a second time round.
The broad response induced by INVAC-1 against numerous hTERT epitopes spanning the entire protein confirmed the advantage of using the full‑length antigen as compared to individual epitopes used in peptide vaccine development.Citation28 By encoding full‑length protein, the whole MHC diversity among the human population is expected to be covered while tumor escape mechanisms should be limited. Indeed, during tumor immunoediting, MHC expression or presentation of tumor antigens decrease on cells leading to the generation of variants resistant to immune effector cell.Citation29
In some cases, the lack of a consistent correlation between the magnitude of the antigen‑specific T‑cell response and the control of tumor growthCitation17,18 indicates that rather than the quantity of specific T‑cells it is necessary to generate polyfunctional effector T‑cells.Citation19 These quality responses depend on their ability to proliferate, migrate, coordinate the immune responses and carry out effector functions by directly killing tumor cells through cytotoxic mechanisms or secretion of cytokines.Citation30 hTERT specific CD8+ T‑cells induced by INVAC‑1 have the capacity to secrete GrB and kill target cells in vivo, as well as to produce IFNγ and TNF (data not shown). These characteristics highlight the quality of hTERT specific CD8+ T‑cells induced by INVAC‑1. Another important consideration is that effector T‑cells must efficiently migrate to the tumor microenvironment in order to control malignant progression.Citation31 Tumor‑infiltrating lymphocytes (TILs) secreting perforin and Th1/CTL cytokines with lytic potential have been shown to improve clinical outcomes by inhibiting tumor growth or tumor recurrence in multiple human cancers.Citation32,33 In the present study, the presence of circulating and tumor‑infiltrating hTERT specific CD8+ T‑cells with functional characteristics were observed. These results showed that these cells are able to reach primary tumors and probably disseminated metastases. Other investigations such as phenotypic characteristics or cytotoxic strength need to be further assessed in order to evaluate their functional status. Indeed, Appay et al. studied tumor-infiltrated lymphocytes on s.c. lesions obtained from two vaccinated patients. They found that Melan‑A‑specific CD8+ T‑cells infiltrated in the tumor, although activated, appeared with suboptimal functional capacities compared with circulating Melan‑A‑specific CTL.Citation34
Therapeutic vaccination with INVAC-1 delayed the tumor growth of Sarc-T2r tumors in HLA‑A2/DR1 transgenic mice expressing human MHC class I and class II. This antitumor effect is likely related to hTERT polyfunctional T-cells induced by INVAC-1. Indeed, the overall amino acid identity between mTERT and hTERT is 64%.Citation35 Moreover, it has been demonstrated that TERT peptides restricted by HLA-A2 can be endogenously processed and presented by human and murine tumor cells, which in turn are recognized by specific CTLs.Citation36,37 Taken together, these observations suggest that hTERT specific T-cells induced by INVAC-1 could recognize mTERT peptides presented by MHC class I HLA-A2 expressed on Sarc-T2r cell surface.
Other studies showed that hTERT DNA vaccines induced similar results against TS/A murine breast cancerCitation38 or murine lung cancerCitation39 in syngeneic tumor models. Tumors often generate systemic immune suppressed or tolerogenic states which reduce the efficacy of immunotherapy.Citation40 FoxP3+CD4+ T‑cells (Treg), myeloid-derived suppressor cells in Sarc-T2r tumors and a downregulation of MHC I expression by tumor cells were evidenced in INVAC‑1‑immunized mice (data not shown). These components of tumor escape mechanisms could prevent complete tumor responses in the Sarc-T2r tumor model. In order to circumvent these hurdles, combination strategies need to be considered. Numerous studies showing synergistic antitumor effects by combining active immunotherapy with other therapies aimed at either reducing the bulk of tumors (such as chemotherapy or radiotherapy) or potentiating the immune response with drugs against immune checkpoint inhibitors (such as anti‑CTLA‑4 or anti-PD‑1 antibodies). For example, it has been shown that a peptide‑based vaccine combined with an anti‑PD‑1 antibody and low‑dose of cyclophosphamide induced synergistic antigen‑specific immune responses.Citation41 Likewise, in colon cancer patients, co‑administration of a platin‑based chemotherapy (oxaliplatin) and a carcinoembryonic antigen peptide pulsed DCs induced a higher level of immune response in patients receiving chemotherapy as compared to patients who did not received.Citation42
In conclusion, the pharmacological results presented in this report showed that treatment with INVAC‑1 induced a reliable antitumor immunity and a moderate survival advantage in murine models. Additional data from safety pharmacology, toxicology and biodistribution studies showed that treatment with INVAC‑1 was safe and well tolerated. Taken together, these data were considered adequate and reliable to support a First‑In‑Human phase I study by French competent authorities. The clinical study, evaluating INVAC‑1 in patients with advanced cancer, is currently ongoing in two clinical centers (https://clinicaltrials.gov/).
Materials and methods
INVAC-1 plasmid DNA
INVAC-1 carries a nine bp deletion that removes three crucial amino resides from the catalytic site along with deletion of the first 47 residues encoding the NoLSCitation43 which was replaced by the ubiquitin polypeptide (Ubi; 76 aa) according to the ubiquitin-fusion approachCitation44 (). The modified hTERT sequence was de novo synthesized by GeneCust (Luxembourg) and subcloned into the NTC8685‑eRNA41H‑HindIII‑XbaI vector backbone designed by Nature Technology Corporation (Lincoln, Nebraska).Citation45 INVAC-1 plasmid was amplified through an antibiotic‑free selection procedure in NTC4862 E. coli cellsCitation46 (DH5α attλ::P5/6 6/68‑RNA‑IN‑SacB, catR). GLP and GMP batchs of INVAC‑1 were manufactured by Eurogentec (Belgium) at used at a final concentration of 2 mg/mL.
Cell lines
HEK293T (from the ATCC) and CrFK (Crandell Rees feline kidney) cells were cultured in RPMI 1,640 medium (Gibco®) supplemented with 10% heat‑inactivated fetal calf serum (FCS) and 1% penicillin/streptomycin. QT6 cells (Japanese quail fibrosarcoma cell line) were cultured in HAM's F10 medium (Eurobio, Courtaboeuf, France) supplemented with 10% FCS, 1% penicillin/streptomycin, 1% chicken serum, 1% L-glutamine and 0.5% tryptose broth. TC-1 is a murine lung epithelial cell line immortalized with HPV16 E6/E7, and transformed with the c-Ha-ras oncogene. This cell line was cultured in RPMI 1,640 medium supplemented with 10% FCS, 1% penicillin/streptomycin, 20 mM Hepes, 0.4 mg/mL G418 and 0.2 mg/mL hygromycin B. The Sarc-T2r cell line was established by in vivo cloning of a spontaneous murine sarcoma that was histologically characterized as a leiomyosarcoma (malignant mesenchymal tumor) obtained from HLA-A2/DR1 mice which expresses HLA‑A2 on its surface. Sarc-T2r cells were cultured in RPMI 1640 medium supplemented with 10% FCS and 1% penicillin/streptomycin at 37°C. To characterize mTERT expression by flow cytometry and protein gel blot, mTERT protein from Sarc-T2r cells was stained using a monoclonal primary rabbit anti-hTERT antibody (Abcam, Cambridge, UK) which cross-reacts with mTERT, followed by secondary goat anti‑rabbit PE or HRP conjugate antibodies (BD Biosciences).
In vitro characterization of INVAC-1 hTERT protein
To assess protein expression, HEK293T cells were transfected either with INVAC-1, the empty NTC vector as negative control and a vector expressing wild type hTERT (pTRIP‑CMV‑hTERT) as positive control using jetPrime transfection reagent (Polyplus‑transfection Inc., France). Cells were harvested from 18 to 96 h post‑transfection, lysed in a specific RIPA buffer (Sigma-Aldrich, St. Louis, USA) and expression assessed by Western blotting assay. hTERT proteins were detected using a primary rabbit monoclonal anti-hTERT antibody (Abcam, Cambridge, UK) followed by a secondary goat anti-rabbit antibody-horseradish peroxidase (HRP) conjugate (Cell Signaling, Danvers, USA). β-actin protein was used as loading control. Peroxidase activity was detected using a chemiluminescence ECL HRP substrate reagent kit (GE Healthcare, Buckinghamshire, UK).
For sub-cellular localization, QT6 cells were transfected for 24 h using Fugene HD transfection reagent (Promega, Charbonnières-les-Bains, France). After fixation, permeabilization and blocking steps, cells were incubated with a rabbit monoclonal anti‑hTERT antibody for 1.5 h followed by a goat anti-rabbit antibody-Alexa Fluor 488® conjugate (Life Technologies, Saint-Aubin, France) for 45 min at room temperature. After washes, samples were mounted in DAPI-containing mounting medium (VECTASHIELD®). Slides were analyzed by fluorescent microscopy (Axio observer Z1 and Axiovision, Carl Zeiss MicroImaging GmbH).
In vitro telomerase activity was assessed on total cell protein extracts from CrFK cells transfected 24 h with DNA plasmids using the TeloTAGGG Telomerase PCR ELISAPLUS kit according manufacturer's instructions (Roche Diagnostic GmbH Mannheim, Germany). Briefly, protein extracts were used to evaluate the telomerase-mediated elongation of telomeric sequences. Products were amplified by PCR (30 cycles) using biotinylated primers. PCR amplification products were transferred to streptavidin pre-coated microplate, incubated with an anti‑digoxigenin HRP linked antibody and revealed using TMB substrate. Absorbance was measured against a blank at 450 nm to determine the level of telomerase activity in each sample. The RTA was obtained using the following formula:where AS: sample absorbance; AS0: heat-treated sample absorbance; AS,IS: internal standard sample absorbance; ATS8: control template absorbance; ATS8,0: lysis buffer (TS8) absorbance; ATS8,IS: TS8 IS absorbance.
Immunization procedure
Female 6-week-old C57BL/6JRj and BALB/cJRj mice were purchased from Janvier laboratories (Saint-Berthevin, France). HLA-B7 and HLA-A2/DR1 transgenic (Tg) mice have been previously described.Citation47,48 and provided from the Institut Pasteur animal breeding facility. All experiments were conducted in strict accordance with the ethical guidelines and good animal practices of the European Committee (Directive 2010/63/EU) and were approved by the registered CETEA of Institut Pasteur committees on ethics in animal experimentation N°89 under the reference 2013–0026. Mice were immunized by intradermal (ID) injection with 100 μg in 50 μL (bilateral injection of 25 μL) of INVAC-1 or NTC empty plasmid or PBS, both as control, at the base of the tail (bilateral injections). Immediately after ID injection, EGT was performed using CLINIPORATOR® 2 (IGEA, Carpi, Italy); one High Voltage (HV) pulse (100 μs duration; 1,250 V/cm) followed 1,000 ms later by one Low Voltage (LV) pulse (400 ms duration; 180 V/cm) were applied with non‑invasive plate electrodes (P-30–8G, IGEA). According to the experiment and vaccine regimen, mice could receive several administrations of DNA or control.
Synthetic hTERT peptides
H2 and HLA‑A*0201 restricted INVAC-1 peptides were determined in silico using four online algorithms (Syfpeithi, http://www.syfpeithi.de/; Bimas, http://www-bimas.cit.nih.gov/; NetMHCpan and SMM, http://tools.immuneepitope.org/main/). The HLA‑B*0702 and HLA‑DRB1 restricted hTERT peptides used in this study have been previously described.Citation20,49,50 Synthetic lyophilized (>90% purity) peptides were purchased from Proimmune (Oxford, UK) or GenScript (Piscataway, USA). The INVAC‑1 hTERT peptide library (>70 % purity) used to ascertain the breadth of immune response was purchased from GenScript. Thirteen pools of peptides spanning the entire INVAC‑1 hTERT protein were used. Each pool was composed of 10 peptides of 15-mers overlapping by 11 amino acids. All peptides were dissolved in sterile water at 2 mg/mL. Details of peptide sequences and MHC restriction are shown in and .
Table 1. hTERT and INVAC‑1 peptides.
Table 2. Pools of INVAC‑1 hTERT overlapping peptides.
IFNγ and Granzyme B ELISpot assay
Murine IFNγ and Granzyme B ELISpot kits were purchased from Diaclone (Eurobio, Courtaboeuf, France) and R&D systems (Bio-Techne, Lille, France) respectively. They were used with Ficoll‑purified lymphocytes from peripheral blood, spleen, tumor or tumor draining lymph nodes following the manufacturer's instructions. Briefly, cells were stimulated in triplicates at 2 × 105 cells/well with pools of restricted hTERT peptides at 5 μg/mL. Serum‑free medium and phorbol-12-myristate-13-acetate (PMA)-ionomycin were used as negative and positive controls respectively. Spots were counted using the Immunospot ELISpot counter and software (Cellular Technology Limited, Bonn, Germany).
Cytokine secretion
Cytometric Bead Arrays Mouse Th1/Th2/Th17 kit (CBA, BD biosciences) was used following manufacturer instructions for simultaneous detection of seven cytokines (IL‑2, IL‑4, IL‑6, IFNγ, TNF, IL‑17A, and IL‑10). Briefly, splenocytes from HLA‑A2/DR1 immunized mice were stimulated with specific HLA‑DRB1-restricted hTERT peptides (UCP2, UCP3 and UCP4) at 5 μg/mL, for 24 h at 37°C. Cell supernatants were collected and processed. Flow cytometry acquisition was done using the FACScan LSR Fortessa flow cytometer (BD Biosciences). Quantitative results were generated using the FCAP Array TM Software version 3.0 (Becton Dickinson).
In vivo cytotoxicity assay
The capacity of CD8+ cytotoxic T‑cells to kill peptide‑loaded target cells in vivo was assessed as described previously.Citation51 Briefly, splenocytes from naive C57BL/6 mice were split into three equal parts and each part was stained with carboxyfluorescein diacetate succinimidyl ester (CFSE) at 5 μM (high concentration), 1 μM (medium) or 0.2 μM (low). Subsequently, CFSEhigh‑labeled cells were pulsed with the immunodominant hTERT p660 peptide and CFSEmedium‑labeled cells were pulsed with the subdominant p1034 hTERT peptide for 1.5 h whereas CFSElow‑labeled cells were left unpulsed. Cells were mixed in a 1:1:1 ratio and 6 × 106 cells were i.v. injected in 50 μL of PBS into control or INVAC‑1 immunized mice 10 d after the second immunization. Fifteen hours later, single‑cell suspensions from spleens were analyzed by MACSQUANT® flow cytometer (Miltenyi, Germany). The percentage of specific killing was determined as follows:
In vivo antitumor assessment
For therapeutic vaccination experiment, 8 to 18-week-old HLA‑A2/DR1 mice were subcutaneously engrafted with 2 × 105 Sarc-T2r cells on the right abdominal flank. When, tumors were palpable, animals were randomized and immunized with INVAC‑1 at D7, D14 and D21 post‑engraftment or left non-treated. Twice a week, tumor growth was monitored using a caliper and mouse weight was individually followed. Mice were euthanized when tumors reached 2,000 mm3. The guidelines for the welfare and use of animals in cancer research were followed, especially for monitoring of clinical signs necessitating immediate intervention.Citation52 Tumor volume was calculated using the following formula: (L*l2)/2 (L = length; l = width). Results are expressed in mm3.
Statistical analysis
Statistical analyses were performed by a two-tailed Mann–Whitney non-parametric test using GraphPad prim 6.0 software (GraphPad Software Inc., USA). p values of ≤0 .05 were considered significant.
Disclosure of potential conflicts of interest
SWH and PLD are founders and shareholders of Invectys. The authors have declared no conflicts of interest for this article
Acknowledgments
We would like to thank Shannon A. Fairbanks and Bernardo Fort Brescia for their unswerving support. The authors would also like to thank the staff of the Institut Pasteur's animal facilities and imaging platform. We also thank Pr. Lluis Mir from Gustave-Roussy for his knowledge on electroporation.
Funding
This work was funded in part by the Paris Region “IDF” and Medicen Cluster in the context of the 12th FUI. EP was supported by an industrial Ph.D. fellowship from the ANRT.
References
- Novellino L, Castelli C, Parmiani G. A listing of human tumor antigens recognized by T cells: march 2004 update. Cancer Immunol Immunother 2005; 54:187-207; PMID:15309328; http://dx.doi.org/10.1007/s00262-004-0560-6
- Collins K, Mitchell JR. Telomerase in the human organism. Oncogene 2002; 21:564-79; PMID:11850781; http://dx.doi.org/10.1038/sj.onc.1205083
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Euro J Cancer 1997; 33:787-91; PMID:9282118; http://dx.doi.org/10.1016/S0959-8049(97)00062-2
- Zhu CQ, Cutz JC, Liu N, Lau D, Shepherd FA, Squire JA, Tsao MS. Amplification of telomerase (hTERT) gene is a poor prognostic marker in non-small-cell lung cancer. Brit J Cancer 2006; 94:1452-9; PMID:16641908; http://dx.doi.org/10.1038/sj.bjc.6603110
- Zanetti M, Hernandez X, Langlade-Demoyen P. Telomerase reverse transcriptase as target for anti-tumor T cell responses in humans. Springer Semin Immunopathol 2005; 27:87-104; PMID:15711953; http://dx.doi.org/10.1007/s00281-004-0197-8
- Beatty GL, Vonderheide RH. Telomerase as a universal tumor antigen for cancer vaccines. Expert Rev Vaccines 2008; 7:881-7; PMID:18767939; http://dx.doi.org/10.1586/14760584.7.7.881
- Harley CB. Telomerase and cancer therapeutics. Nat Rev Cancer 2008; 8:167-79; PMID:18256617; http://dx.doi.org/10.1038/nrc2275
- Berzofsky JA, Terabe M, Oh S, Belyakov IM, Ahlers JD, Janik JE, Morris JC. Progress on new vaccine strategies for the immunotherapy and prevention of cancer. J Clin Investigat 2004; 113:1515-25; PMID:15173875; http://dx.doi.org/10.1172/JCI21926
- Rice J, Ottensmeier CH, Stevenson FK. DNA vaccines: precision tools for activating effective immunity against cancer. Nat Rev Cancer 2008; 8:108-20; PMID:18219306; http://dx.doi.org/10.1038/nrc2326
- Fioretti D, Iurescia S, Fazio VM, Rinaldi M. DNA vaccines: developing new strategies against cancer. J Biomed Biotechnol 2010; 2010:174378; PMID:20368780; http://dx.doi.org/10.1155/2010/174378
- Liu MA. DNA vaccines: a review. J Int Med 2003; 253:402-10; PMID:12653868; http://dx.doi.org/10.1046/j.1365-2796.2003.01140.x
- Calvet CY, Thalmensi J, Liard C, Pliquet E, Bestetti T, Huet T, Langlade-Demoyen P, Mir LM. Optimization of a gene electrotransfer procedure for efficient intradermal immunization with an hTERT-based DNA vaccine in mice. Mol Ther — Methods Clin Dev 2014; 1:14045; PMID:26015983; http://dx.doi.org/10.1038/mtm.2014.45
- Kutzler MA, Weiner DB. DNA vaccines: ready for prime time? Nat Rev Genet 2008; 9:776-88; PMID:18781156; http://dx.doi.org/10.1038/nrg2432
- Drosopoulos WC, Prasad VR. The active site residue Valine 867 in human telomerase reverse transcriptase influences nucleotide incorporation and fidelity. Nucleic Acids Res 2007; 35:1155-68; PMID:17264120; http://dx.doi.org/10.1093/nar/gkm002
- Wang Q, Lei C, Wan H, Liu Q. Improved cellular immune response elicited by a ubiquitin-fused DNA vaccine against Mycobacterium tuberculosis. DNA Cell Biol 2012; 31:489-95; PMID:21905875; http://dx.doi.org/10.1089/dna.2011.1309
- Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med 1999; 5:677-85; PMID:10371507; http://dx.doi.org/10.1038/9525
- Dunbar PR, Smith CL, Chao D, Salio M, Shepherd D, Mirza F, Lipp M, Lanzavecchia A, Sallusto F, Evans A et al. A shift in the phenotype of melan-A-specific CTL identifies melanoma patients with an active tumor-specific immune response. J Immunol 2000; 165:6644-52; PMID:11086110; http://dx.doi.org/10.4049/jimmunol.165.11.6644
- Palucka K, Ueno H, Banchereau J. Recent developments in cancer vaccines. J Immunol 2011; 186:1325-31; PMID:21248270; http://dx.doi.org/10.4049/jimmunol.0902539
- Godet Y, Fabre E, Dosset M, Lamuraglia M, Levionnois E, Ravel P, Benhamouda N, Cazes A, Le Pimpec-Barthes F, Gaugler B et al. Analysis of spontaneous tumor-specific CD4 T-cell immunity in lung cancer using promiscuous HLA-DR telomerase-derived epitopes: potential synergistic effect with chemotherapy response. Clin Cancer Res 2012; 18:2943-53; PMID:22407833; http://dx.doi.org/10.1158/1078-0432.CCR-11-3185
- Andersson HA, Barry MA. Maximizing antigen targeting to the proteasome for gene-based vaccines. Mol Ther 2004; 10:432-46; PMID:15336644; http://dx.doi.org/10.1016/j.ymthe.2004.05.035
- Endmann A, Baden M, Weisermann E, Kapp K, Schroff M, Kleuss C, Wittig B, Juhls C. Immune response induced by a linear DNA vector: influence of dose, formulation and route of injection. Vaccine 2010; 28:3642-9; PMID:20362204; http://dx.doi.org/10.1016/j.vaccine.2010.03.034
- Nicolas JF, Guy B. Intradermal, epidermal and transcutaneous vaccination: from immunology to clinical practice. Expert Rev Vaccines 2008; 7:1201-14; PMID:18844594; http://dx.doi.org/10.1586/14760584.7.8.1201
- Bagarazzi ML, Yan J, Morrow MP, Shen X, Parker RL, Lee JC, Giffear M, Pankhong P, Khan AS, Broderick KE et al. Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci Translat Med 2012; 4:155ra38; PMID:23052295; http://dx.doi.org/10.1126/scitranslmed.3004414
- Gothelf A, Gehl J. What you always needed to know about electroporation based DNA vaccines. Hum Vaccines Immunother 2012; 8:1694-702; PMID:23111168; http://dx.doi.org/10.4161/hv.22062
- Elnekave M, Furmanov K, Hovav AH. Intradermal naked plasmid DNA immunization: mechanisms of action. Expert Rev Vaccines 2011; 10:1169-82; PMID:21854310; http://dx.doi.org/10.1586/erv.11.66
- Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, Palmer DC, Chan CC, Klebanoff CA, Overwijk WW et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol 2005; 174:2591-601; PMID:15728465; http://dx.doi.org/10.4049/jimmunol.174.5.2591
- Parkhurst MR, Riley JP, Igarashi T, Li Y, Robbins PF, Rosenberg SA. Immunization of patients with the hTERT:540-548 peptide induces peptide-reactive T lymphocytes that do not recognize tumors endogenously expressing telomerase. Clin Cancer Res 2004; 10:4688-98; PMID:15269141; http://dx.doi.org/10.1158/1078-0432.CCR-04-0325
- Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007; 121:1-14; PMID:17386080; http://dx.doi.org/10.1111/j.1365-2567.2007.02587.x
- Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol 2008; 8:247-58; PMID:18323851; http://dx.doi.org/10.1038/nri2274
- Fisher DT, Chen Q, Appenheimer MM, Skitzki J, Wang WC, Odunsi K, Evans SS. Hurdles to lymphocyte trafficking in the tumor microenvironment: implications for effective immunotherapy. Immunol Investigat 2006; 35:251-77; PMID:16916754; http://dx.doi.org/10.1080/08820130600745430
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313:1960-4; PMID:17008531; http://dx.doi.org/10.1126/science.1129139
- Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A 2005; 102:18538-43; PMID:16344461; http://dx.doi.org/10.1073/pnas.0509182102
- Appay V, Jandus C, Voelter V, Reynard S, Coupland SE, Rimoldi D, Lienard D, Guillaume P, Krieg AM, Cerottini JC et al. New generation vaccine induces effective melanoma-specific CD8+ T cells in the circulation but not in the tumor site. J Immunol 2006; 177:1670-8; PMID:16849476; http://dx.doi.org/10.4049/jimmunol.177.3.1670
- Greenberg RA, Allsopp RC, Chin L, Morin GB, DePinho RA. Expression of mouse telomerase reverse transcriptase during development, differentiation and proliferation. Oncogene 1998; 16:1723-30; PMID:9582020; http://dx.doi.org/10.1038/sj.onc.1201933
- Hernandez J, Garcia-Pons F, Lone YC, Firat H, Schmidt JD, Langlade-Demoyen P, Zanetti M. Identification of a human telomerase reverse transcriptase peptide of low affinity for HLA A2.1 that induces cytotoxic T lymphocytes and mediates lysis of tumor cells. Proc Natl Acad Sc U S A 2002; 99:12275-80; PMID:12218171; http://dx.doi.org/10.1073/pnas.182418399
- Minev B, Hipp J, Firat H, Schmidt JD, Langlade-Demoyen P, Zanetti M. Cytotoxic T cell immunity against telomerase reverse transcriptase in humans. Proc Natl Acad Sci U S A 2000; 97:4796-801; PMID:10759561; http://dx.doi.org/10.1073/pnas.070560797
- Yamano T, Kaneda Y, Hiramatsu SH, Huang S, Tran AN, Giuliano AE, Hoon DS. Immunity against breast cancer by TERT DNA vaccine primed with chemokine CCL21. Cancer Gene Ther 2007; 14:451-9; PMID:17318199; http://dx.doi.org/10.1038/sj.cgt.7701035
- Yan J, Pankhong P, Shin TH, Obeng-Adjei N, Morrow MP, Walters JN, Khan AS, Sardesai NY, Weiner DB. Highly optimized DNA vaccine targeting human telomerase reverse transcriptase stimulates potent antitumor immunity. Cancer Immunol Res 2013; 1:179-89; PMID:24777680; http://dx.doi.org/10.1158/2326-6066.CIR-13-0001
- De Pas T, Giovannini M, Rescigno M, Catania C, Toffalorio F, Spitaleri G, Delmonte A, Barberis M, Spaggiari L, Solli P et al. Vaccines in non-small cell lung cancer: rationale, combination strategies and update on clinical trials. Crit Rev Oncol/Hematol 2012; 83:432-43; PMID:22366114; http://dx.doi.org/10.1016/j.critrevonc.2011.12.005
- Mkrtichyan M, Najjar YG, Raulfs EC, Abdalla MY, Samara R, Rotem-Yehudar R, Cook L, Khleif SN. Anti-PD-1 synergizes with cyclophosphamide to induce potent anti-tumor vaccine effects through novel mechanisms. Euro J Immunol 2011; 41:2977-86; PMID:21710477; http://dx.doi.org/10.1002/eji.201141639
- Lesterhuis WJ, de Vries IJ, Aarntzen EA, de Boer A, Scharenborg NM, van de Rakt M, van Spronsen DJ, Preijers FW, Figdor CG, Adema GJ et al. A pilot study on the immunogenicity of dendritic cell vaccination during adjuvant oxaliplatin/capecitabine chemotherapy in colon cancer patients. Brit J Cancer 2010; 103:1415-21; PMID:20924373; http://dx.doi.org/10.1038/sj.bjc.6605935
- Yang Y, Chen Y, Zhang C, Huang H, Weissman SM. Nucleolar localization of hTERT protein is associated with telomerase function. Exp Cell Res 2002; 277:201-9; PMID:12083802; http://dx.doi.org/10.1006/excr.2002.5541
- Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science 1986; 234:179-86; PMID:3018930; http://dx.doi.org/10.1126/science.3018930
- Carnes AE, Luke JM, Vincent JM, Anderson S, Schukar A, Hodgson CP, Williams JA. Critical design criteria for minimal antibiotic-free plasmid vectors necessary to combine robust RNA Pol II and Pol III-mediated eukaryotic expression with high bacterial production yields. J Gene Med 2010; 12:818-31; PMID:20806425; http://dx.doi.org/10.1002/jgm.1499
- Luke J, Carnes AE, Hodgson CP, Williams JA. Improved antibiotic-free DNA vaccine vectors utilizing a novel RNA based plasmid selection system. Vaccine 2009; 27:6454-9; PMID:19559109; http://dx.doi.org/10.1016/j.vaccine.2009.06.017
- Pajot A, Michel ML, Fazilleau N, Pancre V, Auriault C, Ojcius DM, Lemonnier FA, Lone YC. A mouse model of human adaptive immune functions: HLA-A2.1-/HLA-DR1-transgenic H-2 class I-/class II-knockout mice. Eur J Immunol 2004; 34:3060-9; PMID:15468058; http://dx.doi.org/10.1002/eji.200425463
- Rohrlich PS, Cardinaud S, Firat H, Lamari M, Briand P, Escriou N, Lemonnier FA. HLA-B*0702 transgenic, H-2KbDb double-knockout mice: phenotypical and functional characterization in response to influenza virus. Int Immunol 2003; 15:765-72; PMID:12750360; http://dx.doi.org/10.1093/intimm/dxg073
- Adotevi O, Mollier K, Neuveut C, Cardinaud S, Boulanger E, Mignen B, Fridman WH, Zanetti M, Charneau P, Tartour E et al. Immunogenic HLA-B*0702-restricted epitopes derived from human telomerase reverse transcriptase that elicit antitumor cytotoxic T-cell responses. Clin Cancer Res 2006; 12:3158-67; PMID:16707616; http://dx.doi.org/10.1158/1078-0432.CCR-05-2647
- Cortez-Gonzalez X, Sidney J, Adotevi O, Sette A, Millard F, Lemonnier F, Langlade-Demoyen P, Zanetti M. Immunogenic HLA-B7-restricted peptides of hTRT. Int Immunol 2006; 18:1707-18; PMID:17077179; http://dx.doi.org/10.1093/intimm/dxl105
- Durward M, Harms J, Splitter G. Antigen specific killing assay using CFSE labeled target cells. J Visualized Exp 2010; PMID:21085108; http://dx.doi.org/10.3791/2250
- Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, Double JA, Everitt J, Farningham DA, Glennie MJ et al. Guidelines for the welfare and use of animals in cancer research. Brit J Cancer 2010; 102:1555-77; PMID:20502460; http://dx.doi.org/10.1038/sj.bjc.6605642