
ABSTRACT
Although immune infiltrates in ovarian cancer are associated with improved survival, the ovarian tumor environment has been characterized as immunosuppressive, due in part to functional shifts among dendritic cells with disease progression. We hypothesized that flux in dendritic cell subpopulations with cancer progression were responsible for observed differences in antitumor immune responses in early and late-stage disease. Here we identify three dendritic cell subsets with disparate functions in the ovarian tumor environment. CD11c+CD11b(−)CD103+ dendritic cells are absent in the peritoneal cavity of healthy mice but comprise up to 40% of dendritic cells in tumor-bearing mice and retain T cell stimulatory capacity in advanced disease. Among CD11c+CD11b+ cells, Lair-1 expression distinguishes stimulatory and immunoregulatory DC subsets, which are also enriched in the tumor environment. Notably, PD-L1 is expressed by Lair-1(hi) immunoregulatory dendritic cells, and may contribute to local tumor antigen-specific T cell dysfunction. Using an adoptive transfer model, we find that PD-1 blockade enables tumor-associated CD103+ dendritic cells to promote disease clearance. These data demonstrate that antitumor immune capacity is maintained among local dendritic cell subpopulations in the tumor environment with cancer progression. Similar dendritic cell subsets are present in malignant ascites from women with ovarian cancer, supporting the translational relevance of these results.
Abbreviations
| DC | = | dendritic cells |
| IFNγ | = | interferon-gamma |
| IL-2 | = | interleukin-2 |
| Lag-3 | = | lymphocyte-activation protein-3 |
| LPS | = | lipopolysaccharide |
| OVA | = | ovalbumin |
| PD-1 | = | programmed cell death protein-1 |
| PD-L1 | = | programmed death protein ligand-1 |
| Tim-3 | = | T cell immunoglobulin and mucin protein-3 |
| TLR | = | Toll-like receptor |
| TNF-α | = | tumor necrosis factor-α |
Introduction
Ovarian cancer affects over 22,000 women annually in the US.Citation1 Despite efforts at early detection, a majority of women with ovarian cancer present at advanced stages with peritoneal carcinomatosis which is associated with a 5-y survival rate below 40%.Citation1 Understanding factors that control the dissemination of tumor cells would facilitate the development of novel therapeutic strategies to improve outcomes for women with ovarian cancer.
Recent efforts have targeted mechanisms of immune escape in ovarian cancer and other solid tumors to enhance antitumor immunity and achieve disease control.Citation2,3 Ovarian cancer can elicit a spontaneous immune response which is associated with improved survival, but the function of effector immune cells is inhibited in malignant ascites and the tumor environment.Citation4-6 Recent studies have described tumor-associated dendritic cells (DC) as incompletely mature, with decreased costimulatory ligand expression and impaired antigen processing and presentation capability leading to incomplete T cell activation and tumor-specific anergy.Citation7,8 Additional evidence that tumor-associated DCs express both co-inhibitory ligands and receptors suggests that DCs may also directly inhibit T cell effector functions.Citation9-11 Finally, Scarlett et al. described a functional transformation among tumor-associated DCs in ovarian cancer based on evidence that DC depletion in early stages of disease accelerated tumor growth, while depletion of late-stage DC had therapeutic benefit in murine models.Citation9,11-15 Despite accumulating evidence of DC-mediated immune escape, the mechanisms responsible for shifts in DC function in ovarian cancer have not been established.
With recent reports identifying functionally distinct DC subpopulations in other solid tumors, we hypothesized that flux in DC subsets in the tumor environment could account for the functional differences observed in early and late stages of ovarian cancer. In particular, CD103+ DC subpopulations maintain a disproportionate capacity to stimulate cytotoxic T cells in the tumor microenvironment, and were required for cross-priming tumor-specific cytotoxic CD8+ T cells in combination with immunotherapy in subcutaneous tumor models.Citation16 In patients, higher ratios of CD103+ DCs have been associated with improved survival in melanoma, breast, lung, and oropharyngeal cancers;Citation17-23 however, a role for CD103+ DCs in ovarian cancer has not been investigated.
Here, we demonstrate accumulation of CD11c(hi)CD11b(–)CD103+ DCs in the peritoneal cavity and within tumor implants during disease progression in ovarian cancer models, and confirm that these cells retain the capacity to activate tumor-specific T cells in advanced disease. In addition, we identify Lair-1 (CD305) coinhibitory receptor expression as a marker which differentiates CD11c(hi)CD11b+ DCs into immunoregulatory Lair1(hi)DCs and stimulatory CD11c(hi)CD11b+Lair-1(lo) DCs. We further demonstrate that increased local expression of co-inhibitory ligands on Lair1(hi) DCs and tumor-associated macrophages correlates with progressive exhaustion among tumor-specific CD8+ T cells. From this we conclude that bona fide DCs in the tumor environment remain immunogenic in late-stage disease, and that effector cell exhaustion, rather than suboptimal stimulation by DC, contributes to the failure of antitumor immunity and the outgrowth of ovarian cancer. We further demonstrate that PD-1 blockade to reverse T cell dysfunction can reveal the endogenous stimulatory capacity of tumor-associated CD103+ DCs. Evidence that CD103+ DC are also present in patient ascites supports the translational relevance of these results.
Results
Dendritic cell subsets accumulate in the tumor environment with ovarian cancer progression in murine models
To determine whether a dynamic interaction between tumor growth and DC function results from shifts in DC subpopulations, we examined progressive changes among DCs in immune competent murine tumor models. Two established ovarian cancer models were used: the implantable syngeneic ID8 ovarian cancer modelCitation24 transfected to express ovalbumin (ID8ova), and a transgenic model of ovarian cancer in which SV40 T antigen is under the control of the Mullerian inhibitory substance II receptor (MISIIR) promoter (TgMISIIRTag mice).Citation25 These are both immune-competent models of high-grade serous epithelial adenocarcinoma, consistent with the most common type of human ovarian cancer. Following intraperitoneal injection, the ID8ova model develops malignant ascites and tumor implants in the omentum and along peritoneal surfaces, mimicking the typical clinical presentation in patients.Citation24 In the TgMISIIRTag model, cancer develops in the ovary and subsequently metastasizes to the omentum and peritoneal cavity, allowing evaluation of early and late stages of disease.Citation25 Results in these models were compared with ascites samples from patients undergoing treatment for ovarian cancer.
Using the ID8ova model, DC subsets were quantified in peritoneal washings from mice at early stages of disease, and malignant ascites from mice with advanced disease. Our results demonstrated rare DCs in the peritoneal cavity of healthy mice; however, in tumor-bearing mice, we observed an influx of DC subsets in the peritoneal cavity with disease progression (). Detailed flow cytometric analysis identified three subpopulations of DCs that accumulated in the peritoneal cavity of tumor-bearing mice: CD11c(hi)CD11b(–)CD103+ DCs, CD11c(hi)CD11b+ Lair1(lo) DCs, and CD11c(hi)CD11b+Lair1(hi)DCs. DCs expressing CD103+ (integrin αEβ7, ITGAE) were found exclusively among CD11c(hi)CD11b(–) cells and comprised the majority of this population ( and S1A). CD103+ cells were confirmed to be bona fide DCs based on a lack of Ly6C, CD115, CD14 and F4/80 expression, and were Clec9A+Sirp-α(−).Citation19 Most CD103+ DCs expressed the chemokine receptor XCR1, but not CX3CR1, characteristic of migratory DCs responsible for cross-priming CD8+ T cells, and exhibited moderate levels of the co-stimulatory marker CD86 (Fig. S1B).Citation19-23
Figure 1. Dendritic cell subsets accumulate in the tumor environment during ovarian cancer progression. (A–C) Flow cytometric analysis of peritoneal DC subpopulations at weekly time points from tumor-free controls (week 0) or tumor-bearing mice injected with 5 × 106 ID8ova cells intraperitoneally on day 0 (n = 3–5 mice/time point). (A) Representative dot plots of CD11c(hi)CD11b+ and CD11c(hi)CD11b(–) DC gating and analysis of CD103+ and Lair-1 expression. (B) Absolute cell number of peritoneal DC subpopulations at each time point. (C) Representative phenotypic analysis of CD11c(hi)CD11b+ and CD11c(hi)CD11b(–) DCs collected at week 8. (D, E) Analysis of DC subsets from the peritoneum, omentum or ovaries of 20-week old TgMISIIRTag mice (n = 3/time point). (D) Absolute cell number of DC subsets. (E) Representative dot plots of CD11c(hi)CD11b+ and CD11c(hi)CD11b(–) gated DC populations for CD103+ and Lair-1 expression.
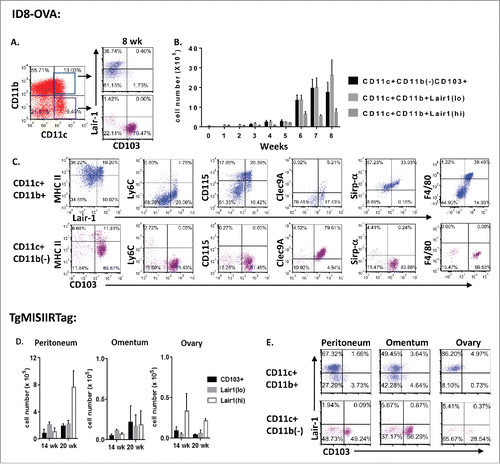
CD103+ expression was absent among CD11c(hi)CD11b+ cells, and this subset was found to express varying levels of the Lair-1 receptor ( and S1A). Lair-1 is a co-inhibitory receptor containing an ITIM domain that binds collagen and the complement component C1q.Citation26-29 In tumor-bearing mice, Lair-1(hi) cells expressed F4/80 and CD115 characteristic of macrophages; however, these markers were rare on Lair-1(lo) DCs (). All three DC subsets (CD103+ DCs, Lair-1(hi) DCs and Lair-1(lo) DCs) were also evident at multiple tumor sites in TgMISIIRTag mice examined at 14 and 20 weeks of age. (). The results of this phenotypic analysis suggest functional differences among DC subsets in the ovarian tumor environment.
An analysis of additional myeloid cell subsets in the peritoneal cavity of ID8ova tumor-bearing mice indicated that peritoneal macrophages (CD11c(lo/–)CD11b(hi)F4/80+ were present in both healthy and tumor-bearing mice at similar levels until very late stages of disease (Fig. S2). Neutrophils (CD11c(–)CD11b(hi)Gr-1+) and myeloid-derived suppressor cells (MDSCs) (CD11c(−)CD11b(mid)Gr-1+) also increased in number with tumor progression but eosinophils (SiglecF+CD11c(−)Gr-1(−)) did not (Fig. S2). Thus, DC accumulation is part of broader shifts in myeloid cell populations in the peritoneal tumor environment with cancer progression.
Similar dendritic cell subsets are present in malignant ascites from women with ovarian cancer
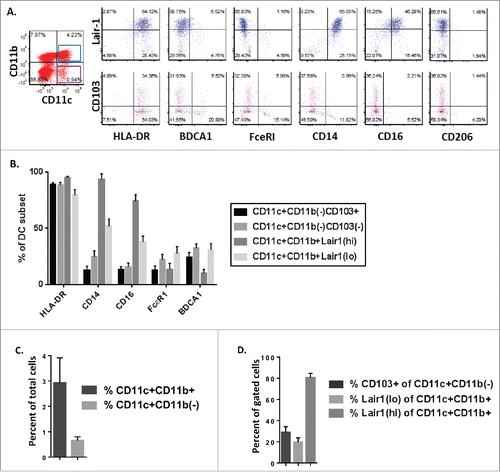
To examine the relevance of these findings to patients, malignant ascites samples from 17 women with advanced serous ovarian cancer were analyzed ( and S3).Citation30,31 In keeping with prior reports, CD11c+ DCs accounted for a minority of total cells retrieved from patient ascites (mean 4.3%, range 0.1–10.4%).Citation32 Compared with the murine models, greater heterogeneity was observed among patients; however, all three DC subsets were identified in ascites samples. In patients, a majority of CD11c(hi)CD11b+ DC were Lair-1(hi) cells (mean 72%, range 29–97% of CD11c(hi)CD11b+; 3% of total ascites cells, range 0.1–9%) which expressed monocytic markers CD14 and CD16. Lair1(lo)DC and CD103+ DC cells comprised a small fraction of total ascites cells in patients (Lair 1(lo)CD11c(hi)CD11b+ mean 1%, CD103+CD11c(hi)CD11b– mean 0.4% of total ascites cells) but a majority of CD11c(hi) DCs in some patients (mean Lair1(lo) of CD11c(hi)CD11b+ 27%, range 3–59%; mean CD103+ cells of CD11c(hi)CD11b− 29%, range 9–63%). As in mice, CD103+ DCs had features of bona fide DCs, expressing HLA-DR+BDCA1(–)FceR1(–)CD14(–)CD16(–)CD206(–) (). Thus, although we observed increased variability in the distribution of DC subsets among patients, Lair1(hi), Lair1(lo), and CD103+ DC were present in the peritoneal tumor environment of women with advanced serous epithelial ovarian cancer.
Figure 2. Similar dendritic cell subsets are present in malignant ascites from women with ovarian cancer. Phenotypic analysis of DC subpopulations in ascites from patients with high grade serous ovarian cancer (n = 17). (A) Representative dot plots of DC subsets gated by CD11c(hi)CD11b+ and CD11c(hi)CD11b(–) expression. (B) Phenotypic analysis of expression of HLA-DR, CD14, CD16, FcεR1 and BDCA1 on indicated DC subpopulations. (C) Dendritic cell populations as a percent of total cells in ascites samples from patients with high-grade serous ovarian cancer. (D) Percent of gated DC populations expressing CD103+ or Lair-1.
Select dendritic cell subsets retain T cell stimulatory capacity in late-stage disease
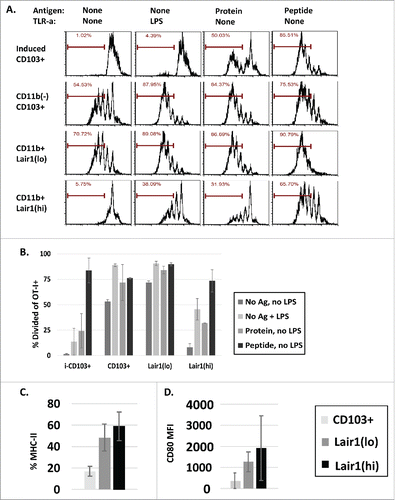
To determine whether phenotypic changes correlated with differences in the functional status of DC subsets, CD103+ DC, Lair-1(hi) DC, and Lair-1(lo) DC subsets were sorted from malignant ascites by FACS for in vitro studies. Following sorting, each DC subset was cultured overnight with or without exogenous tumor-specific antigen (ovalbumin (OVA) protein or peptide) and with or without the Toll-like receptor agonist lipopolysaccharide (LPS). CD103+ DCs induced in vitro from bone marrow (BM) were sorted and used as a tumor-naive control.Citation33 After 18 h, DCs were washed and purified and Thy1.1 congenic naive tumor-specific CD8+ T cells (OT-I) labeled with CFSE were added to culture. T cell proliferation was assessed by CFSE dilution after 72 h, and cytokine production was measured using intracellular staining (). Both CD103+ DCs and Lair-1(lo) DCs from ascites induced tumor-specific T cell proliferation and cytokine production ex vivo. This effect was not significantly enhanced with the addition of exogenous antigen, but proliferation was increased with LPS (). In contrast, CD11b+Lair-1(hi) DCs were unable to activate T cells ex vivo, and remained poorly stimulatory even with innate immune signals from LPS or the addition of exogenous ovalbumin protein. T cell activation was only achieved when Lair-1(hi) DCs were pulsed with ovalbumin peptide, bypassing the need for antigen processing. Phenotypic analysis of DC subsets for MHC II and CD80 demonstrated lower expression of these markers among stimulatory CD103+ and Lair1(lo) DC subsets in comparison to Lair-1(hi) DCs, suggesting that CD103+ and Lair-1(lo) DCs downregulate these markers following activation(). From this, we conclude that CD103+ DC and Lair-1(lo) DC subsets retain tumor-specific T cell stimulatory capacity in advanced disease.
Figure 3. Select dendritic cell subsets retain T cell stimulatory capacity in late-stage disease. (A, B) CD103+ DCs, Lair1(hi) DCs, and Lair1(lo) DCs were sorted from peritoneal samples collected 8 weeks after ID8ova tumor challenge. In vitro-induced CD11c+CD11b(lo)CD103+ DCs used as tumor-naive controls. 2 × 103 DCs were cultured overnight in round bottom plates with or without exogenous tumor antigen (ovalbumin protein (10 ug/mL) or SIINFEKL peptide (100 ng/mL) or LPS (100 ng/mL) stimulation) and washed twice before addition of 2 × 104 CFSE-labeled Thy1.1+OT-I T cells. (A, B) CFSE dilution of OT-I T cells following co-culture with DC subsets under indicated conditions. (C–D) Analysis of DC subsets from mice 8 weeks after tumor challenge for (C) MHC II (%) and (D) CD80 (MFI) expression.
Acquired expression of PD-L1 by Lair1(hi) DCs is associated with progressive T cell dysfunction in the tumor environment
To evaluate alternate mechanisms of immunoregulation by tumor-associated DCs, we examined PD-L1 expression on DC subsets and tumor-associated myeloid cells at early and late stages of disease ( and S4). PD-L1 (B7-H1, CD274) is a ligand for the co-inhibitory receptor PD-1 on T cells, and expression is associated with peripheral suppression of activated T cell activity.Citation34 Nearly 100% of Lair-1(hi) DCs from tumor-bearing mice expressed PD-L1 at all time points examined. In contrast, PD-L1 expression was rare among stimulatory CD103+ DC and Lair1(lo) DC subsets (). Notably, PD-L1 was also highly expressed by macrophages (CD11c(lo/–)CD11b(hi)F4/80+) (Figs. S4A and B), but not by MDSC (CD11b(mid)Gr-1+) in ascites (Figs. S4C and D). In addition, CD45(−) cells from ascites, consisting primarily of tumor cells, did not express Lair-1 or PD-L1 (Fig. S4E). Based on the potent ability of the PD-1/PD-L1 pathway to negatively regulate antitumor immunity,Citation35 these results suggest that inhibitory signaling by Lair-1(hi) DCs and macrophages in the tumor environment may counter the stimulatory effects of CD103+ DCs and Lair-1(lo) DCs.
Figure 4. Acquired expression of PD-L1 by Lair1(hi) DCs is associated with progressive T cell dysfunction in the tumor environment. (A, B) Peritoneal DCs were analyzed for PD-L1 and CD80 expression. (A) Representative dot plots of PD-L1 and CD80 expression by peritoneal DC subsets collected from tumor-naive controls or from mice 8 weeks after tumor challenge. (B) Percent of PD-L1+ cells (top) and PD-L1 expression by MFI (bottom) for each DC subset is shown at weekly intervals (n = 2–3 mice/time point). (C–E) T cells recovered from the peritoneal cavity of healthy or ID8ova tumor-bearing mice at weekly intervals were analyzed by flow cytometry. (C) Intracellular staining for IFNγ, IL-2 and TNF-a production by peritoneal CD8+ T cells following treatment with cell stimulation cocktail. (n = 5/group; week 0 vs. week 8 cytokine (−) CD8+ T cells: p = 0.00002) (D) PD-1 expression by peritoneal CD8+ T cells 2, 5 and 7 weeks after ID8ova tumor challenge. (E) CD8+PD-1+ T cells analyzed for co-expression of Lag-3 and Tim-3. (F–H) Analysis of T cells the peritoneum, omentum, or ovary of TgMISIIRTag mice at 14 weeks of age (n = 3). (F) Cytokine production by CD8+ T cells as in (C). (G) PD-1 expression by CD8+ T cells. (H) CD8+PD-1+ T cells analyzed for Lag-3 and Tim-3 co-expression. All comparisons by two-way ANOVA, ***p < 0.0005.
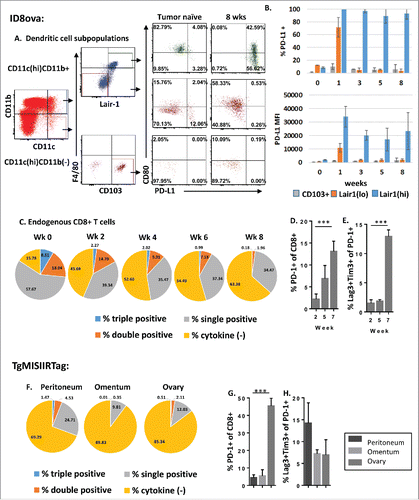
With evidence of acquired PD-L1 expression by cells in the ovarian tumor environment, we next performed a phenotypic and functional analysis of peritoneal T cells collected from ID8ova tumor-bearing mice at serial time points. While both CD8+ and CD4+ T cell numbers increased over time, regulatory T cells (Treg; CD4+FoxP3+) did not accumulate intraperitoneally with cancer progression (Fig. S5). The functional status of endogenous CD8+ T cells in the tumor environment was assessed by flow cytometric analysis of cytokine production (IFNγ, TNF-α and IL-2). In healthy mice, a majority of peritoneal CD8+ T cells produced at least one measured cytokine with over 25% producing multiple cytokines consistent with polyfunctionalityCitation36 (). In contrast, peritoneal CD8+ T cells in tumor-bearing mice showed a decrease in cytokine production as early as week 2 after tumor inoculation, and progressive reduction at each time point, with a majority producing none of the measured cytokines by week 8 (63 +/− 8.5%, p = 0.0002 week 8 vs. week 0) (). This decrease in T cell functional capacity was associated with a significant upregulation of PD-1 among peritoneal CD8+ cells in advanced disease (). Because expression of multiple co-inhibitory receptors has been used to identify and characterize exhausted T cells, we examined the co-expression of Lag-3 and Tim-3 by PD-1+ T cells and found that the proportion of CD8+ T cells expressing all three markedly increased with disease progression ().Citation36,37
In the TgMISIIRTag model, evidence of T cell exhaustion was demonstrated at multiple tumor sites. As in the ID8ova model, cytokine production was diminished in peritoneal T cells as well as T cells from the primary ovarian tumor and from the omentum (). PD-1 was also highly expressed on CD8+ T cells in the primary ovarian tumor, with lower levels on CD8+ T cells in the peritoneal cavity and omentum ();; however, the percentage of PD-1+ CD8+ T cells expressing Lag-3 and Tim-3 (triple positive) was similar at all sites (). From these data, we conclude that an increase in PD-L1 expression by Lair1(hi) DCs and macrophages in the tumor environment may contribute to progressive T cell dysfunction.
Tumor-specific T cells are activated but fail to improve survival in advanced stage disease
To test the impact of local immunoregulatory signaling on tumor-specific T cell activation, we examined proliferation and cytokine production by tumor-specific CD8+ T cells retrieved one week after transfer to tumor-bearing mice at serial time points (). In this experiment, CFSE-labeled naïve thy1.1+ OT-I T cells were injected intraperitoneally into tumor-bearing recipients at the time of tumor challenge or at week 1, 3, 5, or 7, and collected 7 d later for analysis (). Proliferation was measured by CFSE dilution and IFNγ production was measured as a marker of functional capacity (). Results demonstrated that tumor-specific T cells proliferated at all time points in tumor-bearing recipients (). Antigen specificity was confirmed by a lack of proliferation in mice inoculated with the parental ID8 tumor, which does not express ovalbumin. Similarly, IFNγ was selectively produced by T cells transferred to ID8ova tumor-bearing mice at each time point (). These data indicate that tumor-specific T cells localize to the peritoneal tumor environment and are activated at both early and late stages of cancer progression, despite increased expression of co-inhibitory molecules on Lair1(hi) DCs and local macrophages.
Figure 5. Tumor-specific T cells are activated but fail to improve survival in advanced stage disease. (A–D) To analyze tumor-specific T cell activation at different stages of cancer progression, 2 × 106 CFSE-labeled Thy1.1+ naive OT-I T cells were adoptively transferred intraperitoneally to mice at indicated times following ID8 or ID8ova tumor challenge. Thy 1.1 cells were isolated for analysis after 7 d (n = 5 mice/time point/group). (A) Schematic of experiments (B) Representative dot plots of OT-I (CD8+Thy1.1+) gating followed by analysis of CFSE and CD44 expression. (C) Percent of OT-I T cells that had divided at least three times following adoptive transfer to ID8ova or ID8wt tumor-bearing mice at each time point. (D) Percent of IFNγ-producing OT-I T cells at each time point following ex vivo restimulation as in . (E) The impact of adoptive transfer of tumor-specific naive T cells on survival was analyzed at two time points. 2 × 105 or 2 × 106 purified naive OT-I T cells were transferred interaperitoneally at the time of ID8ova tumor challenge or 3 weeks later. Untreated mice served as controls. Survival was measured from the time of tumor challenge until animals reached 30 g (n = 5/group). Kaplan–Meier curves were constructed and pair-wise comparisons were evaluated using the Log Rank test. *p < 0.05.
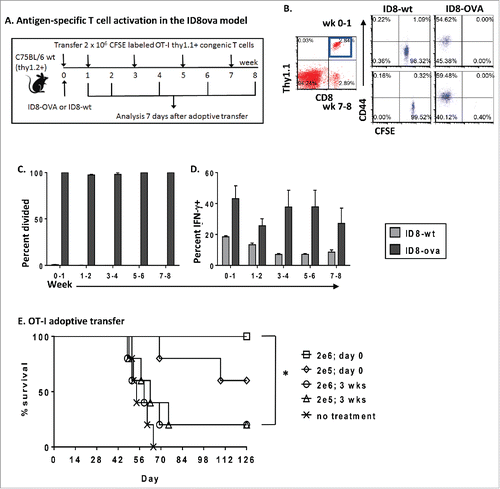
With evidence that T cells can be activated in the established tumor environment, we next tested the protective effect of tumor-specific T cells in vivo at early and late time points. In these experiments, naive OT-I T cells were adoptively transferred into recipients prior to tumor challenge or at week 3, and survival was measured (). Experimental mice received 2 × 105 or 2 × 106 T cells and untreated mice served as controls. We found that T cells transferred at the time of tumor challenge prevented tumor growth. In contrast, T cells transferred as early as 3 weeks after tumor challenge had no survival benefit compared with controls. Thus, despite initial activation and proliferation, progressive changes in the tumor environment disarm antigen-specific T cells and enable cancer progression.
T cell exhaustion is prevalent among tumor-specific T cells
With evidence that tumor-specific T cells failed to protect mice from tumor growth at later time points, we evaluated whether exposure to the established tumor environment resulted in exhaustion following initial activation and proliferation. In these experiments, OT-I T cells were transferred to ID8ova tumor-bearing mice on day 0 or weeks 3 and 5 and retrieved 14 d later to evaluate tumor-specific T cell function following initial activation and expansion (). Similar to endogenous CD8+ T cells, our results demonstrated that tumor-specific T cells exhibited reduced functional capacity at later time points as measured by cytokine production (). The proportion of tumor-specific CD8+ T cells that produced at least one measured cytokine (IFNγ, TNF, or IL2) decreased from a mean of 58% at week 2 to 10% at week 7 (), and IFNγ production decreased from a mean of 60% at week 2 to 5% at week 7 (). As with endogenous CD8+ T cells, this decrease in functional status was associated with an increase in the expression of co-inhibitory receptors with tumor progression. While PD-1 was variably expressed on tumor-specific T cells transferred on day 0 and retrieved at 2 weeks, it was present on nearly all OT-I T cells retrieved at weeks 5 or 7 (). Additionally, a significant increase in the proportion of PD-1+ OT-I T cells co-expressing Lag-3 and Tim-3, characteristic of exhaustion, was evident by week 7 (150-fold increase compared to week 2) ().
Figure 6. T cell exhaustion is prevalent among tumor-specific T cells. (A–F) 2 × 106 naive CD8+Thy1.1+ OT-I T cells were adoptively transferred to C57BL/6 mice 12 h prior to ID8ova tumor challenge or at weeks 3 or 5 weeks and peritoneal cells were analyzed 2 weeks later (n = 5/time point). (A) Experiment schematic. (B) Representative dot plots from early and late time points. (C) Peritoneal CD8+Thy1.1OT-I T cells were analyzed for IFNγ, TNF-a and IL-2 production. (D) Percent of peritoneal CD8+Thy1.1OT-I T cells expressing IFNγ. (E) PD-1 expression by CD8+Thy1.1OT-I T cells. (F) OT-I PD-1+ T cells analyzed for co-expression of Lag-3 and Tim-3. (G-M) 2 × 106 purified Thy1.1+ SV40Tag CD8+ T cells were adoptively transferred to 12 week old TgMISIIRTag recipients (n = 5). Two weeks later (age 14 weeks), peritoneal wash, ovary, omentum and spleen were analyzed by flow cytometry. (G) Experiment schematic. (H) Percent of SV40Tag T cells of total CD8+ cells at each location. (I) Percent of CD44+ activated SV40Tag T cells. (J, K) Analysis of cytokine production by SV40Tag T cells. (J) Mean percent of SV40Tag T cells at each location producing IFNγ, TNF-a and/or IL-2. (K) Percent of SV40Tag T cells expressing IFNg. (L) Percent of SV40Tag T cells that expressed PD-1. (M) Percent of PD-1+SV40Tag T cells co-expressing Lag-3 and Tim-3. Pairwise comparisons by ANOVA. *p < 0.05, ***p < 0.0005.

Performing a similar study in the TgMISIIRTag model allowed an evaluation of tumor-specific T cell homing and location-specific effects. Tumor antigen-specific SV40Tag CD8+ T cells that recognize the SV40 large T antigen epitope I were transferred intraperitoneally into 12-week old recipients and retrieved 2 weeks later for analysis (). In these experiments, tumor-specific T cells were enriched in the primary ovarian tumor in comparison to the peritoneal cavity or omentum (p = 0.007 ovary vs. omentum) (). This is consistent with tumor load at a time point when mice have primary ovarian tumor but metastases are not yet grossly apparent. While a majority of tumor-specific T cells at all sites were CD44+ (), consistent with an activated phenotype, SV40Tag CD8+ T cells from tumor sites demonstrated markedly reduced cytokine production (). In comparison, cytokine production, and particularly IFNγ levels were preserved among adoptively transferred T cells in the spleen (p < 0.05) (). PD-1 expression was also greater on T cells retrieved from tumor sites (). While the proportion of tumor-specific PD-1+ T cells co-expressing Lag-3 and Tim-3 was similar in all tissues, this accounted for approximately 25% of tumor-specific CD8+ cells retrieved from the ovary but less than 2% of cells from the spleen (). From this, we conclude that the exhausted phenotype observed among tumor-specific T cells is mediated by local factors in the tumor environment.
Reversal of T cell exhaustion enables CD103+ dendritic cells to induce protective antitumor immunity in vivo
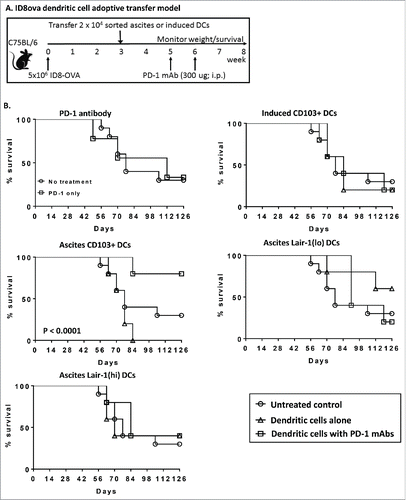
With evidence that CD103+ DC and Lair-1(lo) DC subsets in ascites retain T cell stimulatory capacity, but that progressive T cell exhaustion disables local effector CD8+ T cells, we tested whether selectively enriching stimulatory DC subsets coupled with a reversal of T cell exhaustion could improve survival. DC subpopulations sorted from ID8ova late-stage malignant ascites were adoptively transferred, without manipulation, to tumor-bearing mice 3 weeks after ID8ova tumor challenge (). Mice receiving CD103+ DCs, Lair-1(lo) DCs and Lair-1(hi) DCs were compared with untreated control mice and mice receiving induced CD103+ tumor-naive DCs. A PD-1-blocking antibody was administered at weeks 5 and 6 () to avoid targeting T cells during early stages of activation.Citation36 In all groups, adoptive transfer of DCs alone, or monotherapy with PD-1 blockade, failed to improve survival (). In mice receiving stimulatory CD103+ DCs together with PD-1 blockade, however, a significant improvement in survival was observed in comparison to untreated controls (p < 0.0001) or transfer of CD103+ DC alone (p = 0.0138). In contrast, no significant survival benefit was observed in mice receiving Lair-1(lo) DCs or Lair 1(hi) DCs, even with the addition of PD-1 blockade. We interpret these results as evidence that chronic stimulation by CD103+ DC and Lair1(lo) DC, along with co-inhibitory signaling by Lair1(hi) DC, contribute to local T cell exhaustion in the ovarian tumor environment; however, checkpoint blockade with anti-PD-1 antibodies reverses this phenotype, permitting ongoing activation of tumor-specific T cells by CD103+ DC.
Figure 7. Reversal of T cell exhaustion enables CD103+ dendritic cells to induce protective antitumor immunity in vivo. To determine whether tumor-associated DCs could improve survival when combined with PD-1 blockade, 2 × 104 DCs sorted from mice 8 weeks after tumor challenge were adoptively transferred intraperitoenally to recipients 3 weeks after ID8-OVA injection. Experimental mice received 300 ug of PD-1 mAbs at weeks 5 and 6. Untreated mice served as a control. Survival was measured from the time of tumor challenge until animals reached 30 g. (A) Schematic of experiments. (B) Kaplan–Meier curves for survival following adoptive transfer of DC subsets. (n = 5–10/group). Pairwise comparisons with the untreated controls by Log–rank (Mant–Cox) test. Survival differences were not statistically significant with the exception of mice receiving CD103+ DC and PD-1 mAb (p < 0.001).
Discussion
The ovarian cancer tumor environment has been characterized as both immune stimulatory and suppressive by multiple studies (reviewed inCitation4-6,38). Here, we demonstrate a dynamic flux in DC populations and significant changes in the functional status of tumor-associated lymphocytes with disease progression in murine cancer models. Detailed subclassification of CD11c(hi) DCs indicates that CD11b(–)CD103+ DCs and CD11b+Lair-1(lo) DCs retain T cell stimulatory capacity, even in advanced stages of disease. In contrast, CD11b+Lair-1(hi) DCs do not activate T cells, and acquire expression of the co-inhibitory ligand PD-L1 in the tumor environment. Because Lair-1 is a coinhibitory receptor that blocks cellular responses through dual ITIM signaling,Citation27 it is likely that future studies will show that Lair-1 plays an important role in regulating immune function in ovarian cancer. Here, we demonstrate that inhibition of PD-L1/PD-1 signaling in combination with adoptive transfer of stimulatory CD103+ DC resulted in tumor control and long-term survival in a majority of mice. Importantly, similar DC populations were found in malignant ascites from women undergoing treatment for ovarian cancer.
A strength of our study is the separate analysis of DC subsets which allowed us to distinguish significant differences in the functional capacity of these cells. Prior work describing tumor-infiltrating CD11c(+) DC as suppressive was likely confounded by pooling or depleting all CD11c+ cells.Citation7,9,12-14,39-41 In these studies, immunoregulatory PD-L1+Lair-1(hi) DCs may have obscured immunostimulatory DC subpopulations when whole CD11c(+) populations were pooled or depleted. Both scenarios illustrate a primary role for immunoregulation in directing antitumor immunity, since chronic exposure to stimulatory DCs as well as direct suppression in response to PD-L1 binding may induce T cell exhaustion. Rather than a functional switch among tumor-associated DCs that promotes tumor growth, we find progressive exhaustion among T cells in the tumor environment which impairs their response to the stimulatory DC subsets that accumulate in the ovarian tumor environment. We conclude that co-inhibitory signaling by tumor-associated DC subsets and macrophages contributes to the development of local immune dysfunction in the ovarian tumor environment.
The recent success of immune checkpoint blockade antibodies in melanoma has fueled enthusiasm for reversing immune dysfunction as a central component of immunotherapeutic strategies.Citation3 PD-1 antibodies target T cells in the periphery to reverse the effects of exhaustion.Citation5 While our data indicate that PD-1 blockade enhances survival following adoptive transfer of ascites CD103+ DC, PD-1 blockade alone demonstrated little effect on survival in these or other ovarian cancer models.Citation42 We interpret these results as consistent with there being insufficient numbers of CD103+ DC in early disease to elicit a large and diverse CD8+ T cell response. Adoptive transfer of additional tumor-associated CD103+ DC, however, resulted in tumor rejection when combined with PD-1 checkpoint blockade to sustain CD8+ T cell activity. That a similar effect was not observed with adoptive transfer of stimulatory Lair1(lo) DC suggests that these cells do not optimally stimulate endogenous CD8+ T cells in vivo. Importantly, in these experiments, tumor-associated DC were transferred to mice 3 weeks after tumor challenge, at a time point when the tumor environment is established, and PD-1 blockade was initiated even later, at weeks 5 and 6, when T cell exhaustion is prevalent. This is in contrast to prior studies in which PD-1 antibodies were administered 3 or 10 d after tumor challenge, when their effect could be interpreted as preventing, rather than reversing exhaustion.Citation10 That Lair-1(lo) cells did not have a similar effect is consistent with evidence that CD11c+CD11b+ DCs are not optimal at cross-presentation, and preferentially stimulate CD4+ T cell responses in vivo.Citation19
These data have significant implications for the design of cell-based immune therapy for the treatment of ovarian cancer. CD103+ DC, previously described as crucial for cross-priming of CD8+ T cells, were superior stimulators of antitumor immunity in ovarian cancer models in vivo. This capability was only revealed in the presence of PD-1 checkpoint blockade, indicating that stimulatory CD103+ DCs cannot overcome and may even contribute to CD8+ T cell exhaustion in cancer environments. Alternatively, recent reports describe tumor-infiltrating PD-1+ DC in ovarian cancer models and patient samples which are functionally suppressed through NF-κB inactivation upon binding to PD-L1.Citation11,15 Although these studies did not distinguish among CD11c+ populations, it is possible that the beneficial effect of PD-1 blockade combined with CD103+ DC transfer was due to direct effects on DC as well as T cells in the tumor environment. In either case, evidence that tumor-experienced CD103+ DC subsets have the capacity to promote tumor clearance presents an opportunity to leverage this effect in the development of personalized immune therapy.
Our finding that CD103+ DCs are present in ascites samples from women undergoing surgery for ovarian cancer supports the translational significance of these results. Standard treatment of women with ovarian cancer includes surgical debulking, even in advanced disease, with the goal of resecting all gross tumor. This presents a unique therapeutic window for the incorporation of immune therapy at a time when patients have minimal residual tumor burden. With evidence that transfer of stimulatory tumor-experienced CD103+ DC in the setting of low-volume disease together with checkpoint blockade resulted in tumor control and long-term survival in mouse models, we expect that transfer of CD103+ DC following surgical reduction of the tumor burden would maximize the benefit of a similar approach in patients.
An important finding in our study is that local effects significantly modulate the functional status of adoptively transferred tumor-specific T cells. We found that tumor-specific T cells trafficking to the spleen did not upregulate co-inhibitory receptors and continued to produce cytokines, while T cells that homed to tumor sites adopted an exhausted phenotype. These results indicate that peripheral measures of lymphocyte function following adoptive T cell therapy or other immune regimens may not accurately reflect the functional capacity of T cells in the tumor environment. This finding is in keeping with emerging evidence demonstrating that circulating T cells differ phenotypically from tissue-resident and tumor-infiltrating T cells, and emphasizes the need to develop assays to measure lymphocyte function at tumor sites.Citation43
In sum, our data indicate that ovarian cancer progression is accompanied by marked changes in local leukocyte populations which significantly impact tumor dissemination and survival outcomes. Rather than suboptimal stimulation and induction of anergy, tumor-associated DC activate T cells at all stages of disease, indicating that these cells are not inherently tolerogenic. On the contrary, evidence of prevalent antigen-specific T cell exhaustion suggests that chronic stimulation together with immunoregulatory signaling contributes to immune dysfunction and tumor outgrowth. We expect that a detailed understanding of the conditions established in the ovarian tumor environment with disease progression, along with strategies to distinguish immune stimulatory and regulatory DC subsets, will facilitate the design of effective immunotherapies for ovarian cancer. Finally, we propose that local sampling of tumor environments will be critical to optimize the efficacy of immune therapy for cancer.
Methods
Mice
C57BL/6Ncrl mice were purchased from Charles River Laboratories. C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-I) and B6.PL-Thy1a/CyJ (Thy1.1) were purchased from Jackson Laboratories and were bred in-house. TgMISIIRTag miceCitation25 were a kind gift from Dr Denise Connolly, and were bred in-house. SV40 T antigen transgenic mice (SV40Tag TCR-I; B6.Cg-Tg(TcraY1,TcrbY1)416Tev/J) were a kind gift from Dr Todd Schell and were bred in-house. This transgenic strain carries a rearranged T cell receptor transgene specific for the H2-Db-restricted SV40 large tumor antigen epitope I (residues 206-215).Citation44 OT-I and SV40 Tag were crossed with Thy1.1 mice, and F1 offspring were used for experiments. All in vivo experiments were initiated in mice at 6–8 week of age. All strains were bred and maintained under specific pathogen-free conditions in the University of New Mexico animal facility.
Cell lines
The ID8-wt and ID8ova cell lines were a kind gift from Dr George Coukos and have been previously described.Citation24 Both cell lines were maintained in DMEM (Hyclone) supplemented with 10% Fetalclone III (Hyclone), 10 mM HEPES (Corning), 1 mM sodium pyruvate (Hyclone), 100 U/mL penicillin and 100 ug/mL streptomycin (Gibco) (DMEM-C). For all experiments described, 5 × 106 cells of ID9-wt or ID8ova in 300 uL PBS were injected intraperitoneally.
The PD-1 (clone C1-G4) hybridoma was kindly provided by Dr Lieping Chen. The CD16/CD32 (clone 2.4G2) hybridoma was purchased from ATCC. Hybridomas were grown and monoclonal antibodies (mAbs) were purified as previously described.Citation45
Antibodies
For murine flow cytometry and FACS purification, the following mAbs were used:
BD Biosciences: CD103+ (M290) PE, BV510, and APC, SiglecF (E50-2440) PE-CF594.
Biolegend: F4/80 (BM8) FITC, PD-1 (CD279; 29F.1A12) FITC, CD14 (Sa14.2) FITC, IFNg (XMG1.2) PE, IL-2 (JES6-5H4) PE, CD11c (N418) AlexaFluor700, CD86 (GL-1) PerCP/Cy5.5, CD44 (IM7) PerCP/Cy5.5, CD45 (30-F11) APC/Cy7, Tim-3 (B8.2C12) APC, CD115 (CSF-1R; AFS98) APC, Ki-67 (16A8) APC.
eBioscience: CD8a (53-6.7) AlexaFluor 488, APC and APC-eFluor780, IFNg (XMG1.2) AlexaFluor 488, Ly6C (HK1.4) AlexaFluor 488, CD62L (MEL-14) FITC or eFluor450, Lair-1 (CD305; 113) PE, FoxP3 (FJK-16s) PE and APC, XCR1 (ZET) PE, Ki-67 (SolA15) PE-eFluor610, Gr-1 (RB6-8CS) AlexaFluor 700 and APC-eFluor780, MHCII (I-A/E; MS/114.15.2) PerCP-eFluor710, Lag-3 (CD223; eBioC9B7W) PerCP-eFluor710 and APC, TNFa (MP6-XT22) PerCP-eFluor710, MHCI (H-2Kb; AF6-88.5.5.3) PerCP-eFluor710, PD-1 (J43) PerCP-eFluor710, Clec9A (42D2) PerCP-eFluor710, SIRPa (CD172a; P84) PerCP-eFluor710, CD11c (N418) APC-eFluor780, CD4+ (GK1.5) APC-eFluor780, APC and eFluor450, CD11b (M1/70) eFluor450 and APC, F4/80 (BM8) eFluor450, CD45 (30-F11) eFluor450, Thy1.1 (CD90.1; HISS1) eFluor450.
For human flow cytometry, the following mAbs were used:
BD Biosciences: CD206 (19.2) APC.
Biolegend: PD-1 (EH12.2H7) FITC, IFNg (4S.B3) FITC, IL-2 (MQ1-17H12) PE, CD14 (HCD14) AlexaFluor 700.
eBioscience: CD28 (CD28.2) PE-eFluor610, FoxP3 (PCH101) PE, CD103+ (Ber-ACT8) PE and PerCP-eFluor710, Tim-3 (F38-2E2) APC, Lag-3 (3DS223H) PE-Cy7, CD4+ (OKT4) APC-eFluor780, CD8a (SK1) APC-eFluor780, TNFa (Mab11) eFluor450, FceR1 (AER-37; CRA1) eFluor450, CD11b (ICRF44) FITC, CD16 (eBioCB16) PE-Cy5, CD1c (L161) PerCP-eFluor710, HLA-DR (LN3) APC-eFluor780, CD11c (3.9) PE-Cy7, Lair-1 (NKTA255) PE.
Tissue isolation and processing
Murine ascites samples and peritoneal washings were obtained after CO2 euthanization. Briefly, the peritoneum was exposed through an incision in the abdominal skin while keeping the peritoneal membrane intact. If ascites was present, it was removed with a 10 mL syringe and placed in a 50 mL conical tube (Corning). Then, 5 mL of PBS was injected intraperitoneally, and fluid was recovered following gentle agitation. This process was repeated and ascites and peritoneal washings were combined for analysis.
Following the collection of peritoneal samples, additional tissues (omentum, spleen, and ovary) were removed. For cell analyses, tissues were minced with a razor blade in a 60 mm dish with 5 mL DMEM media (serum free) containing 200 ug of Liberase-TM and 50 ug Dnase I. Tissues were digested at 37°C for 30 min, and then passed through a 70 micron screen (Fisher Scientific) and washed with 10 mL of DMEM-C. For T cell analysis, tissues were not digested, but were disaggregated with the frosted portion of glass slides, and then passed through a 70 micron mesh screen. Following the procedures above, cells were centrifuged at 1500 rpm for 5 min. Red blood cells were lysed with ACK buffer, and samples were resuspened in DMEM-C and passed through a second 70 micron screen. Viable cells were counted by trypan blue exclusion and used for analyses. Cells analyzed for intracellular cytokine production were restimulated at 2–5 × 106 cells/mL in 24 well plates for 5 h with cell stimulation cocktail (eBioscience) containing PMA, ionomycin and Golgi transport inhibitors according to manufacturer's recommendation (500x dilution).
Flow cytometric analysis
Single-cell suspensions of murine or human samples were first incubated with Fc receptor blockers (1 ug anti-CD16/CD32 (clone 2.4G2) and 1 ug mouse IgG (Thermo Sci), or human Fc blocking reagent ((Miltenyi Biotec) in 100 uL FACS buffer (FB; PBS with 1% Fetalclone III) for 15 min at 4°C prior to staining with flourescent antibodies in an additional 100 uL FB (200 uL total). Primary staining of cell surface molecules was performed in the dark at 4°C for 20–30 min followed by washing in 4 mL FB. Cells were fixed in 2% formaldehyde. For intracellular staining after cell surface staining, the FOXP3/Transcription Factor Staining Buffer Set (eBioscience) was used following the manufacturer's recommendations. An LSRFortessa cell analyzer (BD) was used to collect data and FCS Express 4 Flow Cytometry data analysis software (De Novo Software) was used for analysis of flow cytometry data.
Dendritic cell subset isolation and FACS
For cell sorting, samples were passed through a 40 micron screen (Fisher Scientific) prior to Fc receptor blockade. Cells were stained at 1 × 107 per 100 L of FB with CD11c-APC-eF780, CD11b-eF450, CD103-APC and Lair1-PE. Cells were washed and resuspended in FB at 2 × 107 mL and sorted using a Sony sy3200 5-laser cell sorter under sterile conditions to > 95% purity.
In-vitro-induced CD103+ DCs (iCD103+ DCs) were generated as previously described with modifications.Citation32 Briefly, BM was isolated from the femur and tibia of C57BL/6Ncrl mice and passed through a 70 micron screen, followed by washing and ACK lysis. Retrieved cells were resuspended at 1.5 × 106 cells/mL and 10 mL of RPMI-C supplemented with 10 ug/mL GM-CSF (Biolegend) and 100 ng/mL FLt3L (eBioscience) and plated in 100 mm tissue culture dishes (Corning Falcon). 5 mL of RPMI-C was added on day 5. On day 9, cells in suspension were harvested, counted and replated in new 100 mm tissue culture dishes with Fresh RPMI-C+GM-CSF and Flt3L as above. Cells in suspension were harvested on day 15 for analysis and sorting using the same gating as ascites DCs. Following in vitro culture, approximately 80% of cells were CD103+ DCs, which were then sorted to > 95% purity.
In vitro functional assays
For in vitro assessment of DC functional capacity, DCs sorted from ascites or iCD103+ DCs were added to 96-well round bottom plates at 2 × 103 cells per well in triplicate for each subset. Cells were cultured overnight incubation at 37°C in RPMI-C with or without addition of exogenous antigen or LPS (lipopolysaccharide). After 18 h, plates were centrifuged at 1500 rpm for 3 min and washed twice with 200 uL pre-warmed RPMI-C to remove LPS or unprocessed ovalbumin protein or unbound peptide. Naïve OT-I+Thy1.1 T cells were isolated from spleen and peripheral lymph nodes using MACS CD8+ negative selection kit (Miltenyi) and labeled with 5 um CFSE (Life Technologies) for 10 min at 37°C, then washed with excess cold RPMI-C. T cells were added to DC at 2 × 104 cells/well for 72 h. For analysis of intracellular cytokine production, 50 uL of well supernatant was replaced with 50 uL of warmed RPMI-C containing cell stimulation cocktail with Golgi transport inhibitors (eBioscience). At 72 h, cells were harvested and triplicate wells combined for staining of cell surface markers followed by intracellular cytokine staining as above.
In vivo functional studies
For tumor-specific T cell proliferation and activation experiments (also see experiment schematic ), MACS isolated naïve OT-I+Thy1.1+ T cells were labeled with 5 uM CFSE. 2 × 106 purified T cells were injected intraperitoneally into recipient mice at week 0, 1, 3, 5 or 7 following ID8wt or ID8ova tumor challenge. After 7 d, mice were euthanized and peritoneal and tissue samples were collected for analysis.
For analysis of tumor-specific T cell exhaustion (also see experiment schematic for ), 2 × 106 purified unlabeled OT-I+Thy1.1+ T cells were injected intraperitoneally into recipient mice at week 1, 3 or 5 following ID8wt or ID8ova tumor challenge, and harvested 2 weeks later for analysis. In the TgMISIIRTag model, 2 × 106 SV40Tag+Thy1.1+ T cells were transferred to female mice at week 12 and isolated 2 weeks later for analysis.
Survival studies
To evaluate the impact of DC subsets alone or in combination with PD-1 blockade on survival, 2 × 104 peritoneal DCs sorted from tumor-bearing mice were transferred to tumor-bearing recipients at week 3 after ID8ova tumor challenge. Day 15 sorted iCD103+ DCs were used as a tumor-naive control. Mice receiving combined therapy were injected intraperitoneally with 300 ug of PD-1 monoclonal antibody in 300 uL of PBS at weeks 5 and 6. Survival was measured from the day of tumor inoculation until mice reached a weight of 30 g due to ascites accumulation or exhibited evidence of tumor-associated morbidity.
Patient samples
Ascites was obtained from 17 patients with serous ovarian cancer undergoing surgery for the treatment of ovarian cancer at the University of New Mexico Cancer Center under an approved clinical protocol. Ascites was analyzed for cell subsets and phenotype by flow cytometry as described above.
Statistics
Statistical analysis was performed using Excel (Microsoft) and GraphPad Prism 6 (Graphpad Software, Inc.). All data are presented as means with error bars representing standard error of the mean. Statistical comparisons between experimental groups were analyzed by one-way and two-way analysis of variance followed by Tukey's procedure for multiple comparisons. Pairwise comparisons of two groups were made using the student's t-test. Kaplan–Meier survival curves were computed and the log-rank statistic was used to evaluate differences among groups. A p value < 0.05 was taken to indicate statistical significance. In all figures, p values are denoted as the following: ***p < 0.001; **p < 0.025; *p < 0.05.
Study approval
All studies using murine models were approved by the Institutional Animal Care and Use Committee of the University of New Mexico Health Sciences Center, or by the Institutional Animal Care and Use Committee at the University of Pennsylvania. Samples from patients were collected under a clinical trial protocol approved by the Institutional Review Board of the University of New Mexico Health Sciences Center. All patients provided written informed consent for sample collection.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author Contributions
DBF designed and conducted experiments. TH, JCH, and VJ also conducted experiments. DBF, TH, PAG and SFA analyzed the data. DBF and SFA wrote the manuscript, with all authors contributing to the final version.
KONI_A_1185583_s02.pptx
Download MS Power Point (546 KB)Acknowledgments
The authors wish to thank Dr George Coukos for ovarian cancer cell lines, Dr Denise Connolly for the MISIIRTag transgenic mouse model, Dr Todd Schell for the SV40Tag mice, and Dr Dennis McCance and Dr Katherine Morris for critical reading of the manuscript. In addition, we wish to thank Dr Carolyn Muller, Dr Teresa Rutledge, Dr Angela Wandinger-Ness, Dr Yuna Guo, and Elsa Romero for patient sample collection.
Funding
This work was funded by grants from the American Cancer Society (MSRG-12-163-01-LIB, SFA), the University of Pennsylvania Research Foundation (4-000002-4820, SFA), the National Institutes of Health K12 Award for Research in Clinical Oncology (10018886, SFA), the American Society of Clinical Oncology Young Investigator Award (552494, SFA), the Fox Chase Cancer Center Ovarian Cancer SPORE (014001, SFA), the Sandy Rolllman Ovarian Cancer Foundation (551118, SFA), and the Florence and Marshall Schwid Award from the Gynecologic Cancer Foundation (547827). Data was generated in the Flow Cytometry Shared Resource Center and the Keck-UNM Small-Animal Imaging Resource Shared Resource Center supported by the University of New Mexico Health Sciences Center, the UNM Cancer Center, and UNM College of Pharmacy (NIH P30CA118100-06).
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016; 66:7-30; PMID:26742998; http://dx.doi.org/10.3322/caac.21349
- Scarlett UK, Conejo-Garcia JR. Modulating the tumor immune microenvironment as an ovarian cancer treatment strategy. Expert Rev Obstet Gynecol 2012; 7:413-9; PMID:24039628; http://dx.doi.org/10.1586/eog.12.41
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252-64; PMID:22437870; http://dx.doi.org/10.1038/nrc3239
- Yigit R, Massuger LF, Figdor CG, Torensma R. Ovarian cancer creates a suppressive microenvironment to escape immune elimination. Gynecol Oncol 2010; 117:366-72; PMID:20144842; http://dx.doi.org/10.1016/j.ygyno.2010.01.019
- Lavoué V, Thédrez A, Levêque J, Foucher F, Henno S, Jauffret V, Belaud-Rotureau MA, Catros V, Cabillic F. Immunity of human epithelial ovarian carcinoma: the paradigm of immune suppression in cancer. J Transl Med 2013; 11:147; PMID:23763830; http://dx.doi.org/10.1186/1479-5876-11-147
- Nelson BH. The impact of T-cell immunity on ovarian cancer outcomes. Immunol Rev 2008; 222:101-16; PMID:18363996; http://dx.doi.org/10.1111/j.1600-065X.2008.00614.x
- Schlienger K, Chu CS, Woo EY, Rivers PM, Toll AJ, Hudson B, Maus MV, Riley JL, Choi Y, Coukos G et al. TRANCE- and CD40 ligand-matured dendritic cells reveal MHC class I-restricted T cells specific for autologous tumor in late-stage ovarian cancer patients. Clin Cancer Res 2003; 9:1517-27; PMID:12684428
- Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 2015; 161:1527-38; PMID:26073941; http://dx.doi.org/10.1016/j.cell.2015.05.025
- Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med 2003; 9:562-7; PMID:12704383
- Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res 2013; 73:6900-12; PMID:23975756; http://dx.doi.org/10.1158/0008-5472.CAN-13-1550
- Krempski J, Karyampudi L, Behrens MD, Erskine CL, Hartmann L, Dong H, Goode EL, Kalli KR, Knutson KL. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J Immunol 2011; 186:6905-13; PMID:21551365; http://dx.doi.org/10.4049/jimmunol.1100274
- Scarlett UK, Rutkowski MR, Rauwerdink AM, Fields J, Escovar-Fadul X, Baird J, Cubillos-Ruiz JR, Jacobs AC, Gonzalez JL, Weaver J et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J Exp Med 2012; 209:495-506; PMID:22351930; http://dx.doi.org/10.1084/jem.20111413
- Huarte E, Cubillos-Ruiz JR, Nesbeth YC, Scarlett UK, Martinez DG, Buckanovich RJ, Benencia F, Stan RV, Keler T, Sarobe P et al. Depletion of dendritic cells delays ovarian cancer progression by boosting antitumor immunity. Cancer Res 2008; 68:7684-91; PMID:18768667; http://dx.doi.org/10.1158/0008-5472.CAN-08-1167
- Peter S, Bak G, Hart K, Berwin B. Ovarian tumor-induced T cell suppression is alleviated by vascular leukocyte depletion. Transl Oncol 2009; 2:291-9; PMID:19956391; http://dx.doi.org/10.1593/tlo.09190
- Karyampudi L, Lamichhane P, Krempski J, Kalli KR, Behrens MD, Vargas DM, Hartmann LC, Janco JM, Dong H, Hedin KE et al. PD-1 blunts the function of ovarian tumor-infiltrating dendritic cells by inactivating NF-κB. Cancer Res 2016; 76:239-50; PMID:26567141; http://dx.doi.org/10.1158/0008-5472.CAN-15-0748
- Sánchez-Paulete AR, Cueto FJ, Martínez-López M, Labiano S, Morales-Kastresana A, Rodríguez-Ruiz ME, Jure-Kunkel M, Azpilikueta A, Aznar MA, Quetglas JI et al. Cancer immunotherapy with immunomodulatory Anti-CD137 and Anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov 2016; 6:71-9; PMID:26493961; http://dx.doi.org/10.1158/2159-8290.CD-15-0510
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014; 26:638-52; PMID:25446897; http://dx.doi.org/10.1016/j.ccell.2014.09.007
- Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, Coussens LM. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014; 26:623-37; PMID:25446896; http://dx.doi.org/10.1016/j.ccell.2014.09.006
- Dutertre CA, Wang LF, Ginhoux F. Aligning bona fide dendritic cell populations across species. Cell Immunol 2014; 291:3-10; PMID:25262488; http://dx.doi.org/10.1016/j.cellimm.2014.08.006
- Schraml BU, Reis e Sousa C. Defining dendritic cells. Curr Opin Immunol 2015; 32:13-20; PMID:25553392; http://dx.doi.org/10.1016/j.coi.2014.11.001
- Bachem A, Güttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, Salama A, Movassaghi K, Opitz C, Mages HW et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med 2010; 207:1273-81; PMID:20479115; http://dx.doi.org/10.1084/jem.20100348
- Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol 2012; 12:557-69; PMID:22790179; http://dx.doi.org/10.1038/nri3254
- Hartung E, Becker M, Bachem A, Reeg N, Jäkel A, Hutloff A, Weber H, Weise C, Giesecke C, Henn V et al. Induction of potent CD8 T cell cytotoxicity by specific targeting of antigen to cross-presenting dendritic cells in vivo via murine or human XCR1. J Immunol 2015; 194:1069-79; PMID:25520399; http://dx.doi.org/10.4049/jimmunol.1401903
- Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, Persons DL, Smith PG, Terranova PF. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis 2000; 21:585-91; PMID:10753190; http://dx.doi.org/10.1093/carcin/21.4.585
- Connolly DC, Bao R, Nikitin AY, Stephens KC, Poole TW, Hua X, Harris SS, Vanderhyden BC, Hamilton TC. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res 2003; 63:1389-97; PMID:12649204
- Meyaard L, Adema GJ, Chang C, Woollatt E, Sutherland GR, Lanier LL, Phillips JH. LAIR-1, a novel inhibitory receptor expressed on human mononuclear leukocytes. Immunity 1997; 7:283-90; PMID:9285412; http://dx.doi.org/10.1016/S1074-7613(00)80530-0
- Meyaard L. The inhibitory collagen receptor LAIR-1 (CD305). J Leukoc Biol 2008; 83:799-803; PMID:18063695
- Son M, Santiago-Schwarz F, Al-Abed Y, Diamond B. C1q limits dendritic cell differentiation and activation by engaging LAIR-1. Proc Natl Acad Sci U S A 2012; 109:E3160-7; PMID:23093673; http://dx.doi.org/10.1073/pnas.1212753109
- Son M, Diamond B. C1q-mediated repression of human monocytes is regulated by leukocyte-associated Ig-like receptor 1 (LAIR-1). Mol Med 2014; 20:559-68; PMID:25247291; http://dx.doi.org/10.2119/molmed.2014.00185
- Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, Wasan PS, Wang XN, Malinarich F, Malleret B et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity 2012; 37:60-73; PMID:22795876; http://dx.doi.org/10.1016/j.immuni.2012.04.012
- van der Aa E, van Montfoort N, Woltman AM. BDCA3(+)CLEC9A(+) human dendritic cell function and development. Semin Cell Dev Biol 2015; 41:39-48; PMID:24910448; http://dx.doi.org/10.1016/j.semcdb.2014.05.016
- Labidi-Galy SI, Sisirak V, Meeus P, Gobert M, Treilleux AB, Combes J-D, Faget J, Mithieux F, Cassignol A, Tredan O et al. Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer Res 2011; 17:5423-34; PMID:21697280; http://dx.doi.org/10.1158/0008-5472.CAN-11-0367
- Mayer CT, Ghorbani P, Nandan A, Dudek M, Arnold-Schrauf C, Hesse C, Berod L, Stüve P, Puttur F, Merad M et al. Selective and efficient generation of functional Batf3-dependent CD103+ dendritic cells from mouse bone marrow. Blood 2014; 124:3081-91; PMID:25100743; http://dx.doi.org/10.1182/blood-2013-12-545772
- Okazaki T, Honjo T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol 2006; 27:195-201; PMID:16500147; http://dx.doi.org/10.1016/j.it.2006.02.001
- Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat Rev Immunol 2015; 15:45-56; PMID:25534622; http://dx.doi.org/10.1038/nri3790
- Wherry EJ. T cell exhaustion. Nat Immunol 2011; 12:492-9; PMID:21739672
- Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol 2015; 36:265-76; PMID:25797516; http://dx.doi.org/10.1016/j.it.2015.02.008
- Kandalaft LE, Motz GT, Duraiswamy J, Coukos G. Tumor immune surveillance and ovarian cancer: lessons on immune mediated tumor rejection or tolerance. Cancer Metastasis Rev 2011; 30:141-51; PMID:21298574; http://dx.doi.org/10.1007/s10555-011-9289-9
- Nesbeth Y, Scarlett U, Cubillos-Ruiz J, Martinez D, Engle X, Turk MJ, Conejo-Garcia JR. CCL5-mediated endogenous antitumor immunity elicited by adoptively transferred lymphocytes and dendritic cell depletion. Cancer Res 2009; 69:6331-8; PMID:19602595; http://dx.doi.org/10.1158/0008-5472.CAN-08-4329
- Scarlett UK, Cubillos-Ruiz JR, Nesbeth YC, Martinez DG, Engle X, Gewirtz AT, Ahonen CL, Conejo-Garcia JR. In situ stimulation of CD40 and Toll-like receptor 3 transforms ovarian cancer-infiltrating dendritic cells from immunosuppressive to immunostimulatory cells. Cancer Res 2009; 69:7329-37; PMID:19738057; http://dx.doi.org/10.1158/0008-5472.CAN-09-0835
- Cubillos-Ruiz JR, Engle X, Scarlett UK, Martinez D, Barber A, Elgueta R, Wang L, Nesbeth Y, Durant Y, Gewirtz AT et al. Polyethylenimine-based siRNA nanocomplexes reprogram tumor-associated dendritic cells via TLR5 to elicit therapeutic antitumor immunity. J Clin Invest 2009; 119:2231-44; PMID:19620771; http://dx.doi.org/10.1172/JCI37716
- Higuchi T, Flies DB, Marjon NA, Mantia-Smaldone G, Ronner L, Gimotty PA, Adams SF. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol Res 2015; 3:1257-68; PMID:26138335; http://dx.doi.org/10.1158/2326-6066.CIR-15-0044
- Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med 2015; 21:688-97; PMID:26121195; http://dx.doi.org/10.1038/nm.3883
- Staveley-O'Carroll K, Schell TD, Jimenez M, Mylin LM, Tevethia MJ, Schoenberger SP, Tevethia SS. In vivo ligation of CD40 enhances priming against the endogenous tumor antigen and promotes CD8+ T cell effector function in SV40 T antigen transgenic mice. J Immunol 2003; 171:697-707; PMID:12847236; http://dx.doi.org/10.4049/jimmunol.171.2.697
- Flies DB, Wang S, Xu H, Chen L. Cutting edge: A monoclonal antibody specific for the programmed death-1 homolog prevents graft-versus-host disease in mouse models. J Immunol (Baltimore, Md: 1950) 2011; 187:1537-41; http://dx.doi.org/10.4049/jimmunol.1100660