
ABSTRACT
Adoptive cell transfer (ACT) is an emerging anticancer therapy that has shown promise in various malignancies. Redirecting antigen specificity by genetically engineering T cells to stably express receptors has become an effective variant of ACT. A novel extension of this approach is to utilize engineered T cells to produce and deliver anticancer therapeutics that enhance cytotoxic T cell function and simultaneously inhibit immunosuppressive processes. Here, we review the potential of using T cells as therapeutic-secreting vehicles for immunotherapies and present theoretical and established arguments in support of further development of this unique cell-based immunotherapy.
The field of gene therapy has advanced rapidly since its advent in the 1970s. Yet, the in vivo delivery of recombinant genes in humans remains restricted by various technical limitations and safety concerns. One gene therapy niche that circumvents many of these limitations is the development of cell vehicles genetically engineered to secrete bioactive therapeutics. These cell vehicles can be prepared ex vivo and are subsequently infused into individuals. Initially developed in the 1990s, the earliest examples of cell-mediated drug delivery systems centered on mesenchymal stem cells (MSC) and T cells engineered to secrete various cytokines.Citation1-4 Marrying advances in genetic engineering with T cell ACT is a logical step for the improvement of ACT as this approach has the potential to circumvent many of the limitations associated with systemic drug delivery. The therapeutic success of this method hinges on two critical factors: (1) the selection of appropriate cell carriers that are well-suited for target applications and (2) the synthesis of specific products that will exert their intended therapeutic function.
A wide variety of cells have been used as drug-delivery vehicles. Perhaps the most extensively studied cell vehicle system is based on adult stem cells such as MSC (reviewed in refs. 4–6).Citation1,4-6 MSCs have been thoroughly evaluated as therapeutic-delivering cells in cancer models but their ability to promote tumor growth, lack of persistence after transplantation in humans, immunosuppressive qualities, and inability to home to specific targets have tempered support for MSC use in cancer therapy.Citation4,7,8 Nevertheless, therapy-delivering MSCs remain a focus in cancer research.Citation9,10 Meanwhile, endothelial precursors, macrophages, neutrophils, and microglia have also been used or proposed to deliver therapeutics to tumors.Citation8,11-14 However, various challenges limit the use of these cells as therapeutic vehicles.Citation8,11,14 Conversely, T cells have been used for several years as therapeutic-delivering cell vehicles. A seminal study of T cells secreting IL-2 was published in 2001, and in the following years streamlining of the genetic manipulation of T cells has allowed this niche field to evolve and advance rapidly.Citation2 The following review focuses on the advantages and future challenges of using genetically engineered T cells to deliver and secrete products to enhance antitumor immunity, particularly in the context of adoptive T cell transfer for cancer. These T cells, from hereon will be referred to as producer T cells.
Adoptive cell transfer and synthetic T cell receptors
Recent progress in ACT to treat cancer patients has bolstered enthusiasm for therapeutic strategies that utilize the immune system's ability to selectively target and destroy malignant cells. One form of ACT consists of using tumor-specific T cells obtained from tumors, referred to as tumor-infiltrating lymphocytes (TILs), or from circulating peripheral T cells. T cells are then expanded ex vivo and infused back into lymphodepleted patients (). The details of this approach have been refined over several years so that TILs can now be successfully generated in a majority of patients.Citation15 However, expanded TILs represent a heterogeneous population of T cells with T cell receptors (TCR) specific for a variety of antigens.
Figure 1. Schematic of possible T cell vehicle biologics and their therapeutic targets. (A) TIL are isolated from tumors, expanded, and can be genetically engineered using a wide variety of transgenes. (B) Immunosuppressive cells generate a tumor microenvironment conducive to tumor cell growth which limits T cell function. (C) Immunosuppressive cytokines and bioactive molecules suppress T cell function. (D) Immune checkpoints are activated by interactions between T cells, tumor cells, and other cells of the tumor microenvironment and suppress effector cell function. (E) Transgenes can be designed with promotors allowing antigen-dependent expression. (F) A wide variety of transgene products can be selected for various purposes. Abbreviations: APC, antigen presenting cell; BiTE, bi-specific T-cell engager; CAR, chimeric antigen receptor; CTL, cytotoxic T lymphocyte; MDSC, myeloid-derived suppressive cell; NFAT, nuclear factor of activated T-cells; NK, natural killer; PD-1, programmed death-1; PD-L1, programmed death ligand 1; pNFAT, NFAT-responsive promoter; PTD, protein transduction domain; TAM, tumor-associated macrophage; TCR, T cell receptor; TGF-β, transforming growth factor β; TIL, tumor infiltrating lymphocyte; Treg, regulatory T cell; VEGF, vascular endothelial growth factor;.
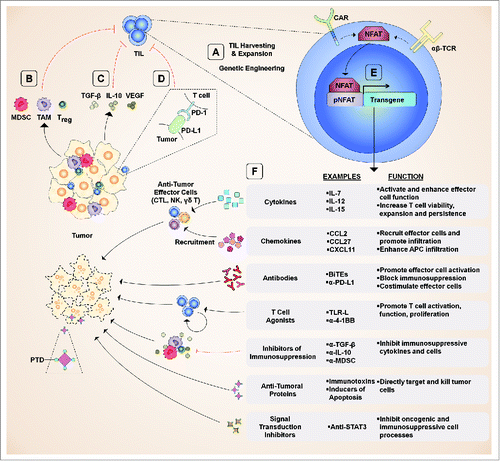
To address the heterogeneity in TILs and improve tumor targeting, genetic engineering has been used to create T cell populations that express not only native TCRs, but also a tumor-specific recombinant α/β-TCR or chimeric antigen receptor (CAR).Citation16–19 CARs are artificial recombinant receptors composed of an extracellular antigen-binding domain and one or more cytosolic T cell signaling domains. The expression of α/β-TCR or CAR artificial receptors allows for the generation of tumor-reactive T cells that have high affinity for tumor antigens. In addition, CARs uniquely bypass the need for T cells to interact with MHC and can bind directly to targets on the cell membranes of tumors. Yet, this form of therapy is not without shortcomings. Generating sufficient numbers of genetically engineered T cells requires that cells remain in ex vivo culture for prolonged periods, which can reduce in vivo T cell function and persistence.Citation20 Additionally, α/β-TCRs and CARs increase the risk for “on-target off-tumor” (the binding of engineered cells to target proteins on non-malignant tissues) toxicities and must be evaluated thoroughly before clinical use.Citation21–24 Finally, designing CARs for solid tumors has proven far more challenging than for hematopoietic malignancies. Nevertheless, encouraging CAR T cell clinical trial results have validated the approach of using genetically engineered T cells for cancer immunotherapy.Citation25–28 In melanoma, ACT objective response rates are approximately 50% and promising rates of complete remission have been observed.Citation29,30 Clinical trials have also demonstrated utility for ACT in several other malignancies.Citation19,31
Barriers to improving ACT efficacy
T cell migration
Despite promising clinical results, several limitations hinder the generation of long-lasting and productive antitumor T cell responses in ACT for solid tumors. One major issue is T cell migration. To engage tumors, T cells must complete a complex process involving extravasation from blood vessels and navigation through interstitial tissues. Several factors limit this process, including loss of adhesion molecules on endothelial cells of the tumor vasculature,Citation32-34 changes to the intratumoral chemokine milieu,Citation33,35,36 and expression of inhibitory molecules such as Fas, transforming growth factor β (TGF-β), and programmed death ligand 1 (PD-L1) by endothelial cells.Citation33,37,38 In combination, these mechanisms limit T cell migration and infiltration to tumor sites.
T cell immunosuppression in ACT
T cells that do reach the tumor environment must persist at the tumor site while maintaining effector function. T cells, in particular CD8+ cytotoxic T lymphocytes (CTL), are well equipped for this task. Unfortunately, numerous tumor-associated immunosuppressive mechanisms reduce antitumor efficacy by inducing T cell dysfunction and/or death.
The results from preclinical and clinical studies have demonstrated that tumor-associated leukocytes such as regulatory T cells (Treg), myeloid-derived suppressor cells (MDSC), tumor-associated macrophages (TAM), and tolerogenic dendritic cells (DC) limit the function of tumor-reactive T cells ().Citation33,39-41 These cells often secrete a variety of immunosuppressive cytokines (e.g. IL-6, IL-10, and TGF-β) and express inhibitory molecules such as PD-L1 and PD-L2 which bind programmed cell death 1 (PD-1) expressed on T cells ().Citation33,42-44 In addition to PD-1, several other immune checkpoints have now been discovered which limit T cell function and survival.Citation45 Tumor cells further restrict antitumor T cell function by promoting the development of immunosuppressive leukocytes and by expressing immune checkpoint ligands. Ultimately, these immunosuppressive mechanisms (among others) result in poor T cell proliferation, maintenance, survival, and cytotoxic function in the tumor microenvironment.
History of therapeutic-delivering T cells
One strategy to overcome the limitations of ACT involves the generation of therapeutic-secreting tumor-targeted T cells that would serve as vehicles producing select molecules at the tumor site. In 2001, for the first time, a human CD8+ T cell clone was transduced with the human IL-2 gene and demonstrated improved survival over untransduced CD8+ T cells.Citation2 This initial study has been followed by several reports which explored the effects of T cells engineered to secrete other endogenous cytokines or therapeutic antibodies.Citation3,46-63 Producer T cells engineered to deliver therapeutics can be used in combination with TCR- or CAR-modified T cells and offer a number of theoretical and proven advantages over the use of other producer cells and systemic cancer therapies ().
Table 1. Advantages of producer T cells.
Advantages of T cells as therapeutic-secreting vehicles
Tumor homing and tumor penetration
Unlike most conventional systemic therapies, T cells possess the ability to specifically home to tumor sites. Despite vasculature-specific immunosuppressive processes which can inhibit tumor invasion, T cells are capable of penetrating into large solid tumors.Citation64 Substantial tumor homing and invasion by T cells is often observed in solid tumors and is typically regarded as a positive prognostic indicator both in untreated cancer patients and those treated with ACT.Citation64 This capability allows T cells to deliver therapeutics throughout solid tumors.
A supplementary T cell asset is the ability to access virtually any anatomical space. T cells are capable of penetrating the blood brain barrier (BBB) to reach the central nervous system (CNS).Citation32,65 The BBB is a complex, multilayered anatomical barrier. That T cells can effectively cross this barrier highlights their ability to migrate throughout the entire body. In addition to the CNS and despite the nomenclature, T cells can also migrate to other immune-privileged sites such as the testes and eyes.Citation66,67 This effective tissue-penetrating characteristic allows for the use of engineered T cells to treat tumors which have been historically difficult to access such as primary brain tumors, brain metastases, and prostate malignancies. In addition, the ability to home to specific targets throughout the body makes T cells ideal for patients with widespread metastatic disease.
Localized and inducible drug production
The ability for T cells to accumulate in tumors offers the unique advantage for localized and continuous production of therapeutics confined within the tumor environment. This is in contrast with the use of systemic drugs that distribute throughout the body exposing both malignant and non-malignant tissues which can result in toxicity and limit the ability to supply an effective dose ().Citation68 Further, the amount of drug required to reach an effective dose can be limited by variations in the tumor vasculature and internal pressures within solid tumors.Citation68 Finally, traditional systemic therapies often suffer from notoriously poor penetration of physiologic barriers such as the BBB.
An additional benefit of localized therapeutic production is that the secreted therapeutic is not subjected to systemic metabolic processes prior to encountering the tumor. These processes often limit the bioavailability and half-life of drugs (). Traditionally administered therapeutics must be titrated to account for tissue distribution, biotransformation, degradation, and renal clearance. In contrast, a therapeutic secreted by producer T cells is initially only exposed to the isolated metabolic pressures of the tumor environment.
The localized production of therapeutics can be further refined using a technique in which only T cells which have engaged tumor cells are able to secrete therapeutics. To this end, multiple studies have shown that a promoter consisting of several nuclear factor of activated T cells (NFAT) binding motifs can be used to drive transgene expression only upon T cell activation ().Citation55,69 In 2000, the NFAT-responsive promoter system was developed to select for antigen-specific T cells from a heterogeneous T cell pool.Citation69 In producer T cells, this strategy offers several advantages over models which use T cells that constitutively express their transgene products. The first and most obvious advantage of this system is that it allows for the production of therapeutics only upon stimulation with tumor antigen. Therefore, secretion is confined within and around tumors. This approach also allows for a much broader selection of therapeutics that would otherwise be toxic if administered systemically. For example, while systemic IL-12 administration is excessively toxic, localized intratumoral IL-12 administration has been shown to be clinically well-tolerated and effective as a cancer immunotherapy.Citation70,71 These findings on IL-12 have been extended to producer T cells.Citation55 In a mouse model of melanoma, the administration of 5×105 T cells constitutively expressing IL-12 resulted in transient body weight loss in mice, but a 6-fold increase (3×106) of NFAT-driven T cells did not result in any observable toxicity or change in body weight.
Potential transgene therapeutics and their ability to enhance T cell immunotherapies
To our knowledge, all of the published reports on producer T cells for ACT have focused on cytokine or bispecific antibody secretion. Yet, countless therapeutic possibilities remain unexplored (). Each of the strategic avenues discussed below has the potential to address major impediments to T cell immunotherapies.
Cytokines
One technique to combat the multitude of redundant immunosuppressive signals in the tumor microenvironment is to tip the balance in favor of T cell proliferation and activity. Cytokines are logical choices to achieve this goal. The seminal preclinical reports on producer T cells secreting IL-2 were established in 2001 and culminated in a 13 patient clinical trial ().Citation2,3,46,47 The clinical trial did not result in significant responses in part due to the fact that IL-2 increases activation-induced cell death (AICD) of T cells.Citation33 Furthermore, because Treg depend on IL-2 for their survival and function, it is likely that IL-2 negatively impacted treatment efficacy by driving Treg function and survival.Citation33,49,72
Table 2. Literature review on producer T cells.
Multiple groups have examined the effects of other cytokines such as IL-15, which acts similarly to IL-2 on T cells, but can also prevent AICD and drive memory CD8+ T cell differentiation.Citation46,48–51,61 IL-15-producing T cells have demonstrated potent antitumor activity in a murine melanoma model and have been examined in both human T cell in vitro experiments and mouse models of cancer ().Citation46,48,49,51,61 Notably, human T cells transduced with constitutively expressed IL-15 did not require exogenous IL-2 for in vitro maintenance and expansion.Citation49 A later study revealed the unintended possibility of generating transformed human T cell clones, which exhibited logarithmic growth independent of exogenous cytokine support in vitro.Citation50 Thus, although IL-2 and IL-15 may not be appropriate biologics, these studies serve as a proof-of-concept that T cells can indeed serve as drug-delivering vehicles.
The most extensively studied cytokine in producer T cells is IL-12. IL-12 is a proinflammatory cytokine which enhances T cell function, drives Th1 immune responses, and inhibits Treg. These attributes have led to systemic administration of IL-12 in cancer clinical trials, but severe dose-dependent toxicities including thrombocytopenia, leukopenia, and hyperbilirubinemia among others have prevented efficacious clinical application of IL-12. The synthesis of producer T cells is a reasonable approach to bypass systemic toxicity and has been pursued comprehensively in various cancer models ().Citation52–59 Initially, IL-12 transgenic Epstein-Barr virus (EBV) specific human T cells were used to target Hodgkin's lymphoma in a preclinical model where they exhibited increased cytotoxic function and resistance to TGF-β.Citation52 Importantly, an early study revealed one of the unique drawbacks of IL-12 by demonstrating that proliferation was reduced in transduced T cells when compared to untransduced counterparts.Citation55 Subsequent studies have elaborated on these results. IL-12 transduced murine T cells had potent preclinical anti-melanoma activity despite administering as few as 1×104 cells.Citation53 Further, the IL-12-modified T cells increased tumor infiltration and recruitment of endogenous CD8+ T cells along with natural killer (NK) cells. In 2011, the transduction of an NFAT-IL-12 transgene into T cells was found to be less toxic and more effective than constitutively expressed IL-12 in a murine model.Citation55 These studies have been extended to human T cells where it appears that this antigen-inducible approach can be used to expand transduced human T cells without interference from constitutively expressed IL-12.Citation56 In addition, constitutively expressing IL-12 transduced murine T cells are effective in ACT without lymphoablative preconditioning, which is typically necessary for efficient engraftment of transferred T cells.Citation57 Finally, IL-12 producer T cells have now been shown to be effective in preclinical cancer models beyond melanoma, including ovarian cancer and leukemia.Citation58,59
The results of these reports led to a phase I clinical trial using NFAT-responsive IL-12-secreting TIL for adoptive transfer.Citation60 Eleven of 32 trial participants experienced objective responses, which appeared to be strongly dependent on the total number of IL-12-engineered TIL infused. Sixty-three percent of patients treated with at least 3×108 transduced TIL experienced objective responses. Notably, the T cell numbers at which objective responses were typically observed were 10- to 100-fold lower than the number required to achieve responses in adoptive transfer with genetically unaltered TIL. Further, IL-2, which is typically infused along with expanded TIL was not used in the clinical trial. Despite relatively high response rates in patients treated with higher numbers of T cells, evidence of T cell persistence was limited. More troubling however, were the high-grade adverse events, particularly in patients infused with higher amounts of TILs, some of which were life-threatening. The poor TIL persistence and high toxicity have been attributed to IL-12. These results highlight the complexity and difficulty of selecting proper transgenes for T cell modification in ACT. Future clinical trials, including a proposed phase I clinical trial for ovarian cancer using MUC-16ecto targeting CAR T cells modified to secrete IL-12 will likely need to refine this therapeutic strategy in humans.Citation73
Antibodies
An additional approach to simultaneously augment T cell targeting and function has been the use of bi-specific T cell engagers (BiTE). BiTEs are recombinant synthetic antibodies which contain two distinct antigen-binding domains, one of which targets a tumor surface antigen while the other binds to T cell activation molecules. This method has been used to generate BiTEs against carcinoembryonic antigen (CEA) and CD3ε using transduced human peripheral blood lymphocytes (PBL).Citation62 Anti-CEA × anti-CD3 antibodies were effective at redirecting T cell cytotoxicity against human colon cancer in vitro and in a xenograft model.Citation62 Additionally, producer T cells secreting BiTEs specific for CD3 and erythropoietin-producing hepatocellular carcinoma A2 (EphA2) were shown to be effective in treating animal models of brain and lung cancer.Citation63 These studies demonstrate that using BiTE antibody-secreting tumor-specific T cells can amplify the antitumor activity of both transferred and endogenous T cells.
Theoretically, producer T cells engineered to secrete antibodies could be used to achieve a diverse set of therapeutic strategies. Antibodies could be used to mask T cell inhibitory molecules such as PD-L1 or costimulate T cells by engaging receptors such as 4–1BB. Alternatively, T cells could be engineered to secrete antibodies to activate DCs (e.g., agonistic anti-CD40 antibody) and in turn potentiate tumor antigen presentation to endogenous T cells.
Chemokines
As discussed above, the disruption of molecules involved in T cell migration often limits T cell tumor infiltration. Thus, T cells engineered to manipulate chemokine signaling pathways may promote T cell tumor homing and accumulation, which is vital for ACT efficacy. To achieve this, T cells could be engineered to produce chemokines such as CCL5, CXCL1, and CXCL16, all of which have been shown to increase T cell tumor infiltration.Citation35 Other chemokines such as CCL19 and CXCL12 have been shown to strengthen T cell interactions with DC, and could be used to promote activation of both endogenous and infused T cells.Citation35 In addition, because homing to different types of malignancies appears to depend on distinct chemokine signatures, T cells could be tailored to different tumor types.Citation35,36 These approaches would allow for T cells which successfully home to tumors to amplify responses by recruiting supplementary tumor-reactive T cells or professional antigen-presenting cells which could further activate endogenous T cells. Notably, the use of NFAT-driven promoters in these approaches would be essential to prevent systemic chemokine release and disordered T cell migration. To date, T cells have only been engineered with chemokine receptors such as CCR2b and CXCR2 in order to augment their target homing abilities.Citation74,75 Yet, using T cells to actively secrete chemokines to either amplify T cell tumor infiltration or recruit other antitumor effector cells has not been examined and remains a promising therapeutic tactic.
Other strategies
While the selection of transgenes in producer T cells has remained limited to cytokines and antibodies, a number of interesting yet unexplored strategies exist. One approach would be to engineer T cells which secrete inhibitors of immunosuppressive cytokines such as IL-6, IL-10, and TGF-β. Indeed, T cells engineered to express a dominant negative of TGF-β to protect them from tumor-derived TGF-β are being examined in clinical trials (NCT00368082, NCT02065362) following promising preclinical results.Citation76,77 Notably, a soluble inhibitor might be more effective by inhibiting TGF-β signaling in T cells along with potentially oncogenic TGF-β signaling in tumor cells or other cells of the tumor microenvironment.
Inhibiting vital intracellular signaling molecules represents another strategy to potentiate antitumor T cell responses. STAT3 signaling in tumor cells as well as immunosuppressive cells such as MDSCs plays a central role in the development of an immunosuppressive tumor environment.Citation78,79 Furthermore, many tumor types rely on STAT3 signaling for survival or proliferation.Citation80 Therefore, identifying relevant molecules that can be produced by T cells to inhibit STAT3 signaling could potentiate antitumor effects by sensitizing tumor cells and simultaneously reshaping the tumor environment toward one that promotes T cell effector function.
An additional possibility includes the secretion of toll-like receptor (TLR) agonists including ligands to TLR1, TLR2, and TLR5. TLR1 and TLR2 bind to bacterial lipoproteins, whereas TLR5 recognizes bacterial flagellin. These ligands are peptide/protein-based, and could be useful in strategies designed to activate potent but localized antitumor responses when produced by tumor-reactive T cells.Citation81 A major advantage to using TLR ligands is their ability to activate antigen presenting cells which can subsequently reciprocally activate tumor-reactive T cells to create an amplifying process. Furthermore, TLR ligands can enhance T cell responses by costimulating TLRs directly on T cells.Citation81 We have shown that producer T cells secreting TLR5 ligand improve antitumor function in part by reshaping the tumor microenvironment. The TLR5 producer T cells were found to limit the expression of T cell exhaustion markers, increase chemokine receptor expression on T cells, and reduce the number of MDSCs in the spleen.Citation82
Other attractive options might include inhibitors of immunosuppressive cells (e.g., MDSC and Treg), immunosuppressive signaling pathway inhibitors, and antitumor proteins (). For example, antibodies to neutralize a variety of factors involved in immunosuppression or the generation of MDSCs or Treg, including GM-CSF, IL-6, IL-10, and VEGF could serve to potentiate antitumor T cell responses. The therapeutics could be designed to target numerous cell types, including T cells, tumor-associated leukocytes, and tumor cells.
Limitations and potential challenges in producer T cells
The field of producer T cells for ACT is still in its infancy, and future studies will undoubtedly reveal unique limitations to this strategy, many of which are predictable (). A major concern with the use of genetically engineered cells is the potential for malignant transformation, which has been observed in HSC clinical trials. Initial studies with IL-15 transduced T cell clones unintentionally generated a seemingly immortalized clone that proliferated without exogenous IL-2 support and was resistant to apoptosis.Citation50 However, studies in mice comparing T cells to HSCs transduced with known T cell oncogenes has revealed that T cell transformation seems unlikely to occur.Citation21 Clinical data supports this notion as no case of malignant transformation has been reported among the hundreds of patients enrolled in clinical trials using engineered T cells.Citation21 Nevertheless, steps to prevent this outcome should be taken, and transgenes should be selected carefully. To this end, producer T cells could be engineered with kill switches such as herpes simplex virus thymidine kinase or inducible apoptosis systems.Citation46,83 Alternatively, producer T cells engineered to express unique surface markers could be depleted using antibodies specific to the markers.
Table 3. Limitations and challenges of producer T cells.
Reaching effective intratumoral drug concentrations may be an additional limitation. The quantity of transgene that can be expressed by T cells may be somewhat limited when compared to other cell types, particularly when using NFAT-based systems. Indeed, the quantity of NFAT-driven IL-12 produced in vitro was decreased in comparison to a similar constitutively expressed vector.Citation55 Comparing human TIL transduced with constitutively produced IL-12 and NFAT-driven IL-12 revealed that NFAT-driven cells produced markedly less IL-12 upon stimulation (˜70,000 pg/mL in constitutively expressing cells vs. ˜20,000 pg/mL in NFAT-responsive cells).Citation56 However, the two transduced populations were not directly comparable due to potential variations in transgene copy number. Future studies may be needed to enhance the potency of NFAT-based promoters. In addition, proteins that require high intratumoral concentrations for efficacy may not be suitable for producer T cells.
A further limitation to the use of producer T cells is that the biologic must be encoded by DNA and the final product must be either a nucleic acid or protein. Thus, the approach excludes the possibility of using chemical small molecules.
To date, all investigations in producer T cells for ACT have examined therapeutics that bind to cell surface molecules or receptors. Therapeutics that must be internalized into target cells will require protein modifications such as internalization signaling domains. Protein transduction domains (PTD) have been used for the delivery of various biologically active molecules, including protein- and nucleic acid-based molecules.Citation84 These PTDs could be easily incorporated into T cell transgenes. Additionally, the identification of PTDs that selectively direct therapeutic internalization into tumor cells could enhance therapeutic efficacy. Nevertheless, the finite efficiency of PTDs or other cell internalization signals will likely limit the quantity of therapeutic that can be delivered to target cells.
Conclusion
ACT is an emerging clinical immunotherapy which holds great promise for cancer therapy. Genetic manipulation of T cells to improve tumor targeting has been a clear advancement for the field. Yet, many biological and practical limitations must be addressed in order to improve ACT efficacy. To this end, using genetically engineered T cells to also improve their own activity, modulate or recruit other effector cells, reduce tumor-dependent immunosuppressive processes, or a host of other opportunities remains unexplored but merits further investigation. This unique approach offers several potential advantages over the use of bulk tumor-reactive T cell populations, and provides a practical avenue to bring T cell adoptive transfer closer to a clinical mainstay for cancer treatment.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Supported by the NIH National Cancer Institute (NCI) R01CA140917, R01CA166961, Melanoma Research Foundation, University of Maryland, Baltimore, Marlene and Stewart Greenebaum Cancer Center, and T32CA154274.
References
- Dao MA, Pepper KA, Nolta JA. Long-term cytokine production from engineered primary human stromal cells influences human hematopoiesis in an in vivo xenograft model. Stem Cells 1997; 15:443-54; PMID:9402657; http://dx.doi.org/10.1002/stem.150443
- Liu K, Rosenberg SA. Transduction of an IL-2 gene into human melanoma-reactive lymphocytes results in their continued growth in the absence of exogenous IL-2 and maintenance of specific antitumor activity. J Immunol 2001; 167:6356-65; PMID:11714800; http://dx.doi.org/10.4049/jimmunol.167.11.6356
- Liu K, Rosenberg SA. Interleukin-2-independent proliferation of human melanoma-reactive T lymphocytes transduced with an exogenous IL-2 gene is stimulation dependent. J Immunother 2003; 26:190-201; PMID:12806273; http://dx.doi.org/10.1097/00002371-200305000-00003
- Meyerrose T, Olson S, Pontow S, Kalomoiris S, Jung Y, Annett G, Bauer G, Nolta JA. Mesenchymal stem cells for the sustained in vivo delivery of bioactive factors. Adv Drug Deliv Rev 2010; 62:1167-74; PMID:20920540; http://dx.doi.org/10.1016/j.addr.2010.09.013
- Liechty KW, MacKenzie TC, Shaaban AF, Radu A, Moseley AM, Deans R, Marshak DR, Flake AW. Human mesenchymal stem cells engraft and demonstrate site-specific differentiation after in utero transplantation in sheep. Nat Med 2000; 6:1282-6; PMID:11062543; http://dx.doi.org/10.1038/81395
- Sanz L, Compte M, Guijarro-Munoz I, Alvarez-Vallina L. Non-hematopoietic stem cells as factories for in vivo therapeutic protein production. Gene Ther 2012; 19:1-7; PMID:21562594; http://dx.doi.org/10.1038/gt.2011.68
- Ankrum JA, Ong JF, Karp JM. Mesenchymal stem cells: immune evasive, not immune privileged. Nat Biotechnol 2014; 32:252-60; PMID:24561556; http://dx.doi.org/10.1038/nbt.2816
- Roth JC, Curiel DT, Pereboeva L. Cell vehicle targeting strategies. Gene Ther 2008; 15:716-29; PMID:18369326; http://dx.doi.org/10.1038/gt.2008.38
- Shah K. Mesenchymal stem cells engineered for cancer therapy. Adv Drug Deliv Rev 2012; 64:739-48; PMID:21740940; http://dx.doi.org/10.1016/j.addr.2011.06.010
- Khorashadizadeh M, Soleimani M, Khanahmad H, Fallah A, Naderi M, Khorramizadeh M. Bypassing the need for pre-sensitization of cancer cells for anticancer TRAIL therapy with secretion of novel cell penetrable form of Smac from hA-MSCs as cellular delivery vehicle. Tumour Biol 2015; 36:4213-21; PMID:25586349; http://dx.doi.org/10.1007/s13277-015-3058-2
- Dudek AZ. Endothelial lineage cell as a vehicle for systemic delivery of cancer gene therapy. Transl Res 2010; 156:136-46; PMID:20801410; http://dx.doi.org/10.1016/j.trsl.2010.07.003
- Lin RZ, Dreyzin A, Aamodt K, Li D, Jaminet SC, Dudley AC, Melero-Martin JM. Induction of erythropoiesis using human vascular networks genetically engineered for controlled erythropoietin release. Blood 2011; 118:5420-8; PMID:21937702; http://dx.doi.org/10.1182/blood-2011-08-372946
- Compte M, Alonso-Camino V, Santos-Valle P, Cuesta AM, Sanchez-Martin D, Lopez MR, Vicario JL, Salas C, Sanz L, Alvarez-Vallina L. Factory neovessels: engineered human blood vessels secreting therapeutic proteins as a new drug delivery system. Gene Ther 2010; 17:745-51; PMID:20336155; http://dx.doi.org/10.1038/gt.2010.33
- Neumann H. Microglia: a cellular vehicle for CNS gene therapy. J Clin Invest 2006; 116:2857-60; PMID:17080190; http://dx.doi.org/10.1172/JCI30230
- Lee S, Margolin K. Tumor-infiltrating lymphocytes in melanoma. Curr Oncol Rep 2012; 14:468-74; PMID:22878966; http://dx.doi.org/10.1007/s11912-012-0257-5
- Kershaw MH, Westwood JA, Slaney CY, Darcy PK. Clinical application of genetically modified T cells in cancer therapy. Clin Transl Immunol 2014; 3:e16; PMID:25505964; http://dx.doi.org/10.1038/cti.2014.7
- Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol 2009; 21:215-23; PMID:19327974; http://dx.doi.org/10.1016/j.coi.2009.02.009
- Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov 2013; 3:388-98; PMID:23550147; http://dx.doi.org/10.1158/2159-8290.CD-12-0548
- Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, Badros AZ, Garfall A, Weiss B, Finklestein J et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 2015; 21:914-21; PMID:26193344; http://dx.doi.org/10.1038/nm.3910
- Zhang Q, Li H, Yang J, Li L, Zhang B, Li J, Zheng J. Strategies to improve the clinical performance of chimeric antigen receptor-modified T cells for cancer. Curr Gene Ther 2013; 13:65-70; PMID:23256743; http://dx.doi.org/10.2174/156652313804806570
- Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity 2013; 39:49-60; PMID:23890063; http://dx.doi.org/10.1016/j.immuni.2013.07.002
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18:843-51; PMID:20179677; http://dx.doi.org/10.1038/mt.2010.24
- Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 2013; 36:133-51; PMID:23377668; http://dx.doi.org/10.1097/CJI.0b013e3182829903
- Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013; 122:863-71; PMID:23770775; http://dx.doi.org/10.1182/blood-2013-03-490565
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 2011; 365:725-33; PMID:21830940; http://dx.doi.org/10.1056/NEJMoa1103849
- Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 2011; 3:95ra73; PMID:21832238; http://dx.doi.org/10.1126/scitranslmed.3002842
- Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012; 119:2709-20; PMID:22160384; http://dx.doi.org/10.1182/blood-2011-10-384388
- Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 2013; 5:177ra38; PMID:23515080; http://dx.doi.org/10.1126/scitranslmed.3005930
- Phan GQ, Rosenberg SA. Adoptive cell transfer for patients with metastatic melanoma: the potential and promise of cancer immunotherapy. Cancer Control 2013; 20:289-97; PMID:24077405
- Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer 2008; 8:299-308; PMID:18354418; http://dx.doi.org/10.1038/nrc2355
- Rosenberg SA. Cell transfer immunotherapy for metastatic solid cancer–what clinicians need to know. Nat Rev Clin Oncol 2011; 8:577-85; PMID:21808266; http://dx.doi.org/10.1038/nrclinonc.2011.116
- Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol 2012; 33:579-89; PMID:22926201; http://dx.doi.org/10.1016/j.it.2012.07.004
- Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity 2013; 39:61-73; PMID:23890064; http://dx.doi.org/10.1016/j.immuni.2013.07.005
- Bouzin C, Brouet A, De Vriese J, Dewever J, Feron O. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J Immunol 2007; 178:1505-11; PMID:17237399; http://dx.doi.org/10.4049/jimmunol.178.3.1505
- Franciszkiewicz K, Boissonnas A, Boutet M, Combadiere C, Mami-Chouaib F. Role of chemokines and chemokine receptors in shaping the effector phase of the antitumor immune response. Cancer Res 2012; 72:6325-32; PMID:23222302; http://dx.doi.org/10.1158/0008-5472.CAN-12-2027
- Oelkrug C, Ramage JM. Enhancement of T cell recruitment and infiltration into tumours. Clin Exp Immunol 2014; 178:1-8; PMID:24828133; http://dx.doi.org/10.1111/cei.12382
- Sata M, Walsh K. TNFalpha regulation of Fas ligand expression on the vascular endothelium modulates leukocyte extravasation. Nat Med 1998; 4:415-20; PMID:9546786; http://dx.doi.org/10.1038/nm0498-415
- Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, Greenfield EA, Liang SC, Sharpe AH, Lichtman AH et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol 2003; 33:3117-26; PMID:14579280; http://dx.doi.org/10.1002/eji.200324270
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID:19197294; http://dx.doi.org/10.1038/nri2506
- Hao NB, Lu MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol 2012; 2012:948098; PMID:22778768; http://dx.doi.org/10.1155/2012/948098
- Ma Y, Shurin GV, Peiyuan Z, Shurin MR. Dendritic cells in the cancer microenvironment. J Cancer 2013; 4:36-44; PMID:23386903; http://dx.doi.org/10.7150/jca.5046
- Burkholder B, Huang RY, Burgess R, Luo S, Jones VS, Zhang W, Lv ZQ, Gao CY, Wang BL, Zhang YM et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim Biophys Acta 2014; 1845:182-201; PMID:24440852; http://dx.doi.org/10.1016/j.bbcan.2014.01.004
- Lippitz BE. Cytokine patterns in patients with cancer: a systematic review. Lancet Oncol 2013; 14:e218–28; PMID:23639322; http://dx.doi.org/10.1016/S1470-2045(12)70582-X
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000; 192:1027-34; PMID:11015443; http://dx.doi.org/10.1084/jem.192.7.1027
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252-64; PMID:22437870; http://dx.doi.org/10.1038/nrc3239
- Quintarelli C, Vera JF, Savoldo B, Giordano Attianese GM, Pule M, Foster AE, Heslop HE, Rooney CM, Brenner MK, Dotti G. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood 2007; 110:2793-802; PMID:17638856; http://dx.doi.org/10.1182/blood-2007-02-072843
- Heemskerk B, Liu K, Dudley ME, Johnson LA, Kaiser A, Downey S, Zheng Z, Shelton TE, Matsuda K, Robbins PF et al. Adoptive cell therapy for patients with melanoma, using tumor-infiltrating lymphocytes genetically engineered to secrete interleukin-2. Hum Gene Ther 2008; 19:496-510; PMID:18444786; http://dx.doi.org/10.1089/hum.2007.0171
- Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, Grewal N, Spiess PJ, Antony PA, Palmer DC et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci U S A 2004; 101:1969-74; PMID:14762166; http://dx.doi.org/10.1073/pnas.0307298101
- Hsu C, Hughes MS, Zheng Z, Bray RB, Rosenberg SA, Morgan RA. Primary human T lymphocytes engineered with a codon-optimized IL-15 gene resist cytokine withdrawal-induced apoptosis and persist long-term in the absence of exogenous cytokine. J Immunol 2005; 175:7226-34; PMID:16301627; http://dx.doi.org/10.4049/jimmunol.175.11.7226
- Hsu C, Jones SA, Cohen CJ, Zheng Z, Kerstann K, Zhou J, Robbins PF, Peng PD, Shen X, Gomes TJ et al. Cytokine-independent growth and clonal expansion of a primary human CD8+ T-cell clone following retroviral transduction with the IL-15 gene. Blood 2007; 109:5168-77; PMID:17353346; http://dx.doi.org/10.1182/blood-2006-06-029173
- Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, Heslop HE, Rooney CM, Brenner MK, Dotti G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010; 24:1160-70; PMID:20428207; http://dx.doi.org/10.1038/leu.2010.75
- Wagner HJ, Bollard CM, Vigouroux S, Huls MH, Anderson R, Prentice HG, Brenner MK, Heslop HE, Rooney CM. A strategy for treatment of Epstein-Barr virus-positive Hodgkin's disease by targeting interleukin 12 to the tumor environment using tumor antigen-specific T cells. Cancer Gene Ther 2004; 11:81-91; PMID:14685154; http://dx.doi.org/10.1038/sj.cgt.7700664
- Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, Yu Z, Palmer DC, Reger RN, Borman ZA, Zhang L et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res 2010; 70:6725-34; PMID:20647327; http://dx.doi.org/10.1158/0008-5472.CAN-10-0735
- Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, Rosenberg SA. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res 2012; 18:1672-83; PMID:22291136; http://dx.doi.org/10.1158/1078-0432.CCR-11-3050
- Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, Rosenberg SA, Morgan RA. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther 2011; 19:751-9; PMID:21285960; http://dx.doi.org/10.1038/mt.2010.313
- Zhang L, Feldman SA, Zheng Z, Chinnasamy N, Xu H, Nahvi AV, Dudley ME, Rosenberg SA, Morgan RA. Evaluation of gamma-retroviral vectors that mediate the inducible expression of IL-12 for clinical application. J Immunother 2012; 35:430-9; PMID:22576348; http://dx.doi.org/10.1097/CJI.0b013e31825898e8
- Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 2012; 119:4133-41; PMID:22354001; http://dx.doi.org/10.1182/blood-2011-12-400044
- Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors. Oncoimmunology 2015; 4:e994446; PMID:25949921; http://dx.doi.org/10.4161/2162402X.2014.994446
- Pegram HJ, Purdon TJ, van Leeuwen DG, Curran KJ, Giralt SA, Barker JN, Brentjens RJ. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia 2015; 29:415-22; PMID:25005243; http://dx.doi.org/10.1038/leu.2014.215
- Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, Nahvi AV, Ngo LT, Sherry RM, Phan GQ et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res 2015; 21:2278-88; PMID:25695689; http://dx.doi.org/10.1158/1078-0432.CCR-14-2085
- Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood 2010; 115:3508-19; PMID:20190192; http://dx.doi.org/10.1182/blood-2009-09-241398
- Compte M, Blanco B, Serrano F, Cuesta AM, Sanz L, Bernad A, Holliger P, Alvarez-Vallina L. Inhibition of tumor growth in vivo by in situ secretion of bispecific anti-CEA x anti-CD3 diabodies from lentivirally transduced human lymphocytes. Cancer Gene Ther 2007; 14:380-8; PMID:17218946; http://dx.doi.org/10.1038/sj.cgt.7701021
- Iwahori K, Kakarla S, Velasquez MP, Yu F, Yi Z, Gerken C, Song XT, Gottschalk S. Engager T cells: a new class of antigen-specific T cells that redirect bystander T cells. Mol Ther 2015; 23:171-8; PMID:25142939; http://dx.doi.org/10.1038/mt.2014.156
- Rahir G, Moser M. Tumor microenvironment and lymphocyte infiltration. Cancer Immunol Immunother 2012; 61:751-9; PMID:22488275; http://dx.doi.org/10.1007/s00262-012-1253-1
- Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev 2006; 213:48-65; PMID:16972896; http://dx.doi.org/10.1111/j.1600-065X.2006.00441.x
- Fijak M, Meinhardt A. The testis in immune privilege. Immunol Rev 2006; 213:66-81; PMID:16972897; http://dx.doi.org/10.1111/j.1600-065X.2006.00438.x
- Cose S. T-cell migration: a naive paradigm? Immunology 2007; 120:1-7; PMID:17233737; http://dx.doi.org/10.1111/j.1365-2567.2006.02511.x
- Kratz F, Warnecke A. Finding the optimal balance: challenges of improving conventional cancer chemotherapy using suitable combinations with nano-sized drug delivery systems. J Control Release 2012; 164:221-35; PMID:22705248; http://dx.doi.org/10.1016/j.jconrel.2012.05.045
- Hooijberg E, Bakker AQ, Ruizendaal JJ, Spits H. NFAT-controlled expression of GFP permits visualization and isolation of antigen-stimulated primary human T cells. Blood 2000; 96:459-66; PMID:10887106
- Rook AH, Wood GS, Yoo EK, Elenitsas R, Kao DM, Sherman ML, Witmer WK, Rockwell KA, Shane RB, Lessin SR et al. Interleukin-12 therapy of cutaneous T-cell lymphoma induces lesion regression and cytotoxic T-cell responses. Blood 1999; 94:902-8; PMID:10419880
- Duvic M, Sherman ML, Wood GS, Kuzel TM, Olsen E, Foss F, Laliberte RJ, Ryan JL, Zonno K, Rook AH. A phase II open-label study of recombinant human interleukin-12 in patients with stage IA, IB, or IIA mycosis fungoides. J Am Acad Dermatol 2006; 55:807-13; PMID:17052486; PMID:12391195; http://dx.doi.org/10.1016/j.jaad.2006.06.038
- Almeida AR, Legrand N, Papiernik M, Freitas AA. Homeostasis of peripheral CD4+ T cells: IL-2R α and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol 2002; 169:4850-60; PMID:12391195; http://dx.doi.org/10.4049/jimmunol.169.9.4850
- Koneru M, O'Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med 2015; 13:102; PMID:25890361; http://dx.doi.org/10.1186/s12967-015-0460-x
- Kershaw MH, Wang G, Westwood JA, Pachynski RK, Tiffany HL, Marincola FM, Wang E, Young HA, Murphy PM, Hwu P. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther 2002; 13:1971-80; PMID:12427307; http://dx.doi.org/10.1089/10430340260355374
- Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, Foster AE. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother 2010; 33:780-8; PMID:20842059; http://dx.doi.org/10.1097/CJI.0b013e3181ee6675
- Foster AE, Dotti G, Lu A, Khalil M, Brenner MK, Heslop HE, Rooney CM, Bollard CM. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-β receptor. J Immunother 2008; 31:500-5; PMID:18463534; http://dx.doi.org/10.1097/CJI.0b013e318177092b
- Bendle GM, Linnemann C, Bies L, Song JY, Schumacher TN. Blockade of TGF-β signaling greatly enhances the efficacy of TCR gene therapy of cancer. J Immunol 2013; 191:3232-9; PMID:23940272; http://dx.doi.org/10.4049/jimmunol.1301270
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009; 9:798-809; PMID:19851315; http://dx.doi.org/10.1038/nrc2734
- Rebe C, Vegran F, Berger H, Ghiringhelli F. STAT3 activation: A key factor in tumor immunoescape. Jak-Stat 2013; 2:e23010; PMID:24058791; http://dx.doi.org/10.4161/jkst.23010
- Sansone P, Bromberg J. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol 2012; 30:1005-14; PMID:22355058; http://dx.doi.org/10.1200/JCO.2010.31.8907
- Kaczanowska S, Joseph AM, Davila E. TLR agonists: our best frenemy in cancer immunotherapy. J Leukoc Biol 2013; 93:847-63; PMID:23475577; http://dx.doi.org/10.1189/jlb.1012501
- Geng D, Kaczanowska S, Tsai A, Younger K, Ochoa A, Rapoport AP, Ostrand-Rosenberg S, Davila E. TLR5 Ligand-Secreting T Cells Reshape the Tumor Microenvironment and Enhance Antitumor Activity. Cancer Res 2015; 75:1959-71; PMID:25795705; http://dx.doi.org/10.1158/0008-5472.CAN-14-2467
- Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 2011; 365:1673-83; PMID:22047558; http://dx.doi.org/10.1056/NEJMoa1106152
- Bae HD, Lee K. On employing a translationally controlled tumor protein-derived protein transduction domain analog for transmucosal delivery of drugs. J Control Release 2013; 170:358-64; PMID:23791976; http://dx.doi.org/10.1016/j.jconrel.2013.06.010