
ABSTRACT
Successful cancer immunotherapy necessitates T cell proliferation and infiltration into tumor without exhaustion, a process closely links optimal maturation of dendritic cells (DC), and adjuvant promotes this process as an essential prerequisite. Poly(I:C) has contributed to adjuvant immunotherapy that evokes an antitumor response through the Toll-loke receptor 3 (TLR3)/TICAM-1 pathway in DC. However, the mechanism whereby Poly(I:C) acts on DC for T cell proliferation and migration remains undetermined. Subcutaneous injection of Poly(I:C) regressed implant tumors (WT1-C1498 or OVA-EG7) in C57BL/6 mice, which coincided with tumor-infiltration of CD8+ T cells. Epitope-specific cytotoxic T lymphocytes (CTLs) were increased in spleen by challenge with Poly(I:C)+Db126 WT-1 peptide but not Poly(I:C) alone, suggesting the need of an exogenous Ag density for cross-priming. In tumor, CXCR3 ligands were upregulated by Poly(I:C), which facilitated recruitment of CTL to the tumor. Thus, Poly(I:C) acts on splenic CD8α+ DC to cross-prime T cells and on intratumor cells to attract CTLs. Besides CD8+ T cell cross-priming, T cell recruitment into tumor was significantly dampened in Batf3−/− mice, reflecting the importance of tumor Batf3-dependent DC rather than macrophages in T cell recruitment. Poly(I:C)-induced XCR1hi CD8α+ DC with high TLR3 levels were markedly decreased in Batf3−/− mice, which hampered the production of IL-12 and IL-12-mediated CD4+/CD8+ T cell proliferation. Subcutaneous administration of Poly(I:C) and adoptive transfer of wild-type CD8α+ DC largely recovered antitumor response in those Batf3−/− mice. Collectively, Poly(I:C) tunes up proper maturation of CD8α+ DC to establish TLR3-mediated IL-12 function and cross-presentation in spleen and lymphocyte-attractive antitumor microenvironment in tumor.
Abbreviations
| APC | = | antigen-presenting cells |
| BATF | = | basic leucine zipper transcription factor ATF-like |
| CFSE | = | carboxyfluorescein succinimidyl ester |
| CTL | = | cytotoxic T lymphocyte |
| DAMP | = | damage-associated molecular pattern |
| DC | = | dendritic cell |
| IFN | = | type I interferon |
| MAVS | = | mitochondrial antiviral signal |
| MDSC | = | myeloid-derived suppressor cells |
| NK | = | natural killer |
| OVA | = | ovalbumin |
| PRR | = | pattern-recognition receptor |
| TAA | = | tumor-associated antigens |
| TAM | = | tumor-associated macrophages |
| TICAM-1 | = | Toll-IL-1R homology domain-containing adaptor molecule 1 |
| TLR | = | toll-like receptor |
| WT1 | = | Wilms's tumor 1 |
Introduction
The essence of immunotherapy for cancer is to promote tumoricidal potential of lymphocytes in tumor. Dendritic cells (DC) play a key role in validating effector cells, antitumor NK cellsCitation1 and CTLs, through their maturation,Citation2 and adjuvants are deeply involved in this process.Citation3 DC particularly participate in proliferation of CTL by the cross-presentation of tumor-associated antigens (TAAs) to lymphocytes. The DC priming of T cells conceptually occurs in draining lymph node (DLN)/spleen in line with complicated orchestration of molecular and cellular interactions.Citation3 On the other hand, a variety of myeloid cells are eventually involved in the regulation of this process. Tumor-associated macrophages (TAM) and myeloid-derived suppressor cells (MDSC) as well as tumor-infiltrating DC are implicated in tumor reprogramming and progression through undefined events including epigenetic modification.Citation4,5 They are also adjuvant-sensitive having pattern-sensing receptors in tumor. Immune modulation by adjuvant immunotherapy works under these complicated conditions.Citation6 Thus, we analyzed what happens in spleen and tumor when adjuvant is subcutaneously (s.c.) injected with TAA in mouse tumor-implant models.
Immune adjuvants represent unusual pattern molecules originating from foreign microbes or denatured host products, which usually accompany microbial infection or tissue damage.Citation7 There are many kinds of pattern-recognition receptors (PRRs), a signal which functions as a strong inducer of maturation of antigen-presenting cells (APCs).Citation3,7 Tumor regression is an outcome of the total function of adjuvant immunotherapy. There appear to be multiple cross-presentation-inducing pathways in DC, and multifarious pattern-sensing signals in innate immunity facilitate DC-mediated proliferation of TAA-specific CTL.Citation2 DC are a group of APCs consisting of many subsets. CD8α+ DC are particularly important in mice for cross-presentation of exogenous Ags in various infections because both Ags and adjuvant are simultaneously provided by other cells infected with microbes to induce CD8α+ DC maturation.Citation8 Tumors with TAA but no adjuvancy have evolved to circumvent the host immune system. CD8α+ DC are the equivalent of human CD141+ CD11c+ DC,Citation9-12 both of which express high levels of TLR3,Citation10,11 and induce efficient cross-priming of CTLs. A desirable quality of an adjuvant is to provide a second signal to DC that upregulates MHC and positive co-stimulators, orchestrates the cytokine network, and contributes to lymphocyte-mediated tumor killing.Citation13
Here, we employed Poly(I:C) as an adjuvant. Poly(I:C) has used for long time in immunotherapy in human and mice.Citation14,15 Poly(I:C) satisfies the above total quality,Citation16,17 but how its function is culminated in administration of Ag has not been well addressed yet. However, due to its endotoxin-like toxicity, only limited doses are used for clinical tests in vaccine immunotherapy in human.Citation18 Mice are relatively resistant to its toxicity, but sometimes accompanied with severe adverse effects including cytokinemia. Poly(I:C) acts as a ligand for endosomal TLR3 as well as many cytoplasmic RNA sensors in DC.Citation16 When Poly(I:C) is i.p. injected in mice, inflammatory cytokines and type I interferons (IFNs) are drastically increased in blood plasma.Citation19 Using the TLR3-specific agonist, we have verified that cross-presentation is induced through the TLR3/TICAM-1 pathway, with minimal participation of IFN/cytokines by the MDA5/MAVS pathway in CD8α+ DC.Citation20 Ultimately, tumor efficiently regresses by treatment with Ag + TLR3 adjuvant in mice without cytokinemia.
It is getting clear that Poly(I:C)-based treatment includes a complex inflammatory response that promotes upregulation in checkpoint molecules (i.e. PD-1 and PD-L1) by tumor cells or stromal cells (including DC) in tumor microenvironment.Citation21 Tumor-associated DC directly participate in proliferation of CTL inside the tumor.Citation22-24 Myeloid cells in tumor are also targets for Poly(I:C) in adjuvant therapy.Citation25 Less-inflammatory TLR3 adjuvant would more benefit for combination therapy with checkpoint blockades than Poly(I:C) in tumor microenvironment having T and tumor cells with PD-1/PD-L1 to further bolster therapeutic efficacy (which will be shown with mouse models elsewhere), in the context that check-point blockades exhibit high therapeutic potential to some tumor types in mouse and clinical tests.Citation26,27 In vaccine immunotherapy, however, tumor regress well in mice by treatment with exogenously added Poly(I:C) and Ag.Citation20,28 What is the role of RNA adjuvant in T cell proliferation, tumor infiltration and tumor cell damage can be analyzed in mouse models.
BATF family proteins are pivotal transcription factors involved in the base for development and maturation for DC, which closely link the effective antitumor adjuvancy. Above all, Batf3−/− mice barely develop CD11c+ DC and lose Ag-presenting capacity of CD8α+ and CD103+ DC.Citation29,30 BATF3 acts cooperatively with IRF8 in the immune system, and is particularly associated with myeloid DC development.Citation31,32 Batf3 is minimally expressed in the common DC precursors,Citation33 while its expression is maximal in terminally differentiated CD8α+ DC in the spleen and their equivalent cells in other tissues.Citation29,31 Since CD8α+ DC are largely diminished in spleen in Batf3−/− mice, BATF3 may be involved in Poly(I:C)-mediated DC maturation and IL-12 production via the TICAM-1 pathway.
Here, we found that Poly(I:C) bi-modally modulates CD8α+ DC in spleen and tumor for cross-priming of CTL and facilitating intratumor CTL infiltration, respectively, to establish antitumor microenvironment.
Results
Batf3 is required for CTL-mediated tumor regression by Poly(I:C)
WT1-C1498 cells, stably expressing WT1,Citation34 were implanted to the back of mice and treated with Poly(I:C) alone 5 and 12 d after implantation. WT1-C1498 tumor regressed in response to Poly(I:C) in wild-type mice (), and this Poly(I:C) effect was abolished if CTL were depleted (). Strikingly, Batf3 knockout completely abrogated the Poly(I:C) antitumor effect in C57BL/6 mice (). NK cells were barely involved in the Poly(I:C)-induced tumor regression (), but CD8+ T cells infiltrated the tumors in wild type, but not in Batf3−/− mice ().
Figure 1. Poly(I:C) induces BATF3-mediated WT1-C1498 tumor regression. WT1-C1498 tumor was implanted to wild-type or Batf3−/− mice (C57BL/6J) and Poly(I:C) was administered around tumor at day 5 and 12 after tumor implantation (A, C). The tumor volume was measured every 2 to 3 d. For depleting CTLs or NK cells, ascites containing anti-CD8α or anti-NK1.1 antibody was i.p. injected the day before Poly(I:C) therapy (B, D). WT1-C1498 tumor was harvested at day 19 as in panels A and C, and the proportion of tumor-infiltrating CD8+ T cells was evaluated by flow cytometer (E). Db126 peptide wrapped in DOTAP with or without Poly(I:C) were administered to tumor-bearing wild-type mice at day 5 and 12 after tumor implantation. At day 20, splenocytes were harvested and cultured in complete medium in the presence of Db126 (5 μg/mL) for 5 d. At day 25, the proportion of WT1-specific CD8+ T cells was evaluated (F). At day 21, splenocytes of Batf3−/− mice treated as in panel F were harvested. The cells were re-stimulated with Db126 and the proportion of WT1-specific CD8+ T cells was evaluated as in panel F (G). Arrows show the day of Poly(I:C) administration. Error bars show ± SEM; n = 3 to 4 per group. Student's t-test was performed to analyze statistical significance. *p < 0.05. ns; not significant. The results are the representatives of more than two independent experiments.
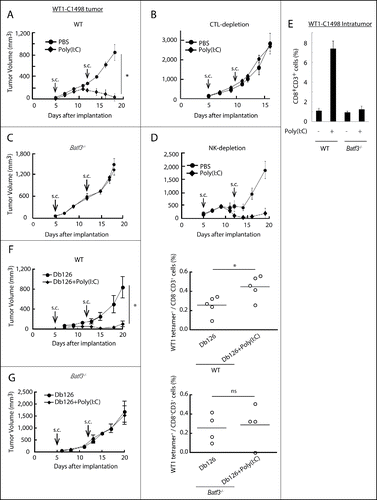
The results indicated that Poly(I:C) induces WT1-directed CTL to regress C1498 tumor. However, the reason remained unknown why the CTL recognizing the Db126 epitope (RMFPNAPYL)Citation34 with the highest avidity to the MHC H-2Db was barely detected in this setting (Nakajima et al., unpublished data). Then, we challenged Db126 peptide + Poly(I:C) to mice bearing WT1-C1498 tumor. The splenocytes were restimulated with the WT1 peptide ex vivo in order to detect specific CTL against WT1 tetramer (). Specific CTL with tumor shrinkage was significantly detected upon early challenge with Poly(I:C) + Db126 in wild-type mice followed by restimulation (). In Batf3−/− mice, neither tumor shrinkage nor specific CTL was detected by Poly(I:C) + peptide challenge under the panel F conditions (). There appears a parallelism between the frequencies of Db126-specific CTLs and tumor regression, and a requirement of BATF3 for Poly(I:C)-dependent optimal antitumor responses in the WT1-C1498 model.
Antigen-specific CTL induction by Poly(I:C) is decreased in Batf3−/− mice
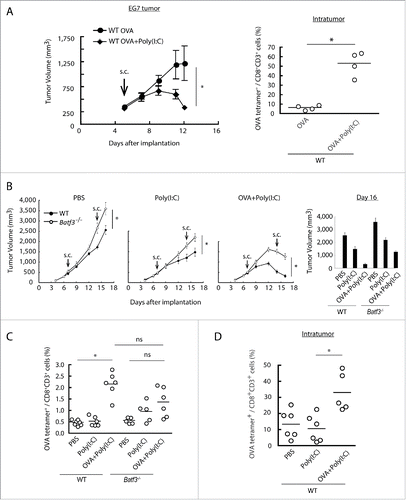
The EG7 implant tumor is a good model for Poly(I:C) therapy in vivo: TLR3/TICAM-1 pathway in DC is crucial for expansion of antigen-specific CTL.Citation28 OVA is an artificial Ag that contains multivalent CD8+ and various CD4+ epitopes. OVA and/or Poly(I:C) were s.c. injected into wild-type or Batf3−/− mice implanting EG7 tumor, and tumor growth was measured. Tumor growth retardation was observed in this model in response to Poly(I:C) + OVA () concomitant with a marked increase of specific CTLs (). Notably, Poly(I:C)/OVA-mediated tumor shrinkage was abrogated in Batf3−/− mice (). 16 d after the tumor implantation with twice therapeutic injection, we found Batf3 expression profoundly linked to Poly(I:C)-mediated growth retardation of implant EG7 (). Basal tumor growth was slightly accelerated in Batf3−/− mice compared to wild-type mice irrespective of Poly(I:C) therapy. 9 d after the Poly(I:C)/OVA injection, OVA(SL8)-specific proliferation of CTL in the spleen was measured by OVA tetramer (). OVA-specific CTLs were obviously expanded in the Poly(I:C)/OVA group in spleen by treatment with Poly(I:C) + OVA (), whereas they were barely detectable in the PBS- or Poly(I:C)-treated groups in wild-type mice (), suggesting the indispensability of exogenous Ag+Poly(I:C) for effective CTL induction. Notably, Tetramer-positive CTL were also increased in tumor in response to OVA+Poly(I:C) in wild-type mice (). A similar tendency was observed in Batf3−/− mice but tetramer-positive T cells were mildly increased in response to Poly(I:C) for unknown reason. Less Ag-specific CTL proliferation was observed in Batf3−/− mice in response to OVA+Poly(I:C) ().
Figure 2. Combined administration of antigen and Poly(I:C) induces EG7 tumor regression. Wild-type mice were inoculated with EG7 tumor and had Poly(I:C) and OVA therapy day 5 after tumor implantation. At day 12, the proportion of tumor-infiltrating OVA-specific CD8+ T cells was evaluated (A). EG7 was implanted to wild-type mice or Batf3−/− mice and Poly(I:C) with or without OVA was administered around the tumor at day 7 and 14. PBS was used as control (B). At day 16, the proportions of OVA-specific CD8+ T cells in spleen were evaluated by flow cytometer (C). EG7-bearing wild-type mice had Poly(I:C) and OVA therapy at day 7, and at day 15, the proportion of tumor-infiltrating OVA-specific CD8+ T cells was evaluated (D). Error bars show ± SEM; n = 4 to 8 per group. Student's t-test (A, B) and Kluskal–Wallis test with Dunn's multiple comparison test (C, D) were performed to analyze statistical significance. *p < 0.05, ns; not significant. Three similar experiments were performed and the results are the representative one.
CD8α+ DC were increased in the spleen and DLN in wild-type C57BL/6 mice in response to Poly(I:C)/OVA (Fig. S1A,B,C). This incremental response of CD8α+ DC was partially abolished in Batf3−/− mice. However, the CD8α+ DC population is reportedly heterologous, not reflecting bona fide CD8α+ DC.Citation30,33 We have gated with XCR1Citation9 and CLEC9A in addition to CD8α and CD11c (Fig. S1A). This CD8α+ DC population was completely diminished and never increased by Poly(I:C)+OVA in Batf3−/− mice (Fig. S1C). The XCR1hi/CLEC9A+ CD8α+ DC became highly mobile in wild-type mice, soon leaving away from the spleen and DLN in response to Poly(I:C)/OVA (Fig. S1C). Notably, this DC fraction was completely abolished in the CD8α+ DC subset in Batf3−/− mice, suggesting the presence of quantitative difference of bona fide CD8α+ DC in Batf3−/− mice. Wild-type and Batf3−/− CD8α+ DC exhibited a little cell death resistance in response to Poly(I:C), which was independent of the reported RIP3 pathway and caspasesCitation35 (Fig. S2A,B).
Induction of T cell cross-priming in CD8α+ DC by OVA and Poly(I:C)
In vivo OVA-tetramer-specific CTLs were scarcely detected in spleen in tumor-unloading wild-type mice by stimulation with Poly(I:C) alone, but became detectable in mice with Poly(I:C)/OVA (). This Poly(I:C)/OVA-mediated CTL induction was completely abrogated in Batf3−/− mice, unlike the tumor-bearing model (). Thus, some tumor factors in concert with Poly(I:C) + OVA facilitate mild induction of tetramer-positive T cells in Batf3−/− mice. OVA tetramer-positive CD8+ T cells barely proliferated in tumor by Poly(I:C) alone treatment (), although total CD8+ T cells were increased in tumor when only Poly(I:C) was s.c. administered in mice.
Figure 3. The cross-priming is partially reduced in Batf3−/− mice. Poly(I:C) and OVA were administered to wild-type and Batf3−/− mice with no tumor-loading. After 7 d, the proportion of OVA-specific CD8+ T cells in spleen was evaluated with tetramer by flow cytometer (A). CD8α+ DCs isolated from spleens of wild-type, Batf3−/− and Tlr3−/− mice were incubated with Poly(I:C) and OVA for 4 h and then co-cultured with CFSE-labeled OT-1 cells. After 60 h, the antigen-specific OT-1 proliferation was evaluated by diminution of CFSE (B) and IFNγ in the culture supernatant (C). Shadow histograms show wild-type specimen treated with PBS without OVA. Student's t-test was performed to analyze statistical significance. *p < 0.05. More than three similar experiments were performed and the results are the representative one.
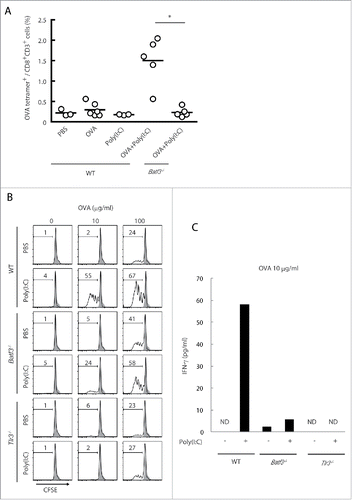
In vitro cross-priming efficacy of CD8α+ DC was tested using OT-1 T cells: CD8α+ DC were isolated from the spleens of wild-type, Batf3−/− or Tlr3−/− mice and co-cultured with OT-1 cells in the presence of OVA and Poly(I:C). Without Poly(I:C) stimulation, none of the CD8α+ DC derived from wild-type, Batf3−/− and Tlr3−/− mice cross-primed OT-1 cells at a low concentration of OVA (10 μg/mL), and almost equally cross-primed OT-1 at a high concentration of OVA (100 μg/mL) (). In the presence of Poly(I:C), however, OT-1 cells co-cultured with wild-type CD8α+ DC robustly proliferated, while OT-1 cells co-cultured with Batf3−/− or Tlr3−/− CD8α+ DC barely proliferated at low concentrations of OVA (). Yet, the cross-priming induced by Poly(I:C) minimally remained in Batf3−/− CD8α+ DC. The IFNγ levels in the co-culture with 10 μg/mL OVA paralleled the proliferation of OT-1 (). These data indicates that the basic level of cross-priming ability in CD8α+ DC is almost the same in wild-type, Batf3−/− and Tlr3−/− genotypes under a high level of Ag input and that BATF3 supplies a basic condition which enables CD8α+ DC to cross-prime OT-1 cells when Poly(I:C) stimulates TLR3/TICAM-1 signaling. Notably, the Ag level is critical for induction of cross-priming: Poly(I:C) is particularly required as a trigger for cross-priming under a low level of Ag input.
TLR3 expression and IL-12p40 production are decreased in Batf3−/− CD8α+ DC
TLR3 and the cytoplasmic RNA sensors RIG-I/MDA5 share the Poly(I:C) signal for cytokine- and type I IFN-induction.Citation36 We tested the steady-state levels of these genes in Batf3−/− CD8α+ DC from C57BL/6 mice. The expression of Tlr3 was decreased, Ddx58 (RIG-I gene) was increased, and Ifih1 (MDA5 gene) and signal adaptors, Ticam1 (TRIF) and Mavs (IPS-1), were unaffected by Batf3 knockout in CD8α+ DC compared to wild-type CD8α+ DC (). The protein expression of TLR3 in Batf3−/− CD8α+ DC was also decreased to half compared to wild-type CD8α+ DC (), suggesting the predominance of the RIG-I pathway in Poly(I:C) sensing in Batf3−/− CD8α+ DC. Of note, no significant upregulation of Batf3 was observed in Poly(I:C)-stimulated CD8α+ DC (Fig. S3), where TLR3 participated in Poly(I:C)-dependent IFN-β induction, but not in Batf3 induction.
Figure 4. TLR3 and inducible IL-12 levels are decreased in CD8α+ DCs in Batf3−/− mice. CD8α+ DCs in spleen were isolated from wild-type and Batf3−/− mice and then the levels of mRNA were evaluated. The expression levels of the indicated genes were measured by qPCR (A). The level of TLR3 protein in CD8α+ DCs was assessed by flow cytometer (B). CD8α+ DCs were isolated from wild-type, Tlr3−/− and Batf3−/− mice, and stimulated with Poly(I:C). After 4 h, mRNA were collected and the expression levels of the indicated genes were evaluated (C). Poly(I:C) was s.c. administered to wild-type and Batf3−/− mice. The blood serum was collected at the times indicated and the amount of IL-12p40 was measure by ELISA (D). Poly(I:C) was s.c. administered to wild-type and Batf3−/− mice. 12 h later, splenocytes were harvested. Collected cells were cultured in the presence of Blefeldin A for 4 h. Then, the proportion of IL-12p35+ p40+ CD8α+ DCs (MHC class II+ CD11chi) was evaluated by flow cytometer (E). Error bars show ± SEM; n = 4 to 6 per group. Kluska–Wallis test with Dunn's multiple comparison test were performed to analyze statistical significance. *p < 0.05, ns; not significant (E). The results are the representatives of three independent experiments.
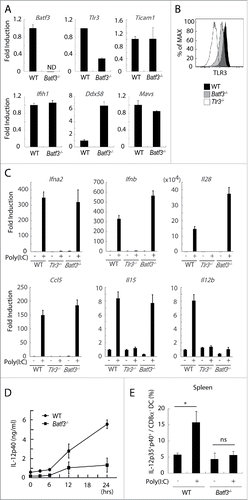
Next, we attempted to determine whether TLR3 signaling worked sufficiently in Batf3−/− CD8α+ DC in response to Poly(I:C). CD8α+ DC isolated from spleens of wild-type, Batf3−/− and Tlr3−/− mice were treated with Poly(I:C) and the mRNA were collected for determination of inducible mediators (Table S1). Ultimately, type I IFNs, Ifna2 and Ifnb, and type III IFN, Il28, were induced by Poly(I:C) in Batf3−/− CD8α+ DC comparable to wild-type CD8α+ DC (). Poly(I:C)-derived Ccl5 and Il15 were unaffected while Il12b was completely abolished in CD8α+ DC by Batf3 knockout (). Although the RIG-I pathway may compensate for cytokine/chemokine production (), Poly(I:C)-derived RIG-I upregulation failed to recover the IL-12p40 level. The RIG-I dominance in Poly(I:C) therapy might explain the remaining CTL induction in Batf3−/− mice with tumor ().
We next tested whether IL-12p40 repression occurs in Poly(I:C)-injected Batf3−/− mice in vivo. When Poly(I:C) was s.c. injected to mice, IL-12p40 was abundantly produced by Poly(I:C) in wild-type, but not in Batf3−/− mice (). IL-12p70 was actually produced in splenic CD8α+ DC in wild-type mice but not Batf3−/− mice (). In line with the IL-12p40 expression, we found the peak of Batf3 signal in the enhancer region of TLR3 in accordance with those of p300, H3K27ac, and H3K4me1 by chip-sequence analysis (Fig. S4A). There was significant Batf3 signal in the 5′-UT region of IL-12p40, which might represent the direct regulation of IL-12p40 by Batf3 (Fig. S4B). No marked changes of the expression levels of membrane molecules, Tnfsf9, Tnfsf10 and Cd40, were observed in response to Poly(I:C) (Fig. S5). These data infer that BATF3 regulates the expression of TLR3, and then IL-12p40 production in CD8α+ DC in Poly(I:C) therapy.
Poly(I:C) endows CD8+ T cells the mobility to infiltrate in tumors by BATF3
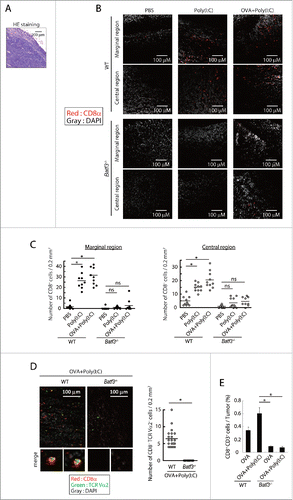
In EG7 tumor specimens, CD8+ cells infiltrated the tumor in wild-type mice 9 d after the first Poly(I:C) or Poly(I:C)/OVA treatment (), while CD8+ cells barely infiltrated in both peripheral and central area in the tumors in mice without Poly(I:C) (). Notably, no CD8+ cells entered into the tumor in Batf3−/− mice in Poly(I:C) therapy (). CD8+ T cells in tumors were counted for quantitative analysis (), where Poly(I:C) treatment confers tumor-infiltrating mobility on CD8+ T cells and BATF3 is indispensable for this Poly(I:C) function.
Figure 5. Poly(I:C)-induced CD8+ cell infiltration into tumor tissues. EG7 tumor-bearing mice had Poly(I:C) + OVA therapy on day 7 and 14, and tumor tissues were excised at day 16. The tumor sections were stained with hematoxylin and eosin (A) or APC-anti CD8α antibody and DAPI (B, C). The confocal microscopy images (×20 magnification) of a margin and a center of the tumor isolated from wild-type mice (Upper panels) or Batf3−/− mice (Lower panels) are shown (Red; CD8α, Gray; DAPI) (B). The numbers of CD8α+ cells in 0.2 mm2 of tissue sections were counted (C). Tumor-bearing mice had Poly(I:C) and OVA therapy at day 9 and tumor tissues were excised at day 15. Tumor sections were stained with APC-anti-CD8α , PE-anti-TCR Vα2 antibody and DAPI (D). The confocal microscopy images are shown (Red; CD8α, Green; TCR Vα2, Gray; DAPI) (Left panel). The numbers of CD8α+ TCR Vα2+ cells in 0.2 mm2 of the tissue sections were counted (Right panel) (D). The proportion of tumor-infiltrating CD8α+ CD3+ cells was evaluated by flow cytometer (E). The results are the representatives of each group (B, C, D). Error bars show ± SEM; n = 5 per group (E). Kluskal–Wallis test with Dunn's multiple comparison test (C, E) and Student's t-test (D) were performed to analyze statistical significance. *p < 0.05, ns; not significant.
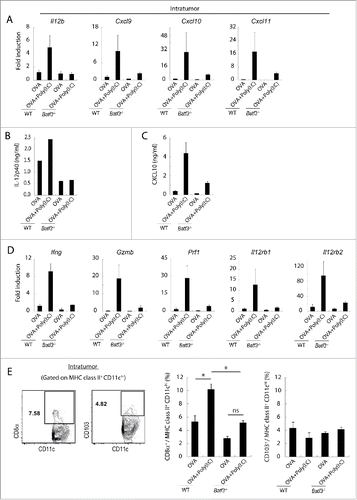
How CTLs are recruited to the tumor by Poly(I:C) therapy is a next matter for analysis. EG7-bearing wild-type and Batf3−/− mice were challenged with Poly(I:C)/OVA therapy and then the mRNA levels of chemokines/cytokines in tumor were assessed in wild-type and Batf3−/− mice by RT-PCR. Chemokines associated with CTL attraction (Cxcl9, Cxcl10 and Cxcl11) and Il12b were upregulated in response to OVA+Poly(I:C) in the tumor in wild-type mice, in contrast to Batf3−/− mice (). A similar tendency was observed in the protein levels of IL-12p40 () and CXCL10 () in the tumors harvested from wild-type vs. Batf3−/− mice. Since tumor cells do not secrete these mediators and macrophages are Batf3-independently activated in tumor (Figs. S6, 7), Batf3-dependent DCs in the tumor would be the main source of these mediators. In the same setting, Ifng, Gzmb, Prf1, Il12rb1 and Il12rb2 were upregulated in the tumor in response to Poly(I:C) in wild-type, but not in Batf3−/− mice (). CD8α+ DC was a dominant subset in intratumor DC compared with CD103+ DC, and increased after treatment with OVA+Poly(I:C) (). The results infer that the CD8α+ DC in tumor modulate antitumor microenvironment to recruit CD8+ T cells, mostly of tumor-specific CTL, by Poly(I:C).
Figure 6. Poly(I:C) changes intratumoral gene expression patterns and IL-12 production in wild-type mice but not in Batf3−/− mice. EG7 tumor-bearing mice were challenged with Poly(I:C) and OVA at day 9 and 14 after tumor implantation. At day 15, tumor tissues were harvested and the levels of mRNA were evaluated (A, D). The quantity of the IL-12p40 and CXCL10 proteins in the fixed volume of tumor tissues was also evaluated by ELISA (B, C). The ratios of tumor-infiltrating CD8α+ DC and CD103+ DC subsets were evaluated by flow cytometer (E). Error bars show ± SEM; n = 4–7 per group. One-way analysis of variance (ANOVA) with Bonferroni's test was performed to analyze statistical significance (E). *p < 0.05, ns; not significant. The results are one of the two independent experiments. In panel B, one representative of each group is shown.
In this context, we checked the possibility that tumor microenvironment other than DC participated in the TLR3/Batf3-mediated T cell infiltration into the tumor. TLR3 levels were essentially low in EG7(OVA) and C1498(WT1) cells, and Poly(I:C) barely affected the expression levels of tumor cell TLR3 (Fig. S6A). Neither the relevant genes for lymphocyte attraction were induced in tumor cells in response to Poly(I:C) (Fig. S6B), nor occurred Poly(I:C)-mediated tumor cell death accordingly (Fig. S6C). Tumor-associated myeloid cells were situated in tumor with similar profiles in wild-type and Batf3−/− mice, and only minimally affected on Poly(I:C) treatment (Fig. S7). Thus, microenvironmental factors marginally regulate T cell infiltration into tumor.
Adoptively transferred CD8α+ DC is responsible for Poly(I:C)-induced tumor regression
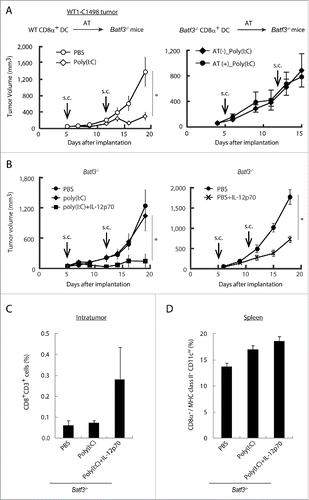
Whether exogenously added CD8α+ DC work for tumor microenvironment in Batf3−/− mice is a question in this context. We next tested if adoptive transfer of wild-type CD8α+ DC can retard WT1-C1498 growth in Poly(I:C)-administered Batf3−/− mice. Tumor growth was significantly repressed by Poly(I:C) in the group of Batf3−/− mice which received transferred wild-type CD8α+ DC (left panel in ). Batf3−/− CD8α+ DC transferred in a similar way did not revert the tumor growth inhibition in this Batf3−/− mouse model (right panel in ). Control studies indicated that Poly(I:C) treatment did not induce tumor regression in Batf3−/− mice in the absence of additional IL-12p70 (left panel in ). Full tumor-repressive activity was induced by treatment with Poly(I:C) + IL-12p70 (). IL-12 has been shown to enhance the rejection of many types of murine tumors,Citation37 but excess IL-12p70 alone exhibited minor potential for WT1 tumor regression (right panel in ). Ultimately, IL-12 induced by TLR3/TICAM-1 from CD8α+ DC is responsible for growth retardation of WT1-C1498 tumors in response to Poly(I:C) treatment, and BATF3 is indispensable for both spleen and tumor DC to evoke Poly(I:C)-derived antitumor effect. Notably, intratumor CD8+ T cells were replenished in response to Poly(I:C) + IL-12p70 treatment in Batf3−/− mice (), and splenic CD8α+ DC density was unaffected in this setting ().
Figure 7. BATF3 plays a key role in CD8α+ DCs for IL-12 production. CD8α+ DCs were isolated from wild-type (Left panel) or Batf3−/− mice (Right panel) and i.v. transferred to WT1-C1498 tumor-bearing Batf3−/− mice at day 4 and 11 after tumor implantation. PBS or Poly(I:C) were administered to the recipient mice at day 5 and 12 (A). WT1-C1498 was implanted to Batf3−/− mice and the mice were treated with Poly(I:C) alone or Poly(I:C) and IL-12p70 (Left panel), PBS or IL-12p70 (Right panel) at day 5 and 12 (B). At day 13, tumor tissues were harvested and the proportion of tumor-infiltrating CD8+ T cells was evaluated by flow cytometer (C). The spleen was also harvested and the proportion of CD8α+ DCs was evaluated (D). Error bars show ± SEM; n = 3 to 5 per group. Kluskal–Wallis test with Dunn's multiple comparison test (the left B) and Student's t-test was performed (A, the right B) to analyze statistical significance. *p < 0.05. Two similar experiments were additionally performed and supported the results shown.
CD4+ T cells promotes cross-priming by CD8α+ DC
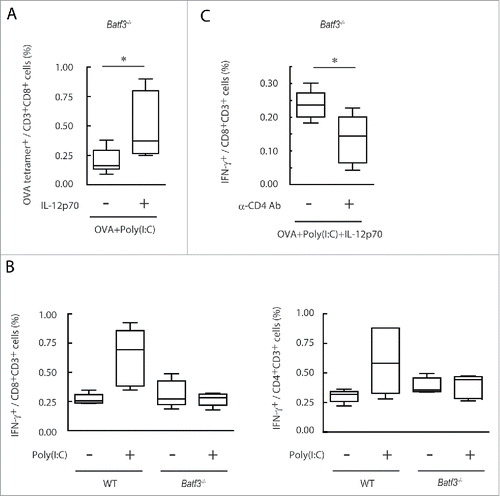
Next, we confirmed whether IL-12p40 produced by CD8α+ DC is involved in effector induction in Batf3−/− mice. When IL-12 was i.p. injected to Batf3−/− mice together with Poly(I:C) and OVA, OVA-tetramer-positive cells were significantly increased in vivo compared to the group treated with Poly(I:C)/OVA without IL-12 (). IFNγ production in OVA-specific CD8+ and CD4+ T cells was upregulated in wild-type, but not Batf3−/− mice in response to Poly(I:C) (). IL-12-mediated IFNγ upregulation was assisted by CD4+ T cells in Batf3−/− mice (), and this IFNγ upregulation was Poly(I:C)-dependent in wild-type mice, but marginal in Batf3−/− mice. CD4+ T cell-involved tumor regression is key in WT1 tumor regression. In vitro cross-priming-enhancing function of OT-II (CD4) cells was reproduced with Batf3−/− DC in the presence of IL-12 (Fig. S8A,B). The XCR1hi CD8α+ DC population, which were mostly in the TLR3hi CD8α+ DC subset, are responsible for production of IL-12 by Poly(I:C) stimulation in wild-type mice (Fig. S9). Hence, the scenario of TLR3 adjuvant is that expression of Batf3 governs TLR3-mediated IL-12p40 production in XCR1hi CD8α+ DC, which in turn activate CD4+ T cells and upregulate IFNγ; then, TICAM-1-mediated cross-priming of TAA-specific CD8+ T cell proliferation is triggered in XCR1hi CD8α+ DC in spleen for establishment of antitumor immunity. Simultaneously, TLR3-high DC release CXCR3 ligands in tumor to recruit the primed CD8+ T cells, resulting in tumor regression. The levels of PD-1 in T cells and PD-L1 in tumor cells are another matter to determine tumoricidal efficacy.
Figure 8. IL-12p70 induces CTL activation and CD4+ T cell assist it in Batf3−/− mice. Poly(I:C) and OVA (1 mg) with or without IL-12p70 were i.p. administered to Batf3−/− mice. 7 d later, splenocytes were harvested and the proportion of OVA-tetramer+ CD8+ T cells was evaluated (A). Poly(I:C) and OVA (60 μg) were administered to wild-type or Batf3−/− mice at day 0 and 7. At day 14, the proportion of IFNγ+ CD8+ T and CD4+ T cells in spleen were evaluated (B). Acsites containing anti-CD4 antibody were injected to Batf3−/− mice at day 0 and 7. 24 h later, Poly(I:C) and OVA (250 μg) with or without IL-12p70 were administered to Batf3−/− mice. At day 15, the proportion of IFNγ+ CD8+ T cells in spleen was evaluated (C). Error bars show ± SEM; n = 4 to 5 per group. Student's t-test (A, C) and Kluskal–Wallis test with Dunn's multiple comparison test (B) were performed to analyze statistical significance. *p < 0.05. Two similar experiments were performed and the results were supported.
Discussion
Our study highlights the importance of TLR3 adjuvant in DC maturation for tumor-specific T cell proliferation and infiltration, which leads to tumor regression. It acts locally in either spleen/DLN or tumor. Poly(I:C) adjuvant not only elevates cross-priming ability of splenic DC, but also induces the liberation of IL-12, type I IFN, chemokines and other cytokines in tumor microenvironment through tumor DC. This is an essential event in T cell migration and exertion of tumoricidal activity in tumor. TLR3 rather than RIG-I/MDA5 participates in evoking Poly(I:C) antitumor immunity in CD8α+ DC, which distinguishes from IFN therapy and minimizes the adverse effect by systemic induction of cytokines/IFNs.Citation14,20 Other RNA adjuvants such as Poly(A:U) might be a candidate for antitumor adjuvant, but their functional properties remained undefined in terms of TLR3-specificity.Citation17,20,38 The point of this study was to clarify by what mechanism Poly(I:C) induces tumor growth retardation in tumor-bearing mice.
In our s.c.-injection setting, Ag and adjuvant limitedly act on both tumor and DLN/spleen. In DLN/spleen, TLR3 adjuvant alone treatment does not allow TAA-specific CTL proliferation, and treatment with both adjuvant and OVA Ag is required for tetramer-positive T cell proliferation in spleen. A number of studies have implicated extrinsic administration of Ag near DLN in Ag-specific CD8+ T cell proliferation,Citation13 and additional TLR3 adjuvant markedly enhances T cell proliferation even under low Ag input.Citation28 Indeed, in our setting only with Poly(I:C), tetramer-positive CD8+ T cells failed to proliferate in DLN/spleen, but plenty of CD8+ T cells migrate into tumor ().
Unless tumor permits T cell infiltration, the PD-1 Ab therapy is ineffective. As shown in melanoma PD-1 (or PD-L1) Ab therapy, metastatic tumors shrink if the CD8+ T cells infiltrate into the tumors.Citation39,40 We show here that Poly(I:C) has ability to provoke T cells to enter the tumor (). CD103+ DC in tumor may make CTL proliferate within tumor,Citation41,42 but this is unlikely in the vaccine therapy, where only a few CD103+ DC reside in the EG7-implant tumor () and OVA tetramer-positive CD8+ T cells barely proliferate in tumor by Poly(I:C) alone treatment (), suggesting that these CD8+ T cells are recruited to tumor from spleen, where Ag-specific T cells markedly proliferate by s.c.-injected Poly(I:C) adjuvant + Ag ().
Importance of Poly(I:C) stimulation in tumor is shown in , where not only CD8α+ DC adoptive transfer but also additional Poly(I:C) is required for tumor regression. Actually, the ratio of CD8α+ subset/intratumor DCs is increased in response to Poly(I:C) and the Poly(I:C)-derived CXCL9, 10 and 11 are all upregulated in wild-type mice. These CXCR3 ligands are diminished in tumor in Batf3−/− mice, suggesting that Batf3-dependent DC rather than MDSC or TAM are the source of T cell-recruiting chemokines. Although the exact subsets of DC for T cell attraction to tumor are undetermined, the results reinforce the idea that Poly(I:C) acts on Batf3-positive DCs in tumor as well.
Recent reports demonstrated that checkpoint blocking of the effector step by PD-1 or PD-L1 Ab brought high tumoricidal activity to CTL in tumor site.Citation27 TLR3 adjuvant synergistically contributes to the PD-L1 therapy by promoting T cell entry into tumor (Kataoka, Takeda et al., submitted for publication). TLR3 adjuvant in DLN/spleen is also indispensable for T cell proliferation (priming step) by Batf3-dependent CD8α+ DC in vaccine therapy since no tumor regression was observed by only adoptive transfer of Batf3−/− CD8α+ DC subset (). Batf3−/− CD8α+ DC have moderate potential for T cell proliferation (presumably, due to high MDA5 expression) but not for TAA-specific T cell priming or IL-12 induction. CD8α+ DC at least work in two distinct modes that sustain T cell priming in spleen and recruitment of effector T cells in tumor to regress tumor. Although tumor microenvironment works like lymphoid organ where intratumor DC might proliferate CTL in the tumor,Citation22 in our vaccine models CD8α+ DC subset play a major role in recruiting CTL to tumor in response to Poly(I:C).
In our study, the Ag density is crucial for priming CTL. Tumor-specific CTL unable to determine by OVA tetramer (targeting other than OVA epitopes) might join Ag-specific T cell priming in spleen or tumor. It is also likely that OVA Ag are released from tumor cells to spread over spleen/DLN and taken up into CD169 macrophages or CD8α+ DCs.Citation43
IL-12 and chemokines are released from myeloid cells in tumor. It is accepted that macrophages and DC in tumor contribute to the formation of tumor microenvironment. IL-12 induces activation of T cells by IFNγ and PD-1 downregulation in CD8+ T cells.Citation21 Transfer of CD8α+ DC or moving CD8α/XCR1hi DC both require Poly(I:C) stimulation for tumor reduction, suggesting that TLR3 signal is critical for the formation of tumor microenvironment to facilitate antitumor immunity. In fact, IL-12 administration with DC transfer results in full tumor regression (). What is the role of IL-12-derived CD4+ T cells (Th1, Th17 or Treg) in the context of antitumor microenvironment yet remains to be further discussed.
Our next question was why Batf3−/− mice have lost the T cell priming and tumor-infiltrating abilities induced by Poly(I:C). Batf3−/− mice are >90% depletion of XCR1hi CLEC9A+ CD8α+ DC in spleen but still possess them in DLN. Batf3−/− mice exhibit minor T cell priming in response to Poly(I:C) or Poly(I:C) + OVA (), but no T cell infiltration into tumor (), which may represents the features of Batf3-independent DC. Alternatively, RIG-I-dependent cross-presentation happens in Batf3−/− CD8α+ DC, which is less efficient than TLR3-dependent one.Citation44 There exist DC in epidermal or tumor region that express high levels of RIG-I/MDA5, which harbor some ability to compensate cross-priming for Batf3−/− CD8α+ DC in response to Poly(I:C) or tumor-derived factors.Citation35 Thus, regarding T cell priming ability CXCR1hi/CD8α+ DC can be replaced with other DC subsets in spleen/DLN with high Ag input, but the ability of T cell infiltration into tumor is dependent on Batf3-positive DC in tumor (). In the EG7 tumor model, tumor-infiltrating CD8α+ DC rather than CD103+ DC may be more important to attract priming T cells from spleen.Citation45 Thus, the mode of T cell proliferation and recruitment to tumor is environment- or tumor-type-dependent, probably in conjunction with DC subsets.Citation41,46 In any case, Batf3 is implicated in TLR3-dependent DC maturation by Poly(I:C) in both spleen/DLN and tumor.
Because OVA is an artificial Ag with nonself epitopes, we challenged C1498-bearing mice with WT-1 peptide (Db126) and compared with Poly(I:C) in this study. It becomes obvious that tumors with mutated Ags of TAA are effective targets for anti-PD-1 therapy than those with differentiated or testis-specific Ags.Citation47 WT-1 likely mimics the latter self-Ag, so that C1498 may represent anti-PD-1 therapy-resistant tumors, yet Poly(I:C) has some therapeutic efficacy toward C1498(WT1). As expected, Db126 + Poly(I:C) less induced tetramer-specific CTL than OVA + Poly(I:C), suggesting the importance of the quality of Ags in vaccine immunotherapy for cancer.Citation48,49 Even in the case with Db126, however, Poly(I:C) endorses proliferation of antitumor CTL in spleen and induces tumor shrinkage. Since C1498 tumor cells do not produce CXCR3 ligands by stimulation with Poly(I:C) in vitro (data not shown), recruiting CTL to tumor will be induced by intratumor DC in WT-1-expressing tumors. In several papers, CXCL10 (or other CXCR3 ligands) allows CXCR3+ CD8+ T cells to direct to inflammatory lesions.Citation50 Although myeloid cells produce CXCR3 ligands by some stimulations, Batf3−/− DC and tumor stroma fail to produce CXCR3 ligands in response to Poly(I:C). Characterization of the tumor-resident DC (CD8α+ DC in our model) is a matter of great interest in this scenario.
Materials and methods
Cells
WT1-C1498 cells were provided by H. Sugiyama (Osaka University, Osaka. Japan) (Nakajima et al., 2012), and EG7 cells were purchased from ATCC (Manassas, VA), where authentication was checked using short tandem repeat analysis. Cells were obtained during 2012–2014 and used within 6 mo after resuscitation. EG7 cells were cultured in RPMI 1640 (GIBCO, the catalog number: 11875–093) supplemented with 10 % heat-inactivated FBS (Thermo Scientific, SH30910.03), 55 μM 2-mercaptoethanol (GIBCO, 21985–023), 10 mM HEPES (GIBCO, 15630–080), 1 mM sodium pyruvate (GIBCO, 11360–070), 50 IU penicillin/50 μg/mL streptomycin (GIBCO, 15070–063) and 0.5 mg/mL G418 (Roche, 04 727 894 001).Citation51 WT1-C1498 cells were cultured in RPMI 1640 supplemented with 10 % heat-inactivated FBS, 55 μM 2-mercaptoethanol, 50 IU penicillin/50 μg/mL streptomycin and 0.5 mg/mL G418. Splenocytes were cultured in complete medium containing 10 % heat-inactivated FBS, 45 % RPMI 1640, 45 % AIM-V (GIBCO, 12055–091), 1 × non-essential amino acid (GIBCO, 11140–050), 55 μM 2-mercapthoethanol and 50 IU penicillin/50 μg/mL streptomycin. 3 and 5 d later, recombinant mouse IL-2 (20 IU/mL) (Pepro Tech, 212–12) was added to the culture.Citation34
Mice
Wild-type C57BL/6J mice were purchased from CLEA. Batf3−/− mice were purchased from Jackson Laboratory (Bar Harbor, ME). Rag2−/− and OT-1 mice, and Tlr3−/− mice were kindly provided by N. Ishii (Tohoku University, Sendai, Japan) and S. Akira (Osaka University, Osaka, Japan), respectively. OT-II mice were kindly provided from Dr Kazuya Iwabuchi (Kitasato University, Kanagawa, Japan). All mice were backcrossed >8 times to C57BL/6 background and maintained under specific pathogen-free conditions in the animal faculty of the Hokkaido University Graduate School of Medicine. Animal experiments were performed according to the guidelines set by the animal safety center, Hokkaido University, Japan.
Reagents and antibodies
Poly(I:C) and EndogGade® Ovalbumin (EndoOVA) were purchased from GE healthcare Life Sciences (the catalog number: 27-4732-01) and Hyglos (321001), respectively. OVA257–264 peptide (SIINFEKL: SL8), OVA323–339 peptide (ISQAVHAAHAEINEAGR: OVA helper peptide), OVA (H2Kb-SL8) Tetramer was purchased from MBL (TS-5001-P, TS-M703-P, TS-5001-1). WT1126–134 peptide (RMFPNAPYL: Db126) and WT1 (H2Db-Db126) Tetramer were kindly provided by H. Sugiyama (Osaka University, Osaka. Japan). Brefeldin A and z-VAD-FMK (z-Val-Ala-Asp fluoromethyl ketone) were purchased from Sigma-Aldrich (B6542-5MG, V116-2MG). Collagenase D and Annexin-V-Fluous Staining Kit were purchased from Roche (11 088 882 001, 1 988 549). Recombinant mouse IL-12p70 (577002) and following antibodies, anti-CD3 (Clone: 17A2, the catalog number: 100215), anti-CD8α (53–6.7, 100706 and 100729), anti-CD11b (M1/70, 101206), anti-CD11c (N418, 117317), anti-CD16/32 (93, 101302), anti-CD103 (2E7, 121405), anti-CLEC9A (7H11, 143503), anti-F4/80 (BM8, 123109), anti-Gr1 (RB6-8C5, 108407), anti-I-Ab (KH74, 115305) and anti-IFNγ (XMG1.2, 505809), anti-IL-12/IL-23p40 (C15.6, 505203), anti-TLR3 (11F8, 141905), anti-XCR1 (ZET, 148205) were purchased from BioLegend. Mouse IFN gamma ELISA KIT (88–7314) and Abs, anti-CD4 (GK1.5, 12-0041-82), anti-IL-12p35 (4D10p35, 50-7352-80), anti-TCR Vα2 (B20.1, 12-5812-82) were from eBiosciences. ViaProbe was from BD Biosciences (555816). Mouse CXCL10/IP10/CRG-2 ELISA KIT was purchased from R and D Systems (515–87921). ProLong® Gold which contains DAPI was purchased from Life technologies (P36931).
Tumor challenge and Poly(I:C) therapy
Mice were s.c. injected with 1 × 106 syngeneic WT1-C1498 or 2 × 106 EG7 cells on the back.Citation28,34 The tumor volume was calculated by the formula: Tumor volume [mm3] = 0.52 × (long diameter [mm]) × (short diameter [mm])2. Poly(I:C) (50 μg) with or without IL-12p70 (100 ng) was s.c. injected around WT1-C1498 tumor at day 5 and 12 after tumor implantation. Poly(I:C) and WT1126–134 peptide (100 μg) were wrapped in 20 μg of DOTAP (Roche, 11202375001) for s.c. injection. In EG7 tumor, Poly(I:C) with or without OVA (100 μg) treatment was started when the tumor volume reached about 200–600 mm3. CD8+ T cells or NK cells were depleted with intraperitoneal (i.p.) administration of hybridoma ascites of anti-CD8α or anti-NK1.1 mAb to mice a day prior to Poly(I:C) treatment.Citation28,52 CD8α+ DCs were isolated from spleens of wild-type or Batf3−/− mice for CD8α+ DC-adoptive transfer by CD8+ DC isolation kit (Miltenyi Biotec, 130-091-169). The purity of the cells was routinely ≥90 %. For adoptive transfer, 5 × 105 CD8α+ DC was intravenously (i.v.) administered to Batf3−/− mice 24 h before the Poly(I:C) treatment.
Analysis of intratumoral microenvironment
WT1-C1498 and EG7 tumors were excised from tumor-bearing mice. For flow cytometer analysis, tumors were cut finely and treated with 0.05 mg/mL collagenase I (Sigma-Aldrich, C0130-100 MG), 0.05 mg/mL collagenase IV (Sigma-Aldrich, C5138-1G), 0.025 mg/mL hyaluronidase (Sigma-Aldrich, H6254-500MG) and 0.01 mg/mL DNase I (Roche, 10 104 159 001) in Hanks' balanced salt solution (Sigma-Aldrich, H9269-500ML) at 33°C for 10 min. Intratumor IL-12 and CXCL10 was measured by ELISA of the supernatant obtained from fixed volume of tumors. Tumor-infiltrating CD8+ T cells and myeloid cells were analyzed by FACS AriaII (BD Biosciences). For the exclusion of false-positive, dead cells were excluded by removing of FSClo SSChi and 7-AAD+ populations. Furthermore, the isotype control antibodies were used as negative controls. For microscopic analysis, 4% paraformaldehyde-fixed, frozen tumor sections of thickness of 10 μm were stained with hematoxylin (Sigma-Aldrich, H9627) and eosin (Merck, 1.15935.0025) or APC-anti-CD8α mAb, PE-anti-TCR-Vα2 and DAPI.Citation40 The stained specimens were analyzed under BZ-9000 (KEYENCE) or LSM510 META microscopy (Zeiss).
OT-1 proliferation assay
OT-1 T cells were prepared from spleens of Rag2−/−/OT-1 mice by CD8+ microbeads (Miltenyi Biotec, 130-049-401). OT-1 cells were labeled with 1 μM CFSE for 10 min at room temperature. 5 × 104 CD8α+ DCs were seeded in a 96-well flat bottom plate in the presence of OVA and Poly(I:C) (50 μg/mL). The CD8α+ DCs were incubated for 3 h and then co-cultured with CFSE-labeled 2.5 × 104 OT-1 cells. In some experiments, IL-12p70 (100 ng/mL) was added simultaneously with OT-1 cells to the wells with DC. After 60 h, these cells were stained with anti-TCR Vα2 and anti-CD8α and OT-1 proliferation was determined with diminution of CFSE by FACS AriaII. Additionally, IFNγ in the culture supernatant was measured by ELISA. In OT-II co-culture assay, 2.5 × 104 OT-II T cells were added to the conditioned wells with CD8α+ DCs together with OT-1 cells.
Reverse transcription-PCR and real-time PCR
Total RNA was prepared using TRIzol Reagent (Ambion, 15596018) following the manufacturer's instructions. Reverse transcription-PCR was carried out using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems, 4368813) according to the manufacturer's instructions. Real-time PCR was performed using a Step One real-time PCR system (Applied Biosystems). Sequences of primers in this study are shown in Table S1. Levels of target mRNAs were normalized to Gapdh and fold-induction of transcripts was calculated using the ddCT method relative to unstimulated cells.
Antigen-specific T cell expansion in vivo
Mice were i.p. immunized with OVA, Poly(I:C) (150 μg) and IL-12p70 (100 ng) once or twice at weekly intervals. For CD4+ cell depletion, hybridoma ascites of anti-CD4 mAb (∼100 μg) was i.p. injected to mice a day prior to immunization. Spleens were collected on day 7 after the last immunization, and Ag-specific CTL activation was analyzed by tetramer assay or intracellular IFNγ staining. In some cases, splenocytes were pulsed with SL8 (100 nM) (for OVA-specific CD8+ T cells) or helper peptide (1 μM) (for CD4+ T cells) for 6 h and Blefeldin A (10 μg/mL) was added during the last 4 h.
Statistical analysis
p values were calculated by the following statistical analysis. For the multiple comparisons, one-way analysis of variance (ANOVA) with Bonferroni's test or Kluskal–Wallis test with Dunn's multiple comparison test were performed. For the comparison between two groups, Student's t-test was performed. Error bar represent the SD or SEM between samples.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1188244_s02.zip
Download Zip (376.6 KB)Acknowledgments
We are grateful to Drs Naoto Ishii and K. Sugamura (Tohoku University, Sendai) for their kind gift of Rag2−/− mice. We thank Drs H. Shime, H. Takaki, K. Funami, M. Tatematsu, A. Maruyama and J. Kasamatsu in our laboratory for their invaluable discussions and Ms N. Ishii, Ms A. Morii-Sakai, and Ms H. Sato for technical support.
Funding
This work was supported in part by the Grants-in-Aid from the Ministry of Education, Science, and Culture (MEXT), “the Carcinogenic Spiral” a MEXT Grant-in-Project (T. Seya), the Ministry of Health, Labor, and Welfare of Japan (T. Seya, M. Matsumoto), the Uehara Memorial Foundation (T. Seya), the Yasuda Cancer Research Foundation (T. Seya), and the Kato Memorial Bioscience Foundation (H. Oshiumi).
References
- Matsumoto M, Seya T, Kikkawa S, Tsuji S, Shida K, Nomura M, Kurita-Taniguchi M, Ohigashi H, Yokouchi H, Takami K et al. Interferon gamma-producing ability in blood lymphocytes of patients with lung cancer through activation of the innate immune system by BCG cell wall skeleton. Int Immunopharmacol 2001; 1:1559-69; PMID:11515819; http://dx.doi.org/10.1016/S1567-5769(01)00071-6
- Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol 2012; 12:557-69; PMID:22790179; http://dx.doi.org/10.1038/nri3254
- Woo SR, Corrales L, Gajewski TF. Innate Immune Recognition of Cancer. Annu Rev Immunol 2012; 33:445-74; http://dx.doi.org/10.1146/annurev-immunol-032414-112043
- Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol 2015; 98:913-22; PMID:26337512; http://dx.doi.org/10.1189/jlb.4RI0515-204R
- Mantovani A, Allavena P. The interaction of anticancer therapies with tumor-associated macrophages. J Exp Med 2015; 212:435-45; PMID:25753580; http://dx.doi.org/10.1084/jem.20150295
- Desch AN, Gibbings SL, Clambey ET, Janssen WJ, Slansky JE, Kedl RM, Henson PM, Jakubzick C. Dendritic cell subsets require cis-activation for cytotoxic CD8 T-cell induction. Nat Commun 2014; 5:4674; PMID:25135627; http://dx.doi.org/10.1038/ncomms5674
- Goutagny N, Estornes Y, Hasan U, Lebecque S, Caux C. Targeting pattern recognition receptors in cancer immunotherapy. Target Oncol 2012; 7:29-54; PMID:22399234; http://dx.doi.org/10.1007/s11523-012-0213-1
- Schulz O, Diebold SS, Chen M, Nauslund TI, Nolte MA, Alexopoulou L, Azuma YT, Flavell RA, Liljeström P, Reis e Sousa C. Toll-like receptor 3 promotes cross- priming to virus-infected cells. Nature 2005; 433:887-92; PMID:15711573; http://dx.doi.org/10.1038/nature03326
- Bachem A, Güttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, Salama A, Movassaghi K, Opitz C, Mages HW et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med 2010; 207:1273-81; PMID:20479115; http://dx.doi.org/10.1084/jem.20100348
- Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, Vu Manh TP, Baranek T, Storset AK, Marvel J et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med 2010; 207:1283-92; PMID:20479118; http://dx.doi.org/10.1084/jem.20100223
- Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, Chen CJ, Dunbar PR, Wadley RB, Jeet V et al. Human CD141+ (BDCA-3)+ dendritic cells (DC) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med 2010; 207:1247-60; PMID:20479116; http://dx.doi.org/10.1084/jem.20092140
- Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, Keller AM, Joffre O, Zelenay S, Nye E et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med 2010; 207:1261-71; PMID:20479117; http://dx.doi.org/10.1084/jem.20092618
- Seya T, Shime H, Takeda Y, Tatematsu M, Takashima K, Matsumoto M. Adjuvant for vaccine immunotherapy of cancer - focusing on Toll-like receptor 2 and 3 agonists for safely enhancing antitumor immunity. Cancer Sci 2015; 106:1659-68; PMID:26395101; http://dx.doi.org/10.1111/cas.12824
- Seya T, Azuma M, Matsumoto M. Targeting TLR3 with no RIG-I/MDA5 activation is effective in immunotherapy for cancer. Expert Opin Ther Targets 2013; 17:533-44; PMID:23414438; http://dx.doi.org/10.1517/14728222.2013.765407
- Vacchelli E, Eggermont A, Sautès-Fridman C, Galon J, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: Toll-like receptor agonists for cancer therapy. Oncoimmunology 2013; 2:e25238; PMID:24083080; http://dx.doi.org/10.4161/onci.25238
- Matsumoto M, Seya T. TLR3: interferon induction by double-stranded RNA including Poly(I:C). Adv Drug Deliv Rev 2008; 60:805-12; PMID:18262679; http://dx.doi.org/10.1016/j.addr.2007.11.005
- Galluzzi L, Vacchelli E, Eggermont A, Fridman WH, Galon J, Sautès-Fridman C, Tartour E, Zitvogel L, Kroemer G. Trial Watch: Experimental Toll-like receptor agonists for cancer therapy. Oncoimmunology 2012; 1:699-716; PMID:22934262; http://dx.doi.org/10.4161/onci.20696
- Sabbatini P, Tsuji T, Ferran L, Ritter E, Sedrak C, Tuballes K, Jungbluth AA, Ritter G, Aghajanian C, Bell-McGuinn K et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin Cancer Res 2012; 18:6497-508; PMID:23032745; http://dx.doi.org/10.1158/1078-0432.CCR-12-2189
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006; 441:101-5; PMID:16625202; http://dx.doi.org/10.1038/nature04734
- Matsumoto M, Tatematsu M, Nishikawa F, Azuma M, Shime H, Seya T. Defined TLR3-specific adjuvant that induces NK and cytotoxic T cell activation without significant cytokine production in vivo. Nat Commun 2015; 6:6280; PMID:25692975; http://dx.doi.org/10.1038/ncomms7280
- Gerner MY, Heltemes-Harris LM, Fife BT, Mescher MF. Cutting edge: IL-12 and type I IFN differentially program CD8 T cells for programmed death 1 re-expression levels and tumor control. J Immunol 2013; 191:1011-5; PMID:23804712; http://dx.doi.org/10.4049/jimmunol.1300652
- Zitvogel L, Kroemer G. CD103+ dendritic cells producing interleukin-12 in anticancer immunosurveillance. Cancer Cell 2014; 26:591-3; PMID:25517740; http://dx.doi.org/10.1016/j.ccell.2014.10.008
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252-64; PMID:22437870; http://dx.doi.org/10.1038/nrc3239
- Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med 2012; 209:1201-17; PMID:22641383; http://dx.doi.org/10.1084/jem.20112741
- Shime H, Matsumoto M, Oshiumi H, Tanaka S, Nakane A, Iwakura Y, Tahara H, Inoue N, Seya T. Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proc Natl Acad Sci U S A 2012; 109:2066-71; PMID:22308357; http://dx.doi.org/10.1073/pnas.1113099109
- Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 2002; 99:12293-7; PMID:12218188; http://dx.doi.org/10.1073/pnas.192461099
- Kamphorst AO, Ahmed R. Manipulating the PD-1 pathway to improve immunity. Curr Opin of Immunol 2013; 25:381-8; http://dx.doi.org/10.1016/j.coi.2013.03.003
- Azuma M, Ebihara T, Oshiumi H, Matsumoto M, Seya T. Cross-priming for antitumor CTL induced by soluble Ag + polyI:C depends on the TICAM-1 pathway in mouse CD11c(+)/CD8α(+) dendritic cells. Oncoimmunology 2012; 1:581-92; PMID:22934250; http://dx.doi.org/10.4161/onci.19893
- Edelson BT, KC W, Juang R, Kohyama M, Benoit LA, Klekotka PA, Moon C, Albring JC, Ise W, Michael DG et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med 2010; 207:823-36; PMID:20351058; http://dx.doi.org/10.1084/jem.20091627
- Edelson BT, Bradstreet TR, KC W, Hildner K, Herzog JW, Sim J, Russell JH, Murphy TL, Unanue ER, Murphy KM. Batf3-dependent CD11b(low/-) peripheral dendritic cells are GM-CSF-independent and are not required for Th cell priming after subcutaneous immunization. PLoS One 2011; 6:e25660; PMID:22065991; http://dx.doi.org/10.1371/journal.pone.0025660
- Hildner K, Edelson BT, Purtha WE, Diamond MS, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 2008; 322:1097-100; PMID:19008445; http://dx.doi.org/10.1126/science.1164206
- Murphy TL, Tussiwand R, Murphy KM. Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat Rev Immunol 2013; 13:499-509; PMID:23787991; http://dx.doi.org/10.1038/nri3470
- Mildner A, Jung S. Development and function of dendritic cell subsets. Immunity 2014; 40:642-56; PMID:24837101; http://dx.doi.org/10.1016/j.immuni.2014.04.016
- Nakajima H, Oka Y, Tsuboi A, Tatsumi N, Yamamoto Y, Fujiki F, Li Z, Murao A, Morimoto S, Hosen N et al. Enhanced tumor immunity of WT1 peptide vaccination by interferon-β administration. Vaccine 2012; 30:722-9; PMID:22133512; http://dx.doi.org/10.1016/j.vaccine.2011.11.074
- Zou J, Kawai T, Tsuchida T, Kozaki T, Tanaka H, Shin KS, Kumar H, Akira S. Poly IC triggers a cathepsin D- and IPS-1-dependent pathway to enhance cytokine production and mediate dendritic cell necroptosis. Immunity 2013; 38:717-28; PMID:23601685; http://dx.doi.org/10.1016/j.immuni.2012.12.007
- Sasai M, Shingai M, Funami K, Yoneyama M, Fujita T, Matsumoto M, Seya T. NAK-associated protein 1 participates in both the TLR3 and the cytoplasmic pathways in type I IFN induction. J Immunol 2006; 177:8676-83; PMID:17142768; http://dx.doi.org/10.4049/jimmunol.177.12.8676
- Colombo MP, Trinchieri G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev 2002; 13:155-68; PMID:11900991; http://dx.doi.org/10.1016/S1359-6101(01)00032-6
- Sugiyama T, Hoshino K, Saito M, Yano T, Sasaki I, Yamazaki C, Akira S, Kaisho T. Immunoadjuvant effects of polyadenylic:polyuridylic acids through TLR3 and TLR7. Int Immunol 2008; 20:1-9; PMID:17981792; http://dx.doi.org/10.1093/intimm/dxm112
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EM, Robert L, Chmielowski B1, Spasic M, Henry G, Ciobanu V et al. PD-1 blockage induces responses by inhibiting adaptive immune resistance. Nature 2014; 515:568-73; PMID:25428505; http://dx.doi.org/10.1038/nature13954
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014; 515:563-7; PMID:25428504; http://dx.doi.org/10.1038/nature14011
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014; 26:638-52; PMID:25446897; http://dx.doi.org/10.1016/j.ccell.2014.09.007
- Krueger PD, Kim TS, Sung SJ, Braciale TJ, Hahn YS. Liver-resident CD103+ dendritic cells prime antiviral CD8+ T cells in situ. J Immunol 2015; 194:3213-22; PMID:25712214; http://dx.doi.org/10.4049/jimmunol.1402622
- Asano K, Nabeyama A, Miyake Y, Qiu CH, Kurita A, Tomura M, Kanagawa O, Fujii S, Tanaka M. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity 2011; 34:85-95; PMID:21194983; http://dx.doi.org/10.1016/j.immuni.2010.12.011
- Miyake T, Kumagai Y, Kato H, Guo Z, Matsushita K, Saitoh T, Kawagoe T, Kumar H, Jang MH, Kawai T et al. Poly I:C-induced activation of NK cells by CD8 α+ dendritic cells via the IPS-1 and TRIF-dependent pathways. J Immunol 2009; 183:2522-8; PMID:19635904; http://dx.doi.org/10.4049/jimmunol.0901500
- Zhang Y, Chen G, Liu Z, Tian S, Zhang J, Carey CD, Murphy KM, Storkus WJ, Falo LD Jr, You Z. Genetic vaccines to potentiate the effective CD103+ dendritic cell-mediated cross-priming of antitumor immunity. J Immunol 2015; 194:5937-47; PMID:25972487; http://dx.doi.org/10.4049/jimmunol.1500089
- Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, Longhi MP. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol 2014; 12:e1001759; PMID:24409099; http://dx.doi.org/10.1371/journal.pbio.1001759
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348:69-74; PMID:25838375; http://dx.doi.org/10.1126/science.aaa4971
- Melief CJ. Cancer immunotherapy by dendritic cells. Immunity 2008; 29:372-83; PMID:18799145; http://dx.doi.org/10.1016/j.immuni.2008.08.004
- Ali OA, Verbeke C, Johnson C, Sands RW, Lewin SA, White D, Doherty E, Dranoff G, Mooney DJ. Identification of immune factors regulating antitumor immunity using polymeric vaccines with multiple adjuvants. Cancer Res 2014; 74:1670-81; PMID:24480625; http://dx.doi.org/10.1158/0008-5472.CAN-13-0777
- McNally B, Willette M, Ye F, Partida-Sanchez S, Flaño E. Intranasal administration of dsRNA analog poly(I:C) induces interferon-α receptor-dependent accumulation of antigen experienced T cells in the airways. PLoS One 2012; 7:e51351; PMID:23236482; http://dx.doi.org/10.1371/journal.pone.0051351
- Ebihara T, Azuma M, Oshiumi H, Kasamatsu J, Iwabuchi K, Matsumoto K, Saito H, Taniguchi T, Matsumoto M, Seya T. Identification of a polyI:C-inducible membrane protein, that participates in dendritic cell-mediated natural killer cell activation. J Exp Med 2010; 207:2675-87; PMID:21059856; http://dx.doi.org/10.1084/jem.20091573
- Akazawa T, Ebihara T, Okuno M, Okuda Y, Shingai M, Tsujimura K, Takahashi T, Ikawa M, Okabe M, Inoue N et al. Antitumor NK activation induced by the Toll-like receptor 3-TICAM-1 (TRIF) pathway in myeloid dendritic cells. Proc Natl Acad Sci U S A 2007; 104:252-7; PMID:17190817; http://dx.doi.org/10.1073/pnas.0605978104