
ABSTRACT
Notch signaling is crucial for lymphocyte effector and memory differentiation. While tumor suppress Notch signaling in antitumor lymphocytes, recent studies show that the pharmacological Delta-like ligand-1 multivalent cluster or proteasome inhibitor bortezomib can restore Notch–NF-κB signaling in T cells of tumor-bearing hosts with a potential to overcome cancer cell resistance to therapy.
Tumors suppress Notch signaling in T lymphocytes to escape from immune surveillance. Three recent studies show that it is possible to restore the cross-regulatory loops of Notch–NF-κB signaling in lymphoid cells of tumor-bearing hosts by Delta-like Notch ligand-1 (DLL1)Citation1 and proteasome inhibitor bortezomib.Citation2,3 Such pharmacological manipulations of host immunity enhanced antitumor T cell responses that could improve the outcome of adoptive cell therapy and avoid resistance of cancer cells to chemotherapeutic agents such as tyrosine kinase inhibitors (TKIs).
Notch is a highly conserved signaling system important for the development and differentiation of multiple organs during embryogenesis. The Notch pathway includes a complex repertoire of four transmembrane Notch receptors (Notch 1–4), five ligands such as Delta-like ligands (DLL)-1, 3, 4 and Jagged-1 and 2 as well as multiple non-canonical signaling partners.Citation4 Engagement of Notch with their ligands brings about receptor conformational changes that results in the cleavage of Notch intracellular domain (NICD) by γ-secretase leading to the release of NICD and its translocation into the nucleus where it interacts with various transcription factors such as recombination signal binding protein for immunoglobulin κJ region (RBP-Jκ) unleashing the so-called “canonical” signaling pathway.Citation4 In a “non-canonical” route, NICD drives transcription via other signaling molecules such as NF-κB, PI3K, mTORC2, Akt, β-catenin, etc.Citation5
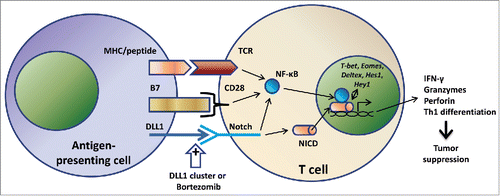
Upon activation, T cells upregulate Notch molecules, whereas the inhibition of Notch signaling results in decreased proliferation and effector function of CD4+ and CD8+ T lymphocytes. Further, Notch enhances IL-2 synthesis and upregulates CD25, the high affinity IL-2 receptor α chain, CD25. Activation of Notch can regulate Th1, Th2, and Th17 differentiation. It can also promote differentiation of naive CD8+ T cells into cytotoxic and memory T lymphocytes via upregulation of the transcription factor Eomesodermin responsible for guiding expression of lytic molecules such as IFNγ, granzymes, and perforins.Citation6 Therefore, interplay of Notch pathway in the transcription and translation of important cellular proteins effectively translates into regulating the differentiation of immune cells and development of the antitumor immunity ().
Dual roles of Notch in tumor development have been established with tumor-promoting implications in T cell acute lymphoblastic leukemia (T-ALL) and various solid tumors including breast cancer, medulloblastoma, colorectal cancer, non-small cell lung carcinoma, and melanoma. Tumor-suppressive properties of Notch have also been described in a wide variety of cells such as hematopoietic cells, skin, pancreatic epithelium, and hepatocytes.Citation7 A novel tumor escape mechanism from T cell immunity includes downregulation of DLL1 and DLL4 in hematopoietic microenvironment whereas restoration of the cognate Notch receptor signaling leads to the suppression of tumor.Citation8
The study by Biktasova et al.Citation1 extended the relevance of Notch signaling for cancer immunotherapy by demonstrating that Notch agonists may offer therapeutic benefits by overcoming tumor-induced T cell immunosuppression. Authors used DLL1-Fc fusion proteins composed of the extracellular domain of mouse or human DLL1 and the Fc part of mouse IgG2A or human IgG1, respectively. To formulate DLL1 clusters, DLL1-Fc, biotinylated anti-IgG antibodies, and NeutrAvidin were utilized. Authors found that clustered DLL1 administered systemically in Lewis lung carcinoma (LLC)-bearing mice promoted infiltration of T cells into tumors and increased T cell activation and function as measured by CD25 upregulation, Th1 differentiation, antigen-specific INFγ production, and the pool of central memory T cells. Further, mice treated with clustered DLL1 displayed diminished tumor vascularization, thereby restricting tumor growth and metastasis. A strong immune response was elicited following adoptive transfer of tumor antigen-specific T cells into immunodeficient SCID-NOD mice bearing palpable tumors. Notably, lymphocytes isolated from DLL1-treated animals attenuated tumor growth in recipients in contrast to naive T cells in the control group showing no sign of therapeutic intervention. Thus, restoration of Notch signaling in antitumor lymphocytes orients the immune response toward strong tumor-inhibitory effects.
Authors further confirmed potential effectiveness of DLL1-based therapy in an EGFRL858R transgenic mouse model, clinically relevant to the condition associated with L858R replacement in exon 21 of EGFR gene, broadly distributed in lung adenocarcinomas as somatic mutation. EGFRL858R replacement is associated with sensitivity and rapid clinical response to the EGFR TKI gefitinib and erlotinib.Citation9,10 However, patients with the EGFRL858R genotype demonstrate residual disease after TKI treatment.Citation9,10 This calls for additional therapeutics necessary to control TKI resistance. In this regard, combined TKI/clustered DLL1 therapy showed promising results with decreased lung tumor burden and significantly improved progression-free survival (40% animals survived beyond 5 weeks in the group of combined therapy versus 100% mice dead by the end of 2nd week in the group of TKI treatment alone). These results correlated with enhanced infiltration of INFγ-producing T cells and CD11b+CD11chigh dendritic cells in lungs of mice, which received combined TKIs/clustered DLL1 therapy.
Despite detailed analysis of the role of Notch activation in resuscitating immune response compromised by tumor growth, mechanistic pathways involved in this process remain unclear. It has been shown in this study that DLL1-mediated activation of immune cells infiltrating tumor is associated with the expression of downstream Notch targets Hes1, Hey1 and Deltex1 along with the upregulation of T-bet, a transcriptional mediator of Stat1 necessary for Th1 differentiation (). In other studies investigating the immune-stimulatory potential of anticancer proteasome inhibitor drug bortezomib, our group demonstrated that bortezomib mediates its therapeutic effects by enhancing cross-regulatory loops of Notch1/2 signaling with NF-κB in tumor-infiltrating CD8+ T lymphocytes as revealed by the use of inhibitors of γ-secretase (blocking canonical Notch pathway) and NF-κB ().Citation2,3 Although the molecular mechanisms underlying these effects need to be dissected, these studies highlight the clinical relevance of augmenting non-canonical downstream Notch signaling pathways by pharmacological agents such as DLL1 clusters or bortezomib in adoptive T cell immunotherapies.
Figure 1. Proposed scheme for co-stimulatory signals generated from Notch receptor stimulation on T cells. T cell stimulatory signals can be provided by Notch ligand DLL1 on juxtaposing antigen-presenting cells or by pharmacological DLL1 multivalent cluster in the presence or absence of drugs such as bortezomib. The Notch signal compliments the standard two-signal scheme of T lymphocyte activation, the first from the TCR engagement by its cognate peptide-MHC complex along with the second costimulatory signal from B7/CD28 interaction. Signaling pathways induced by Notch stimulation include canonical and non-canonical cascades resulting in NICD–NF-κB cross-regulation. The resulting activation can impel T cell effector functions necessary for tumor suppression.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
AS is supported by funds from the following National Institutes of Health grants: U54 CA163069, P50 CA090949, U54 MD007593, SC1 CA182843, and R01 CA175370.
References
- Biktasova AK, Dudimah DF, Uzhachenko RV, Park K, Akhter A, Arasada RR, Evans JV, Novitskiy SV, Tchekneva EE, Carbone DP et al. Multivalent Forms of the Notch Ligand DLL-1 Enhance Antitumor T-cell Immunity in Lung Cancer and Improve Efficacy of EGFR-Targeted Therapy. Cancer Res 2015; 75(22):4728-41; PMID:26404003; http://dx.doi.org/10.1158/0008-5472.CAN-14-1154.
- Thounaojam MC, Dudimah DF, Pellom ST, Jr., Uzhachenko RV, Carbone DP, Dikov MM, Shanker A. Bortezomib enhances expression of effector molecules in anti-tumor CD8+ T lymphocytes by promoting Notch-nuclear factor-kappaB crosstalk. Oncotarget 2015; 6:32439-55; PMID:26431276; http://dx.doi.org/10.18632/oncotarget.5857
- Shanker A, Pellom ST, Dudimah DF, Thounaojam MC, de Kluyver RL, Brooks AD, Yagita H, McVicar DW, Murphy WJ, Longo DL et al. Bortezomib improves adoptive T cell therapy by sensitizing cancer cells to FasL cytotoxicity. Cancer Res 2015; 75(24):5260-72; PMID:26494122; http://dx.doi.org/10.1158/0008-5472.CAN-15-0794
- Minter LM, Osborne BA. Canonical and non-canonical Notch signaling in CD4(+) T cells. Curr Top Microbiol Immunol 2012; 360:99-114; PMID:22695917; http://dx.doi.org/10.3389/fimmu.2014.00054
- Ayaz F, Osborne BA. Non-canonical notch signaling in cancer and immunity. Front Oncol 2014; 4:345; PMID:25538890; http://dx.doi.org/10.3389/fonc.2014.00345
- Radtke F, Fasnacht N, Macdonald HR. Notch signaling in the immune system. Immunity 2010; 32:14-27; PMID:20152168; http://dx.doi.org/10.1016/j.immuni.2010.01.004
- Lobry C, Oh P, Aifantis I. Oncogenic and tumor suppressor functions of Notch in cancer: it's NOTCH what you think. J Exp Med 2011; 208:1931-5; PMID:21948802; http://dx.doi.org/10.1084/jem.20111855
- Huang Y, Lin L, Shanker A, Malhotra A, Yang L, Dikov MM, Carbone DP. Resuscitating cancer immunosurveillance: selective stimulation of DLL1-Notch signaling in T cells rescues T-cell function and inhibits tumor growth. Cancer Res 2011; 71:6122-31; PMID:21825014; http://dx.doi.org/10.1158/0008-5472.CAN-10-4366
- Pao W, Miller VA. Epidermal growth factor receptor mutations, small-molecule kinase inhibitors, and non-small-cell lung cancer: current knowledge and future directions. J Clin Oncol 2005; 23:2556-68; PMID:15767641; http://dx.doi.org/10.1200/JCO.2005.07.799
- Wang Y, Schmid-Bindert G, Zhou C. Erlotinib in the treatment of advanced non-small cell lung cancer: an update for clinicians. Ther Adv Med Oncol 2012; 4:19-29; PMID:22229045; http://dx.doi.org/10.1177/1758834011427927