
ABSTRACT
Programmed death one (PD-1) is a well-established co-inhibitory regulator that suppresses proliferation and cytokine production of T cells. Despite remarkable progress in delineating the functional roles of PD-1 on T lymphocytes, little is known about the regulatory role of PD-1 expressed on myeloid cells such as dendritic cells (DCs). Here, we show that CD8+ T cells can be more potently activated to secrete IL-2 and IFNγ by PD-1-deficient DCs compared to wild-type DCs. Adoptive transfer of PD-1-deficient DCs demonstrated their superior capabilities in inducing antigen-specific CD8+ T cell proliferation in vivo. In addition, we provide first evidence demonstrating the existence of peripheral blood DCs and CD11c+ tumor-infiltrating myeloid cells that co-express PD-1 in patients with hepatocellular carcinoma (HCC). The existence of PD-1-expressing HCC-infiltrating DCs (HIDCs) was further supported in a mouse model of HCC. Intratumoral transfer of PD-1-deficient DCs rendered recipient mice resistant to the growth of HCC by promoting tumor-infiltrating CD8+ effector T cells to secrete perforin and granzyme B. This novel finding provides a deeper understanding of the role of PD-1 in immune regulation and has significant implications for cancer immunotherapies targeting PD-1.
Abbreviations
| APCs | = | antigen presenting cells |
| DC | = | dendritic cell |
| HCC | = | hepatocellular carcinoma |
| HIDCs | = | HCC-infiltrating DCs |
| PD-1 | = | Programmed death one |
| TILs | = | tumor-infiltrating T lymphocytes |
Introduction
PD-1, a member of the immunoglobulin superfamily, is inducibly expressed on activated T and B lymphocytes.Citation1,2 The critical role of PD-1 in the control of peripheral tolerance and lymphocyte activation is supported by the development of various autoimmune phenotypes in PD-1-deficient mice.Citation3-8 Co-inhibitory signals in lymphocytes are initiated by binding of PD-1 to its ligands PD-L1 (B7-H1) and PD-L2 (B7-DC).Citation9,10 Engagement of PD-1 results in the recruitment of the tyrosine phosphatase SHP-2, which dephosphorylates and inactivates proximal effector molecules such as Zap70 in T cells. Citation11-14 The immediate outcomes of PD-1-mediated signaling in T cells are the inhibition of both T cell proliferation and secretion of cytokines such as IFNγ and IL-2.
PD-1 acts as a rheostat to modulate immunity by increasing the threshold of antigen responses and reducing the cytotoxic lymphocyte activity of CD8+ T cells.Citation15 Such unique properties endow PD-1 with immunoregulatory functions during chronic viral infectionsCitation16-19 and tumor progression. A critical observation that the blockade of PD-1:PD-L1 interactions can restore the capacity of exhausted CD8+ T cells to attack tumorsCitation20,21 led to clinical trials using antibodies targeting PD-1Citation22 and PD-L1.Citation23 These studies have produced impressive results, reporting significant and durable responses in several types of cancer. Thus, PD-1 blockade is considered a promising target for cancer immunotherapy,Citation24,25 with a number of therapeutic antibodies targeting PD-1, including nivolumab (Opdivo; BristolMyers Squibb/Ono Pharmaceuticals) and pembrolizumab (Keytruda; Merck & Co.), have recently been approved by the FDA for the treatment of metastatic melanoma.
Although PD-1 is known to be expressed on T lymphocytes, evidence has emerged indicating that other non-lymphoid innate cells also express PD-1, suggesting that it may play additional roles in immune regulation that are mediated by these cells.Citation1 A number of recent studies demonstrated that expression of PD-1 on DCs impedes DC survival,Citation26 reduces pro-inflammatory cytokine production,Citation27 and diminishes innate immunity against bacterial infection.Citation28 Whether expression of PD-1 on DCs also regulates CD8+ T cell function or plays a role in immune surveillance remains to be seen.
In this study, we show that expression of PD-1 on DCs plays a crucial role in the suppression of IL-2 and IFNγ secretion in vitro and of antigen-specific CD8+ T cell proliferation in vivo. Our study provides the first evidence demonstrating the existence of PD-1-expressing DCs in peripheral blood and CD11c+ myeloid cells in cancerous tissues derived from patients with HCC. Intratumoral transfer of PD-1-deficient DCs was beneficial in suppressing tumor growth in a mouse model of HCC, and deficiency of PD-1 expression in DCs rendered mice resistant to tumor progression through increased CD8+ T cell function. Our findings provide a deeper conceptual understanding of the regulation of CD8+ T cell function and antitumor immunity by DC-expressed PD-1.
Results
PD-1 expression on DCs suppresses T cells responses in vitro and in vivo
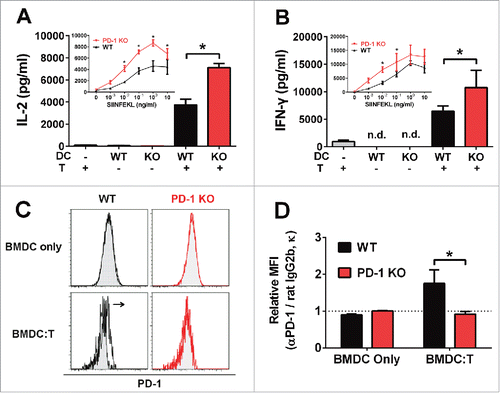
To evaluate whether T cell activation was influenced by expression of PD-1 on DCs, we measured cytokine production from OT-I T cells stimulated with WT or PD-1 KO bone marrow-derived dendritic cells (BMDCs), pre-pulsed with antigenic OVA peptide (SIINFEKL) at concentrations ranging from 1 pg to 10 ng/mL. We observed significantly higher production of IL-2 () and IFNγ () from T cells activated by antigen-pulsed PD-1 KO BMDCs compared with WT BMDCs (p < 0.05). Induction of IL-2 and IFNγ secretion required the presence of antigen-presenting DCs, as DCs or T cells alone failed to stimulate cytokine responses (, ). Consistent with previous reports,Citation28 we observed that WT BMDCs do not upregulate surface PD-1 expression following Poly I:C stimulation (), unlike splenic DCs (Fig. S1). However, the differential regulation of T cell activation by WT and PD-1 KO BMDCs indicates that PD-1 is functional in this system. We hypothesized that the induction of PD-1 expression in BMDCs is crucially dependent on their intercellular contacts with T cells. Indeed, PD-1 expression was significantly upregulated in WT BMDCs (gated on DAPI-, CD3ϵ-, TCR-Vα2-, CD11c+, MHC-II+ cell population), following co-culture with OT-I T cells (p = 0.018) (, ). BMDCs from WT and PD-1 KO mice displayed similar levels of peptide-linked MHC complexes at the cell surface (Fig. S2). Furthermore, the expression of several other cell surface signaling molecules, such as CD40, CD80 and CD86 were expressed at similar levels in WT and PD-1 KO BMDCs (Fig. S3), indicating that these factors did not contribute to the differential capacity of PD-1 KO BMDCs to modulate T cell function.
Figure 1. PD-1 expression on BMDCs inhibits IL-2 and IFNγ secretion by T cells. Cytokine secretion of (A) IL-2 and (B) IFNγ after a 2 d co-culture of CD8+ OT-I T cells with or without WT (black) or PD-1 KO (red) BMDCs pre-pulsed with 0.1 μg/mL OVA peptide (SIINFEKL). Inset figures show cytokine secretion from CD8+ OT-I T cells co-cultured with WT (black) or PD-1 KO (red) BMDCs pre-pulsed with different concentrations of OVA peptide (0–1 μg/mL) in the presence (solid line) of Poly I:C (1 μg/mL). BMDC:T co-culture ratio is 1:1. *p < 0.05, unpaired t-test. Bars indicate mean ± SD. N = 3. (C–D) PD-1 expression of WT (black) or PD-1 KO (red) BMDCs after a 2 d co-culture with (BMDC:T) or without (BMDC only) CD8+ OT-I T cells in the presence of 0.1 μg/mL OVA peptide (SIINFEKL) and 10 μg/mL Poly I:C. BMDCs were gated on DAPI-, CD3ϵ-, and TCR-Vα2- CD11c+, MHC-II+ cell populations. Open histogram: rat anti-mouse PD-1 antibody; Filled histogram: isotype control rat IgG 2b, κ. Quantification of PD-1 expression was determined from mean fluorescent intensity (MFI) fold change with respect to isotype control rat IgG 2b, κ. *p = 0.018, unpaired t-test. Bars indicate mean ± SD (N = 3, data was pooled from three independent experiments).
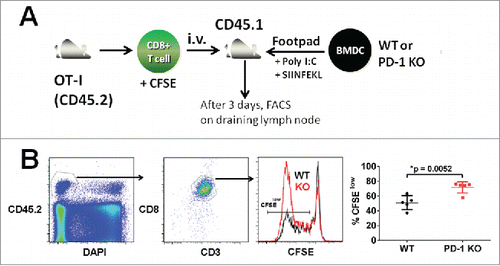
To establish the role of PD-1 on DCs in regulating T cell proliferation in vivo, we stimulated intravenously transferred CFSE-labeled, CD45.2+ OT-I T cells by adoptive transfer of SIINFEKL-pulsed WT or PD-1 KO BMDCs into the footpad of WT CD45.1+ recipient mice (). To minimize the variation of T cell responses triggered by different recipient mice, adoptive transfer of BMDCs was performed into different footpads of the same recipient mice. At three days post adoptive T cell transfer, a significantly higher level of T cell proliferation (gated on DAPI-, CD45.2+, CD3ϵ+, CD8α+, and CFSElow cell population) was measured in the draining lymph node in the presence of PD-1 KO BMDCs compared with WT BMDCs (71.7 ± 7.7% vs. 50.9 ± 9.5%, N = 5, p = 0.0052) (). These results indicate that DCs are able to elicit antigen-specific T cell proliferation to a greater extent in the absence of PD-1 expression.
Figure 2. PD-1 deficient DCs augment antigen-specific T cell proliferation. (A) Enhanced antigen-specific T cell proliferation induced by PD-1-deficient DCs. BMDCs (WT or PD-1 KO) were pre-pulsed with or without 1 μg OVA peptide (SIINFEKL) in medium containing 10 μg/mL Poly I:C for 4 h and washed twice with PBS before injecting them in the presence of 50 μg Poly I:C into the footpads of WT CD45.1 recipient mice. CD8+ OT-I T cells (CD45.2 background) were labeled with CFSE and adoptively transferred by intravenous injection into recipient mice. (B) Three days post transfer, proliferation of antigen-specific OT-I T cells in draining popliteal lymph nodes was calculated by analyzing the dilution of CFSE by flow cytometry. OT-I T cells were gated on CD45.2+, DAPI-, CD3ϵ+, and CD8α+ cell populations derived from draining popliteal lymph nodes in individual recipient mice. Quantification of proliferation of antigen-specific OT-I T cells was determined from the percentage of CFSElow OT-I T cells. *p = 0.0052, unpaired t-test, N = 5, data was pooled from two independent experiments.
Human peripheral blood dendritic cells co-express PD-1
To establish the relevance of DC expression of PD-1 in human disease, we characterized PD-1 expression in DC subsets from human peripheral blood using flow cytometry (). In the steady state, a low level of PD-1 was expressed in CD141hi (or BDCA3hi) myeloid DCs (mDCs), but not in CD1c+ (or BDCA1+) mDCs and CD123+ plasmacytoid DCs (pDCs) in healthy donors (, ). However, expression of PD-1 was found in all DC subsets (mDCs and pDCs) in the peripheral blood of HCC (HCC) patients. CD14+ monocytes from both healthy donors and HCC patients were negative for PD-1 expression (, ).
Figure 3. PD-1 expression on human DC subsets in peripheral blood. (A) Gating strategy for flow cytometric sorting of human blood DC subsets. Freshly isolated singlet PBMC were gated on DAPI- and CD45+ to identify live immune cells. Immune cells, including T, B, and NK populations, were stained with a panel of lineage markers (Lin: CD3, CD7, CD19, CD20, and CD56) and excluded from further analysis. Dendritic cell subsets and monocytes were identified within the MHC-II+ (HLA-DR) Lin- population. Monocytes were identified as CD14+/CD16- and CD14low/CD16+ populations. CD123 was used to identify plasmacytoid DC (pDC). Myeloid DCs (mDCs) were characterized as CD141hi or CD1c+ populations. Red boxes: DC subsets and CD14+/CD16- monocytes were sorted and cultured in medium (B) PD-1 expression on freshly isolated human blood DC subsets and CD14+/CD16- monocytes. Open histogram: mouse anti-human PD-1 antibody; Filled histogram: mouse IgG1, κ. Representative histogram from five different donors (C) PD-1 expression (MFI fold change with respect to isotype control) on each DC subset and CD14+/CD16- monocytes in human peripheral blood samples of healthy or hepatocellular carcinoma (HCC) patients. DC subsets were identified based on the gating strategy shown in Fig. 3A. Bar charts show data (mean ± SD) pooled from five individual donors. (D) PD-1 expression (MFI fold change with respect to isotype control) on each DC subset in human blood samples following culture in the presence of 10 μg/mL Poly I:C or PBS for 18 h. Bar charts show data (mean ± SD) from five healthy donors.
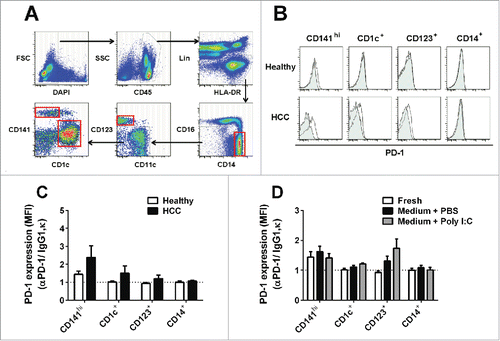
To investigate if PD-1 expression could be induced upon activation, we cultured human blood DC subsets and CD14+ monocytes from healthy donors in medium with Poly I:C or PBS for 18 h (red boxes, ). PD-1 expression in CD141hi DCs, which exhibited low levels of PD-1 expression ex vivo, was unchanged by Poly I:C stimulation. However, we observed that PD-1 expression in CD1c+ DCs and CD123+ pDCs could be induced by Poly I:C stimulation. CD14+ monocytes remained negative under all test conditions ().
Human and murine liver cancer-infiltrating CD11c+ myeloid cells co-express PD-1
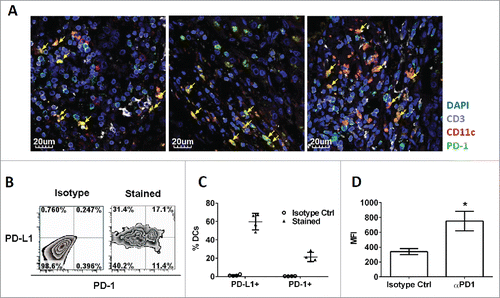
It was previously reported that tumor-associated myeloid cells express PD-L1, which transmits co-inhibitory signals to PD-1-expressing tumor-infiltrating T lymphocytes (TILs), thus suppressing the antitumor activity of TILs.Citation20,29 However, little is known about the functional role of PD-1 expressed on myeloid cells in the regulation of antitumor immunity. To assess whether PD-1 was present on liver cancer-associated myeloid cells, we performed immunofluorescence staining on cancerous tissues derived from patients with HCC. Consistent with previous findings, a large fraction of HCC-infiltrating CD3+ T cells expressed PD-1. Notably, we also observed expression of PD-1 on CD3- CD11c+ myeloid cells in the same tissue (). Given that CD11c is expressed on all conventional myeloid DCs, we hypothesized that these cells represented HIDCs that expressed PD-1 in the tumor microenvironment.
Figure 4. Existence of tumor-infiltrating PD-1+ CD11c+ myeloid cells in cancerous liver tissue of patient and mice. (A) Immunofluorescence (IF) images showing co-localization of PD-1 (green) with either CD11c (red) or CD3 (white). Cell nuclei were stained with DAPI (blue). CD11c+ myeloid cells expressing PD-1 were identified in yellow color (arrow head). Each IF image was captured from cancerous liver tissue from individual HCC patients (N = 3). (B) HIDCs co-express PD-1 and PD-L1 in the spontaneous HCC mouse model. Quadrants were established with reference to the isotype controls. The inset values are the percentage of HIDCs within each quadrant. All HIDCs were gated on GR1-, CD3-, F4/80-, CD11chi, MHC-II+ cells. (C) Percentages of HIDCs which co-express PD-1 or PD-L1. Each data point was derived from individual HCC mice (N = 4). (D) Quantification of PD-1 expression on HIDCs by flow cytometry. Mean fluorescent intensity (MFI) values were determined from individual HCC mice (N = 4). HCC: hepatocellular carcinoma; HIDC: HCC-infiltrating DC. *p < 0.02, unpaired t-test.
To better understand the surface phenotype and function of HIDCs, we developed spontaneous HCC murine models. Multispectral flow cytometry analysis demonstrated that a large population of HIDCs (gated on live, CD3-, F4/80-, GR1-, CD11chi, MHC-II+ cells) co-expressed both PD-L1 (59.9 ± 9% PD-L1+ HIDCs) (, ) and PD-1 (21.4 ± 5%) ().
Effects of PD-1 on DCs in regulating antitumor effects on primary established HCC
Although expression of PD-L1 on tumor-infiltrating DCs is known to regulate immune suppression, the function of PD-1 expression on these cells remains elusive. To investigate the role of PD-1 expressed on DCs in T cell suppression in the tumor microenvironment, we performed an adoptive transfer of WT or PD-1 KO BMDCs into Hepa1-6 transplantable tumor-bearing mice. Although intratumoral transfer of DCs from both WT and PD-1 KO mice resulted in a significant suppression of growth of primary established subcutaneous HCC (as early as 3–7 d post DC transfer), PD-1 KO DCs suppressed tumor growth more efficiently than WT DCs, suggesting that immune surveillance against tumors is tightly regulated by PD-1 expression on DCs ().
Figure 5. Enhanced antitumor effect on primary established HCC by intratumoral transfer of PD-1 KO DCs. (A) Hepa1-6 cells were transplanted subcutaneously into the flanks of WT recipient mice. Intratumoral injection with WT (black), PD-1 KO (red) BMDCs or PBS only (white) were performed 1 week later. Day 0 is defined as the day of DC injection. Sizes of transplanted HCC tumors were measured for 14 d post DC transfer, and the data were normalized to the tumor sizes at day 0. *p < 0.05, unpaired t-test, N = 7, data was pooled from two independent experiments. (B) Increased perforin- and granzyme B-secreting tumor-infiltrating CD8+ T cells in primary established HCC after transfer of PD-1 KO BMDCs. Mice with established subcutaneous Hepa1-6 tumors were sacrificed on day 7 after intratumoral transfer of WT and PD-1 KO BMDCs. Perforin- and granzyme B-secreting CD8+ T cells were evaluated by intracellular staining followed by flow cytometry. All cells were gated on the CD45+, CD3ϵ+, and CD8+ population. Quadrants were established with reference to the isotype controls. A representative density plot from one mouse is shown. (C) Percentages of perforin- and granzyme B-expressing CD8+ T cells and total tumor-infiltrating CD8+ T cells 7 d post intratumoral transfer of WT and PD-1 KO BMDCs. Cell percentages are relative to total CD45+ live singlet T cells; *p < 0.05, unpaired t-test, N ≥ 3 per group.
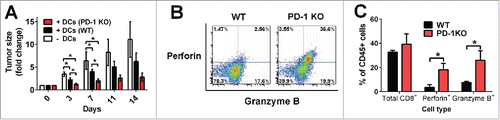
To further examine the mechanism by which PD-1 expression on DCs suppressed tumor growth, we monitored the secretion of perforin and granzyme B from CD8+ effector TILs by intracellular flow cytometry analysis (). One week after intratumoral transfer of WT or PD-1 KO DCs, 30–40% of CD45+ leukocytes were CD8+ in the tumor-bearing recipient mice (). Intratumoral transfer of PD-1 KO DCs resulted in an approximately 4.9-fold increase in perforin+ CD8+ TILs (18.3 ± 5.1% vs. 3.7 ± 2.1%) and an approximately 3.4-fold increase in granzyme B+ CD8+ TILs (26.1 ± 7.7 % vs. 7.7 ± 0.9%) compared with WT DCs ().
Discussion
The co-inhibitory functions of T cell-expressed PD-1 have been studied extensively over the last decade, but the functional role of PD-1 expressed on myeloid cells, including DCs, was unclear. Using PD-1 deficient mice,Citation4 we demonstrate that expression of PD-1 on BMDCs plays a crucial role in suppressing both IL-2 and IFNγ cytokine responses in vitro and antigen-specific CD8+ T cell proliferation in vivo.
PD-1 expression is regulated by distinct pathways in T cells and DCs. In T cells, PD-1 expression is induced by TCR triggering;Citation30 however in murine splenic DCs, it is regulated by TLR signaling instead.Citation28 Consistent with previous findings,Citation28 PD-1 expression was induced in murine splenic DCs after activation by Poly I:C in vivo (Fig. S1), but was not inducible in activated BMDCs per se. Poly I:C upregulated surface expression of other co-stimulatory molecules, including CD40, CD80, and CD86, in BMDCs (Fig. S3), indicating that appropriate test conditions were used. The inducible PD-1 expression in WT BMDCs was crucially dependent on their intercellular contacts with T cells, but was not due to the acquirement of PD-1 molecules from neighboring T cells through trogocytosis,Citation31,32 since PD-1 surface expression level was not detected in PD-1 KO BMDCs in the same DC:T co-culture system.
The ability of PD-1 in BMDCs to modulate T cell function was not related to differences in antigen presentation or the expression of other cell surface signaling molecules because these were unchanged between WT and PD-1 KO BMDCs. The suppression of T cell proliferation mediated by PD-1 on BMDCs may be caused by ligation of PD-1 to PD-L1, which is also expressed on the surface of T cells.Citation1 Alternatively, T cell inhibition may be caused by reverse signaling in DCs triggered by PD-1 engagement, which reduces DC survival signalsCitation26 and pro-inflammatory cytokine production.Citation27 In addition, antibodies targeting PD-1 in co-culture experiments with T cells and PD-1 KO DCs significantly promoted IL-2 and IFNγ cytokine secretion, confirming the role of PD-1 expression on T cells in suppressing cytokine responses (Fig. S4). Taken together, these results suggest that both PD-1 and PD-L1 on DCs are capable of delivering bidirectional and synergistic signaling to inhibit T cell responses.
Although the co-inhibitory functions of PD-1 expressed on tumor-infiltrating T cells have been studied extensively, the functional role of PD-1 on tumor-infiltrating myeloid cells, the presence of which we report for the first time in HCC patients, requires further investigation. In an ovarian tumor mouse model, it was recently shown that PD-1-expressing tumor-associated DCs inhibited T cell proliferation and activation through downregulation of NF-κB activation, cytokine production, and co-stimulatory molecule expression.Citation27 However, the ability of PD-1 expressed on DCs to regulate tumor growth in vivo has not been demonstrated. Selective deletion of PD-1 on DCs in mouse tumor models allows a precise evaluation of the roles of PD-1 in antitumor immunity. Unfortunately, DC-specific PD-1-deficient mice are not yet available. Nevertheless, our in vivo DC transfer experiments provide the first functional insights into the role of PD-1 expression in DCs in modulating T cell responses directly in the tumor microenvironment. Using a mouse model of HCC, we show that tumor growth can be effectively suppressed following the transfer of PD-1-deficient DCs. This is accompanied by an expansion of perforin- and granzyme B-secreting tumor-infiltrating CD8+ T cells, which are crucial in suppressing tumor growth. Thus, in addition to the previously understood functions of PD-1 in inhibiting innate immunity,Citation28 cytokine productionCitation27 and survival signalsCitation26 in DCs, our results suggest a novel role for PD-1 expression in DCs in regulating effector functions of CD8+ TILs and antitumor immunity.
Cancer immunotherapy targeting the PD-L1:PD-1 pathway aims to restore the function of exhausted T cells in the tumor microenvironment.Citation25,33 Therapeutic antibodies targeting PD-1Citation22 for cancer immunotherapy have shown remarkable results in clinical trials, with two agents (nivolumab and pembrolizumab) recently gaining FDA approval. It is well established that immunosuppressive signals can be transmitted via PD-1 on T cells to inhibit their antitumor activity.Citation20,29 Thus, one of the predicted mechanisms of action of these agents is to block PD-1:PD-L1 interactions by directly binding to PD-1 on TILs in the tumor microenvironment. However, many studies have suggested that PD-1 blockade could potentially invoke other mechanisms of action.Citation34 In this study, we provide the first evidence of the existence of blood-circulating DCs and CD3- CD11c+ tumor-infiltrating myeloid cells that co-express PD-1 in HCC patients, suggesting that a clinical blockade targeting PD-1 may have an important functional impact directly on circulating DCs and tumor-infiltrating myeloid cells in cancer patients.
Our study provides novel functional insights into the regulation of immune responses in the tumor microenvironment by expression of PD-1 on DCs, and will contribute to a better understanding of the consequences of cancer immunotherapy targeting PD-1.
Materials and methods
Mice
C57BL/6 (CD45.2 or CD45.1 background) mice were purchased from the Biological Resource Center (BRC, A*STAR, Singapore). OT-I.Rag1-/- miceCitation35,36 were provided by Taconic from the National Institute of Allergy and Infectious Diseases Exchange Program (# 004175; Bethesda, MD). PD-1 KO (C57BL/6-Pdcd1-/-) miceCitation4 were kindly provided by Prof. Tasuku Honjo (Kyoto University) and were imported from the RIKEN Bioresource Center, Japan. All mice were maintained in the SPF animal facility of the Biological Resource Center (BRC) of Biopolis in Singapore.
Ethics statement
Mice studies were carried out in strict accordance with the recommendations of the Institutional Animal Care and Use Committee (IACUC) of the Biological Resource Center (BRC) of Biopolis in Singapore. The BRC IACUC protocol was approved by the National Advisory Committee for Laboratory Animal Research in Singapore (Permit Numbers: 110626 and 120773). Peripheral blood and resected tumor samples were obtained with Ethics Committee approval from HCC patients who underwent curative resection. Formalin-fixed paraffin-embedded tissue sections were used for immunofluorescence staining.
Cell isolation and culture
Splenic CD8+ T cells were purified from OT-I mice by negative selection using the CD8α+ T cells isolation kit II (Miltenyi Biotec). Splenic DCs were purified from WT or PD-1 KO mice by positive selection using CD11c+ microbeads (Miltenyi Biotec). All cell isolations were performed according to the manufacturer's protocols. OVA peptide (SIINFEKL) was purchased from Anaspec Corporation. Freshly isolated splenic T cells were maintained at 37ºC in RPMI-1640 medium containing 10% fetal bovine serum, 100 IU/mL Penicillin, 100 μg/mL Streptomycin, 2 mM Glutamine, and 50 μM β-mercaptoethanol (all from GIBCO). Bone marrow-derived DCs (BMDCs) from 8- to 12-week-old WT and PD-1 KO mice were prepared as previously described.Citation37 After 7–10 d of culture, BMDCs were analyzed for CD11c expression and used in assays when > 85% were CD11c positive. The Hepa1-6Citation38 HCC cell line was prepared and maintained as described previously. PBMCs were isolated from blood using Ficoll-Paque PREMIUM (GE Healthcare). Human DC subsets were sorted by flow cytometry and cultured at 37°C in X-Vivo 15 medium (Lonza) with 2% human serum (Off the Clot; Gemini), 100 IU/mL Penicillin (GIBCO) and 100 μg/mL Streptomycin (GIBCO).
Flow cytometry
BMDCs, splenic DCs and OT-I T cells were stained with antibodies against cell surface molecules, and were analyzed using a BD FACS LSR flow cytometer. BMDCs were co-cultured with or without CD8+ OT-I T cells in the presence of 0.1 μg/mL OVA peptide (SIINFEKL) and 10 μg/mL Poly I:C (Invivogen) in 96-well plates (5 × 104 BMDC and 5 × 104 OT-I T cells per well) for 2 d before FACS analysis. Human DC sorting was performed using a BD FACS Aria flow cytometer. Intracellular staining was performed using BD GolgiPlug according to the manufacturer's protocol. Dead cells were discriminated from live cells using DAPI (Sigma-Aldrich) or the LIVE/DEAD® Fixable Blue Dead Cell Stain Kit (Molecular Probes). Antibodies purchased from eBioscience were as follows: hamster anti-mouse CD11c-PE-Cy7 (N418), CD3ϵ-PerCP5.5 (145–2C11), CD279-FITC (RMP1-30), perforin-PE (eBioOMAK-D), granzyme B-PE-Cy7 (16G6), mouse anti-mouse SIINFEKL-H-2Kb-APC (25-D1.16), mouse anti-human CD7-FITC (4H9), mouse anti-human HLA-DR-e605NC (LN3), mouse IgG1,κ-APC isotype control (P3.6.2.8.1), and hamster IgG-PE-Cy7 and -PerCP5.5 isotype controls (eBio299Arm). Antibodies purchased from BD PharMingen were as follows: FITC-conjugated murine (BALBC/c) anti-mouse H-2Kb (AF6-88.5), hamster anti-mouse CD54-FITC (3E2), CD80-PE (16-10A1), mouse anti-human CD11c-V450 (B-ly6), CD45-V500 (HI30), rat anti-mouse CD40-FITC (3/23), CD86-PE (GL1), I-A/I-E-V500 (M5/114.15.2), CD274-PE (MIH5), CD8α-APC-Cy7 (53-6.7) and Vα2 TCR-APC (B20.1). Isotype controls, also from BD PharMingen, included hamster IgG1,κ-APC (A19-3), hamster IgG2,κ-PE (B81-3), mouse IgG2a,κ-FITC (G155-178), rat IgG2a,κ-FITC, PE and APC-Cy7 (R35-95), rat IgG2a,λ-PE, and APC (B39-4), rat IgG2b,κ-FITC, PE, and APC-Cy7 (A95-1). Mouse anti-human CD14-ECD (RMO52) was purchased from Beckman Coulter and mouse anti-human CD141-APC (AD5-14H12) and human Fc receptor blocking reagent were both from Miltenyi Biotec. Mouse anti-human CD3-FITC (UCHT1),CD19-FITC (HIB19),CD20-FITC (2H7),CD56-FITC (MEM-188), CD123-PerCp-Cy5.5 (6H6), CD1c-PE-Cy7 (L161), CD16-APC-Cy7 (3G8), CD279-PE (EH12.2h7), and mouse IgG1,κ-PE isotype control (MOPC-21) were all from BioLegend (Genomax).
ELISA cytokine assay
BMDCs were pre-pulsed for 4 h with different concentrations of OVA peptide (SIINFEKL) in the presence or absence of 10 μg/mL Poly I:C (Invivogen) and were then co-cultured with OT-I T cells in 96-well plates (5 × 104 DC and 5 × 104 T cells per well) without washing. IL-2 and IFNγ secretion were measured by ELISA 48 h later. For blocking experiments, 10 μg/mL of purified antibodies (functional grade) against PD-1/CD279 (RMP1-30) (all from eBiosciences) or their corresponding isotype controls were added into DC-T co-cultures.
In vivo T cell proliferation assay
BMDCs from WT or PD-1 KO mice were pre-pulsed with or without 1 μg OVA peptide (SIINFEKL) in medium containing 10 μg/mL Poly I:C for 4 h and washed twice with PBS, before injecting them together with 50 μg Poly I:C into the footpads of CD45.1+ WT congenic recipient mice (3 × 106 BMDCs per footpad). OT-I T cells (CD45.2+ background) were labeled with 5 μM CellTraceTM CFSE (Molecular Probes, Invitrogen) and transferred intravenously into recipient mice (106 T cells per mouse). After three days, draining popliteal lymph nodes (LNs) were collected and T cell proliferation was analyzed by flow cytometry.
Transplanted tumor mouse model
The Hepa1-6 transplanted tumor mouse model was established as described previously.Citation38 Briefly, 5 × 106 Hepa1-6 cells were subcutaneously injected into each flank of 6- to 8-week-old WT C57BL/6 mice (day 7). One week post Hepa1-6 tumor transplantation (day 0), tumor-bearing mice were injected intratumorally with 100 μL PBS containing 1 × 106 WT or PD-1 KO BMDCs or 100 μL PBS only (control with no DC transfer). Tumor growth was monitored on the indicated days by palpation or using a digital caliper from day 1 to 14 before the tumors were harvested and analyzed by flow cytometry.
Spontaneous liver tumor mouse model
Mice expressing the hepatitis virus B surface antigen in their hepatocytes to promote chronic inflammationCitation39 and the liver-specific Sleeping Beauty transposon to drive spontaneous mutagenesisCitation40 were generated. Aged mice (11- to 18-month-old) with liver tumors were sacrificed and livers were harvested for analysis by flow cytometry.
Confocal microscopy
Immunofluorescence (IF) staining was performed on paraffin-embedded HCC samples as described before.Citation41 IF images were captured with an Olympus FlourView FV1000 confocal microscope. Antibodies were as follows: rabbit anti-human CD11c (EP1347Y, Abcam); goat CD3ϵ (M-20 SantaCruz), and mouse anti-human PD-1 (NAT105, Abcam).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1085146_supplemental_data.docx
Download MS Word (175.2 KB)Acknowledgments
We thank Prof. Tasuku Honjo (Kyoto University) for providing PD-1 KO mice (C57BL/6-Pdcd1-/-) and Dr. Han Chong Toh (National Cancer Center, Singapore) for providing paraffin-embedded hepatocellular carcinoma samples. We would like to thank Kerry McLaughlin of Insight Editing London for critical review of the manuscript.
Funding
This work was supported by research funding from the Biomedical Research Council (BMRC), Agency for Science, Technology & Research (A*STAR) and grant support (reference number: NMRC/MOHIAFCAT2/005/2015) from National Medical Research Council (NMRC), Singapore.
References
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008; 26:677-704; PMID:18173375; http://dx.doi.org/10.1146/annurev.immunol.26.021607.090331.
- Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 1992; 11:3887-95; PMID:1396582.
- Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999; 11:141-51; PMID:10485649; http://dx.doi.org/10.1016/S1074-7613(00)80089-8.
- Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001; 291:319-22; PMID:11209085; http://dx.doi.org/10.1126/science.291.5502.319.
- Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U S A 2005; 102:11823-8; PMID:16087865; http://dx.doi.org/10.1073/pnas.0505497102.
- Wang J, Okazaki IM, Yoshida T, Chikuma S, Kato Y, Nakaki F, Hiai H, Honjo T, Okazaki T. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int Immunol 2010; 22:443-52; PMID:20410257; http://dx.doi.org/10.1093/intimm/dxq026.
- Yoshida T, Jiang F, Honjo T, Okazaki T. PD-1 deficiency reveals various tissue-specific autoimmunity by H-2b and dose-dependent requirement of H-2g7 for diabetes in NOD mice. Proc Natl Acad Sci U S A 2008; 105:3533-8; PMID:18299579; http://dx.doi.org/10.1073/pnas.0710951105.
- Okazaki T, Otaka Y, Wang J, Hiai H, Takai T, Ravetch JV, Honjo T. Hydronephrosis associated with antiurothelial and antinuclear autoantibodies in BALB/c-Fcgr2b-/-Pdcd1-/- mice. J Exp Med 2005; 202:1643-8; PMID:16352741; http://dx.doi.org/10.1084/jem.20051984.
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000; 192:1027-34; PMID:11015443; http://dx.doi.org/10.1084/jem.192.7.1027.
- Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2001; 2:261-8; PMID:11224527; http://dx.doi.org/10.1038/85330.
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004; 173:945-54; PMID:15240681; http://dx.doi.org/10.4049/jimmunol.173.2.945.
- Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005; 25:9543-53; PMID:16227604; http://dx.doi.org/10.1128/MCB.25.21.9543-9553.2005.
- Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med 2012; 209:1201-17; PMID:22641383; http://dx.doi.org/10.1084/jem.20112741.
- Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, Qiu Y, Jussif JM, Carter LL, Wood CR et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett 2004; 574:37-41; PMID:15358536; http://dx.doi.org/10.1016/j.febslet.2004.07.083.
- Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol 2013; 14:1212-8; PMID:24240160; http://dx.doi.org/10.1038/ni.2762.
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006; 439:682-7; PMID:16382236; http://dx.doi.org/10.1038/nature04444.
- Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006; 443:350-4; PMID:16921384; http://dx.doi.org/10.1038/nature05115.
- Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, Boulassel MR, Delwart E, Sepulveda H, Balderas RS et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med 2006; 12:1198-202; PMID:16917489; http://dx.doi.org/10.1038/nm1482.
- Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, Ferrari C. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol 2006; 80:11398-403; PMID:16956940; http://dx.doi.org/10.1128/JVI.01177-06.
- Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 2002; 99:12293-7; PMID:12218188; http://dx.doi.org/10.1073/pnas.192461099.
- Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol 2005; 17:133-44; PMID:15611321; http://dx.doi.org/10.1093/intimm/dxh194.
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443-54; PMID:22658127; http://dx.doi.org/10.1056/NEJMoa1200690.
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366:2455-65; PMID:22658128; http://dx.doi.org/10.1056/NEJMoa1200694.
- Yao S, Zhu Y, Chen L. Advances in targeting cell surface signalling molecules for immune modulation. Nat Rev Drug Discov 2013; 12:130-46; PMID:23370250; http://dx.doi.org/10.1038/nrd3877.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252-64; PMID:22437870; http://dx.doi.org/10.1038/nrc3239.
- Park SJ, Namkoong H, Doh J, Choi JC, Yang BG, Park Y, Chul Sung Y. Negative role of inducible PD-1 on survival of activated dendritic cells. J Leukoc Biol 2014; 95(4):621-9; PMID:24319287; http://dx.doi.org/10.1189/jlb.0813443
- Krempski J, Karyampudi L, Behrens MD, Erskine CL, Hartmann L, Dong H, Goode EL, Kalli KR, Knutson KL. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J Immunol 2011; 186:6905-13; PMID:21551365; http://dx.doi.org/10.4049/jimmunol.1100274.
- Yao S, Wang S, Zhu Y, Luo L, Zhu G, Flies S, Xu H, Ruff W, Broadwater M, Choi IH et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood 2009; 113:5811-8; PMID:19339692; http://dx.doi.org/10.1182/blood-2009-02-203141.
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002; 8:793-800; PMID:12091876; http://dx.doi.org/10.1038/nm0902-1039c.
- Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, Collins M, Honjo T, Freeman GJ, Carreno BM. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol 2002; 32:634-43; PMID:11857337; http://dx.doi.org/10.1002/1521-4141(200203)32:3%3c634::AID-IMMU634%3e3.0.CO;2-9.
- Davis DM. Intercellular transfer of cell-surface proteins is common and can affect many stages of an immune response. Nat Rev Immunol 2007; 7:238-43; PMID:17290299; http://dx.doi.org/10.1038/nri2020.
- Dhainaut M, Moser M. Regulation of immune reactivity by intercellular transfer. Front Immunol 2014; 5:112; PMID:24734030; http://dx.doi.org/10.3389/fimmu.2014.00112.
- Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med 2012; 366:2517-9; PMID:22658126; http://dx.doi.org/10.1056/NEJMe1205943.
- Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat Rev Immunol 2015; 15:45-56; PMID:25534622; http://dx.doi.org/10.1038/nri3790.
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell 1994; 76:17-27; PMID:8287475; http://dx.doi.org/10.1016/0092-8674(94)90169-4.
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 1992; 68:869-77; PMID:1547488; http://dx.doi.org/10.1016/0092-8674(92)90030-G.
- Granucci F, Zanoni I, Pavelka N, Van Dommelen SL, Andoniou CE, Belardelli F, Degli Esposti MA, Ricciardi-Castagnoli P. A contribution of mouse dendritic cell-derived IL-2 for NK cell activation. J Exp Med 2004; 200:287-95; PMID:15289500; http://dx.doi.org/10.1084/jem.20040370.
- Chew V, Tow C, Huang C, Bard-Chapeau E, Copeland NG, Jenkins NA, Weber A, Lim KH, Toh HC, Heikenwalder M et al. Toll-like receptor 3 expressing tumor parenchyma and infiltrating natural killer cells in hepatocellular carcinoma patients. J Natl Cancer Inst 2012; 104:1796-807; PMID:23197495; http://dx.doi.org/10.1093/jnci/djs436.
- McCartney S, Vermi W, Gilfillan S, Cella M, Murphy TL, Schreiber RD, Murphy KM, Colonna M. Distinct and complementary functions of MDA5 and TLR3 in poly(I:C)-mediated activation of mouse NK cells. J Exp Med 2009; 206:2967-76; PMID:19995959; http://dx.doi.org/10.1084/jem.20091181.
- Chisari FV, Klopchin K, Moriyama T, Pasquinelli C, Dunsford HA, Sell S, Pinkert CA, Brinster RL, Palmiter RD. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell 1989; 59:1145-56; PMID:2598264; http://dx.doi.org/10.1016/0092-8674(89)90770-8.
- Chew V, Tow C, Teo M, Wong HL, Chan J, Gehring A, Loh M, Bolze A, Quek R, Lee VK et al. Inflammatory tumour microenvironment is associated with superior survival in hepatocellular carcinoma patients. J Hepatol 2010; 52:370-9; PMID:19720422; http://dx.doi.org/10.1016/j.jhep.2009.07.013.