
ABSTRACT
Natural killer (NK) cells are promising antitumor effector cells, but the generation of sufficient NK cell numbers for adoptive immunotherapy remains challenging. Therefore, we developed a method for highly efficient ex vivo expansion of human NK cells.
Ex vivo expansion of NK cells in medium containing IL-2 and irradiated clinical-grade feeder cells (EBV-LCL) induced a 22-fold NK cell expansion after one week that was significantly increased to 53-fold by IL-21. Repeated stimulation with irradiated EBV-LCL and IL-2 and addition of IL-21 at the initiation of the culture allowed sustained NK cell proliferation with 1011-fold NK cell expansion after 6 weeks. Compared to naive NK cells, expanded NK cells upregulated TRAIL, NKG2D, and DNAM-1, had superior cytotoxicity against tumor cell lines in vitro and produced more IFNγ and TNF-α upon PMA/Iono stimulation. Most importantly, adoptive transfer of NK cells expanded using feeder cells, IL-2 and IL-21 led to significant inhibition of tumor growth in a melanoma xenograft mouse model, which was greater than with NK cells activated with IL-2 alone. Intriguingly, adoptively transferred NK cells maintained their enhanced production of IFNγ and TNF-α upon ex vivo restimulation, although they rapidly lost their capacity to degranulate and mediate tumor cytotoxicity after the in vivo transfer.
In conclusion, we developed a protocol for ex vivo NK cell expansion that results in outstanding cell yields. The expanded NK cells possess potent antitumor activity in vitro and in vivo and could be utilized at high numbers for adoptive immunotherapy in the clinic.
Abbreviations
| APC | = | allophycocyanin |
| CAR | = | chimeric antigen receptor |
| EBV-LCL | = | Epstein–Barr virus-transformed lymphoblastoid cell line |
| FITC | = | fluorescein isothiocyanate |
| IFN | = | interferon |
| Ig | = | immunoglobulin |
| IL | = | interleukin |
| Iono | = | ionomycin |
| i.p. | = | intraperitoneal |
| i.v. | = | intravenous |
| NK | = | natural killer |
| NSG | = | NOD-scid IL-2Rgammanull |
| PBMC | = | peripheral blood mononuclear cells |
| PBS | = | phosphate-buffered saline |
| PE | = | phycoerythrin |
| PMA | = | phorbol myristate acetate |
| REA | = | REAfinity antibody |
| RPMI | = | Roswell Park Memorial Institute |
| TNF | = | tumor necrosis factor |
Introduction
Adoptive transfer of NK cells is a promising treatment option for patients with cancer. Although still in its early stages of development, pilot trials evaluating the adoptive infusion of peripheral blood-derived human NK cells have established the safety of NK cell-based immunotherapy.Citation1 Further, increasing evidence suggests NK cells may be particularly efficacious for the treatment of hematological malignancies, such as AML, where adoptive NK cells transfer may reduce the risk of disease relapse and improve long-term survival.Citation2 However, successful NK cell therapy is confronted with several challenges, in particular the hurdle of being able to obtain only limited numbers of NK cells from donors, which are too low to have an efficacious effect in patients following adoptive transfer.Citation3 To overcome this limitation, suitable methods to generate high NK cell numbers ex vivo in a way that is applicable in the clinics are absolutely essential.
Most protocols utilized to date implement interleukin (IL)-2 containing media for ex vivo NK cell activation and expansion. Although IL-2-containing medium can activate NK cells before their adoptive transfer, it only results in a minor 5–20-fold NK cell expansion in 2–4 weeks.Citation4,5 In contrast, co-culture of NK cells with clinically approved Epstein–Barr virus-transformed lymphoblastoid cell line (EBV-LCL) feeder cells in IL-2-containing medium allows for a 3637-fold expansion in 24–27 d, has proven therapeutic applicability in the clinic and can be incorporated into protocols fully automated for clinical use.Citation6-8 Despite this advance, expansion of NK cells by stimulation with irradiated EBV-LCL feeder cells is limited to a duration of only 2 to 4 weeks, as NK cells ultimately become senescent with more prolonged culturing.Citation8 Thus, even with the improved ability to expand NK cells using feeder cells, the achievable yield of NK cells remains limited, potentially restricting clinical trials that seek to incorporate multiple repeated NK cell infusions. Therefore, additional advances and improvements to expand even greater numbers of clinical-grade NK cells for adoptive immunotherapy are needed.
The cytokine IL-21 may play an important role for NK cell expansion, since it has been reported that continuous stimulation of NK cells with K562 feeder cells bearing membrane bound IL-21 results in constant and sustained ex vivo proliferation of NK cells.Citation9 However, at present, the role that IL-21 will play in NK cell expansion for clinical use remains to be determined. IL-21 has been originally discovered as cytokine that plays a role in the development of NK cells from bone marrow progenitors.Citation10 IL-21 is predominantly produced by CD4+ T cells and its heterodimeric receptor shares the common γ chain with the IL-2 cytokine family.Citation11 In mice, IL-21 acts inhibitory on the expansion of NK cells but induces functional NK cell maturation.Citation12,13 Although Wendt et al. published that human IL-21 appears to increase the proliferation of human CD56bright NK cells,Citation14 others did not observe an impact of IL-21 on the proliferation of NK cells from healthy human donors or HIV patients.Citation15 Taken together, IL-21 differentially affects the expansion of NK cells dependent on the experimental setup and its properties make it a good candidate to investigate for its potential to enhance EBV-LCL feeder cell-driven NK cell expansion.
Here, we investigated the effect of IL-21 on the proliferation and expansion of NK cells in co-culture with irradiated EBV-LCL feeder cells. Further, we evaluated the expanded NK cells for their antitumor activity in vitro and in vivo using a therapeutic melanoma xenograft mouse model. Our findings resulted in the development of an optimized and highly efficient method for ex vivo NK cell expansion for potential clinical use.
Results
IL-21 significantly increases the expansion of NK cells in the presence of irradiated EBV-LCL
First, to establish an optimized protocol for NK cell expansion, NK cells purified from buffy coats of healthy donors were cultured with IL-2 in the presence or absence of irradiated EBV-LCL feeder cells with or without IL-21. Low NK cell expansion was observed in the absence of feeder cells despite the addition of IL-21 (). Irradiated EBV-LCL and IL-2 induced a 22-fold mean NK cell expansion after one week that was further increased to 53-fold by adding IL-21 to the medium. To test whether addition of IL-21 affected the proliferation of NK cells, we performed a proliferation assay by monitoring the CellTrace Violet dye dilution. Of note, NK cells did not start to proliferate until day 3 after initiation of culture (). Thereafter, IL-21 enhanced the proliferation of NK cells in the presence of irradiated EBV-LCL, while IL-21 had no effect on the proliferation of NK cells by itself when feeder cells were absent. Furthermore, there was a pronounced positive correlation between the concentration of supplemented IL-21 and the increasing expansion of NK cells in co-culture with irradiated EBV-LCL feeder cells (). Intriguingly, we observed that it was sufficient to add IL-21 only at the beginning of the culture to drive EBV-LCL-mediated NK cell expansion. Culturing in medium supplemented continuously with IL-21 did not enhance NK cell expansion compared to when IL-21 was added only at the beginning of the culture. Further, permanently adding IL-21 to the culture actually resulted in lower NK cell numbers after prolonged culture compared to an initial single exposure to IL-21 ().
Figure 1. The combination of IL-21 supplementation at start of cultivation, IL-2 and irradiated EBV-LCL feeder cells enabled a long-lasting, pronounced expansion of NK cells. (A) NK cells were cultivated with IL-2 (black bars) or with IL-2 and 100 ng/mL IL-21 (gray bars) in the presence or absence of irradiated EBV-LCL feeder cells. The expansion of NK cells was measured after 7 d. Displayed are mean values and standard deviation of eight NK cell donors and statistical significance was tested by paired Student's t-test. (B) NK cells were labeled with CellTrace Violet Proliferation Dye and cultivated with IL-2 (dark gray histograms) or IL-2 and 100 ng/mL IL-21 (light gray histograms) in the presence or absence of irradiated EBV-LCL feeder cells. The dilution of CellTrace Violet corresponding to cell proliferation was analyzed by flow cytometry at day 3 and day 5. A representative donor out of three donors is shown. (C) NK cells were co-cultured with irradiated EBV-LCL feeder cells and IL-2 at different concentrations of IL-21. Mean NK cell fold expansion and standard deviation are displayed for three donors. (D) NK cells were co-cultured with irradiated EBV-LCL feeder cells and IL-2. IL-21 (100 ng/mL) was supplemented only at day 0 (gray dots) or permanently (gray squares), meaning at day 0, 7, 9, 11, 13, 16, 18, 20, and 26 when fresh medium was added. Mean NK cell expansion fold and standard deviation of six donors are shown at different time points. (E) NK cells were cultured with IL-2 and 100 ng/mL IL-21 was added at day 0. NK cells were co-cultured with irradiated EBV-LCL and re-stimulated with EBV-LCL at day 13, 26, and 39 (gray line and dots). In addition, the expansion without EBV-LCL restimulation at day 13, 26, or 29 was determined and monitored for 7 d (gray dashed line and triangles). Shown are mean and range of the NK cell expansion folds of six donors at different time points.
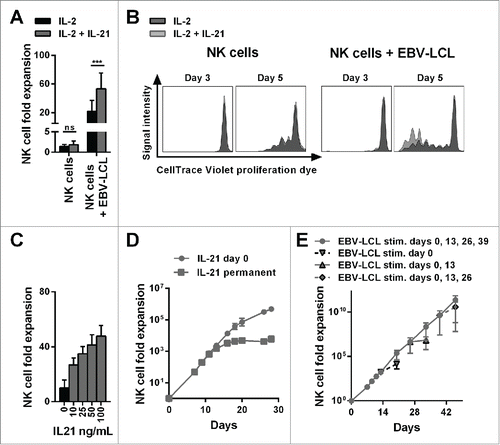
Nevertheless, under all conditions the rate of proliferation of NK cells declined after 2 weeks, limiting the ex vivo time period during which NK cell expansion occurred to 2–4 weeks, precluding further NK cell expansion past this time point (). This limitation could be overcome by adding of IL-21 at the start of culturing followed by repeated stimulation with irradiated EBV-LCL feeder cells every 2 weeks. This allowed for the sustained expansion of NK cells for a longer time period, with a 2.7 × 1011-fold mean increase in NK cell numbers being achievable after 46 d (). Importantly, at later time points, NK cell expansion declined without repeated EBV-LCL stimulations, indicating that NK cells had not acquired an uncontrolled ability to proliferate, which would raise safety concerns for their clinical application. Taken together, we developed a highly efficient method for expansion of NK cells using repeated exposure to irradiated EBV-LCL feeder cells in the presence of IL-2 and a single dose IL-21 supplementation at the start of culture. This protocol, which results in superior NK cell expansion than using EBV-LCL and IL-2 alone, is further referred as the “optimized expansion method.”
Ex vivo-expanded NK cells are highly functional in vitro
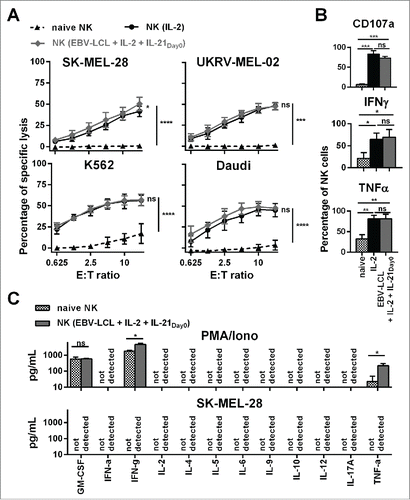
Next, we investigated the functional competence of ex vivo-expanded NK cells in vitro. The optimized expansion method generated NK cells that were highly cytotoxic in a dose-dependent manner against four leukemic and melanoma target cell lines (). To date, IL-2 activated NK cells have been explored most extensively in the clinic and have proven clinical applicability.Citation4 NK cells generated by the optimized expansion method showed similar comparable cytolytic activity as IL-2 activated NK cells that were cultured without feeder cells. In contrast, freshly isolated, naive NK cells possessed only low cytotoxicity against K562 and Daudi leukemic cells and lacked cytotoxicity against SK-MEL-28 and UKRV-Mel-02 melanoma cells, confirming that ex vivo culturing led to NK cell activation and enhanced NK cell-mediated antitumor activity. Compared to naive NK cells, ex vivo-expanded NK cells displayed a significantly higher degranulation and the intracellular production of the immune stimulatory cytokines IFNγ and TNF-α was significantly increased upon stimulation with phorbol myristate acetate (PMA) and ionomycin (Iono) (). Of note, when we stimulated the expanded NK cells with SK-MEL-28 melanoma cells, that are later used in in vivo experiments, neither IFNγ nor TNF-α were detectable intracellularly (data not shown). Furthermore, upon stimulation with PMA/Iono, but not with SK-MEL-28 melanoma cells, expanded NK cells secreted GM-CSF, and elevated levels of IFNγ and TNF-α, whereas NK cells did not produce detectable levels of IFN-α, IL-2, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12, and IL-17A as determined by cytokine bead array (). Thus, the expanded NK cells show an increased potential to produce cytokines, as observed after PMA/Iono stimulation. However, SK-MEL-28 target cells did not elicit cytokine production.
Figure 2. Ex vivo-expanded NK cells were highly cytotoxic against different tumor cell lines and showed enhanced degranulation and production of IFNγ and TNF-α. (A) Different NK cells were tested for cytotoxicity against four tumor cell lines using a standard 51Chromium release assay. Specific lysis at different effector-to-target (E:T) ratios is shown for freshly isolated NK cells (black triangles and dashed line) and NK cells that have been expanded for 13 or 14 d, either with IL-2 (back line and dots) or by use of IL-2, irradiated EBV-LCL and IL-21 supplemented at day 0 (gray line and diamonds). Displayed are mean values and standard deviation of NK cells from 4–8 donors per target cell line and statistical significance was tested by Student's t-test. (B) NK cells were tested for degranulation and production of IFNγ and TNF-α by flow cytometry upon stimulation with PMA/Iono. Shown are data for freshly isolated, naive NK cells (gray and white dotted bars) and NK cells that have been expanded for 13 or 14 d, either with IL-2 (black bars) or by use of IL-2, irradiated EBV-LCL and IL-21 supplementation at day 0 (gray bars). Displayed are mean values and standard deviation of NK cells from five donors. Statistical significance was tested by paired Student's t-test. (C) NK cells were expanded ex vivo for 14 d by use of IL-2, irradiated EBV-LCL and IL-21 supplementation at day 0. Then, NK cells were stimulated for 24 h with PMA/Iono or by co-culture with SK-MEL-28 cells. Supernatants of the cell cultures were harvested and analyzed for the concentrations of 12 different cytokines using a multiplex bead-array assay. Mean and standard deviation for NK cells from three different donors are displayed. Statistical significance was tested by paired Student's t-test.
In conclusion, our optimized expansion protocol not only increased the numbers of NK cells, but also enhanced their in vitro antitumor activity compared to freshly isolated NK cells from peripheral blood.
Ex vivo NK cells expanded by the optimized method control tumor growth in vivo
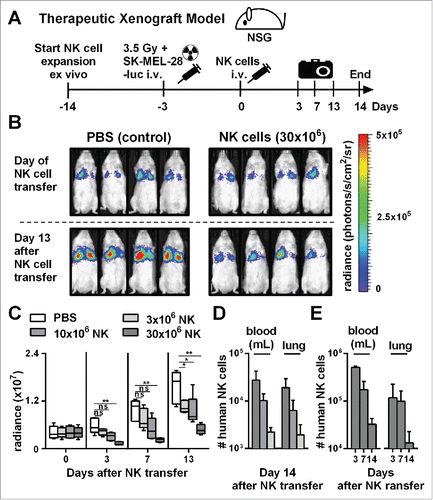
Next, we evaluated the antitumor activity of NK cells obtained by our optimized protocol in an in vivo therapeutic xenograft mouse model (). We established human melanoma-derived metastases in the lungs of immunocompromised NOD-scid IL-2Rgammanull (NSG) mice by intravenous injection of human melanoma cells (SK-MEL-28) expressing luciferase (, top). Successful engraftment of the human tumor cells was confirmed by luciferase activity in the lungs 3 d after injection. Next, we injected either phosphate-buffer saline (PBS), as a control, or NK cells that were expanded using the optimized expansion method into tumor-bearing mice. IL-2 was injected repeatedly to support the in vivo persistence of transferred NK cells. Single doses of up to 30 × 106 NK cells per mouse were administered and were tolerated without any noticeable side effects. In the control group without transfer of NK cells, the tumor load significantly increased within the 2 weeks follow-up period (, bottom). In contrast, treatment with expanded NK cells significantly controlled tumor growth, indicating a potent antitumor activity of the ex vivo generated NK cells in vivo. Furthermore, there appeared to be a dose response relationship between the number of injected NK cells and the control of tumor growth, with the highest dose of 30 × 106 injected NK cells showing the best therapeutic effect (). Injection of higher human NK cell numbers also correlated with higher numbers of NK cells that could be retrieved from blood and lungs 14 d after NK cell transfer (). Nevertheless, NK cell numbers continuously declined after adoptive transfer (), indicating that transferred NK cells were unable to sustain continuous expansion in vivo. In conclusion, adoptive transfer of NK cells generated with the optimized protocol resulted in efficient control of tumor in a xenograft mouse model without noticeable side effects.
Figure 3. Adoptive transfer of NK cells expanded by the optimized protocol in tumor-bearing mice resulted in pronounced tumor growth control. (A) Scheme is shown for evaluation of expanded NK cells in vivo using a xenograft model. Mice were irradiated and received human SK-MEL-28 melanoma cells-expressing luciferase by intravenous (i.v.) injection. Three days later, after tumor engraftment, the mice were treated (i.v.) with human NK cells expanded with the optimized protocol and the tumor load was monitored by luciferase activity. IL-2 was repeatedly injected intraperitoneally. (B) Tumor-bearing mice, as described in A, were treated with PBS, as a control, or with 30 × 106 NK cells that were expanded for 14 d using the optimized expansion protocol. The pictures display the bioluminescence (radiance) showing the in vivo luciferase activity of four representative mice of each group at the day of NK cell transfer (top) and 13 d thereafter (bottom). (C) Tumor-bearing mice were treated with NK cells as described in B using different NK cell doses. Mean and range of the tumor burden, measured by bioluminescence, is shown at different time points for one representative experiment with 4–5 mice per group. (D) Mice were treated as described in C, and the transferred human NK cells were re-isolated from blood and lungs of the mice 14 d after NK cell injection and were enumerated using flow cytometry. Mean and standard deviation of NK cell numbers per lung and per mL of blood are shown for four mice per group. (E) Tumor-bearing mice were treated with 30 × 106 expanded NK cells as described in B and at day 3, 7, and 14 the mice were sacrificed and human NK cells were re-isolated from blood and lungs. Mean and standard deviation of NK cell numbers per lung or mL of blood are shown for four mice per group. Statistical significance in all experiments was tested by Student's t-test.
Ex vivo-expanded NK cells change their phenotype and function in vivo
Next, we monitored changes in phenotype and function of ex vivo-expanded NK cells after adoptive transfer into tumor-bearing animals. NK cells isolated from the lungs shortly after adoptive transfer were compared to naive NK cells and ex vivo-expanded NK cells that remained in culture (). Based on our recently published work, showing that NK cells greatly change at gene expression and phenotype level upon ex vivo activation,Citation7 we selected TRAIL, DNAM-1, and NKG2D surface markers to assess NK cell activation. We observed that ex vivo-expanded and activated NK cells exhibited upregulated TRAIL, DNAM-1, and NKG2D compared to naive NK cells (). Intriguingly, 3 d after adoptive transfer of ex vivo-expanded NK cells, these cells expressed again low levels of TRAIL, DNAM-1, and NKG2D similar to naive NK cells.
Figure 4. Already three days after transfer into NSG mice, ex vivo-expanded NK cells changed their phenotype and lost their cytotoxicity and potential for degranulation, while they retained an enhanced ability to produce IFNγ and TNF-α. (A) Scheme for characterization of three types of NK cells from the same donor. First, freshly isolated, naive NK cells were analyzed (black). Second, NK cells were expanded ex vivo for 17 d by use of short-term IL-21 stimulation, IL-2 and irradiated EBV-LCL feeder cells (red). Third, NK cells were expanded in the same way for 14 d before they were transferred to tumor-bearing mice (30 × 106 NK cells per mouse) (blue). Three days after the transfer, the transferred human NK cells were recovered from the mouse lungs and NK cells from 3–4 mice were pooled per donor (blue). (B) The differentially prepared NK cells, as described in A, were analyzed for the surface markers NKG2D, TRAIL, and DNAM-1 by flow cytometry. Histograms for one representative NK cell donor are shown (back) together with isotype controls (gray). (C) The flow cytometric analysis, as described in B, is applied for three different donors. For each marker the mean and standard deviation of all donors are shown for the mean fluorescence intensity (MFI) corrected by isotype subtraction (top) and the frequency of NK cells expressing the marker (bottom). (D) The differentially prepared NK cells, as described in A, were analyzed for cytotoxicity against K562 and SK-MEL-28 target cell lines at a 3:1 effector-to-target ratio in triplicates per donor. One out of two representative experiments is shown using two different NK cell donors. (E) The differentially prepared NK cells, as described in A, were analyzed for degranulation and production of IFNγ and TNF-α upon stimulation with PMA/Iono. Mean and standard deviation of three different NK cell donors are displayed. Statistical significance was tested by paired Student's t-test in all experiments.
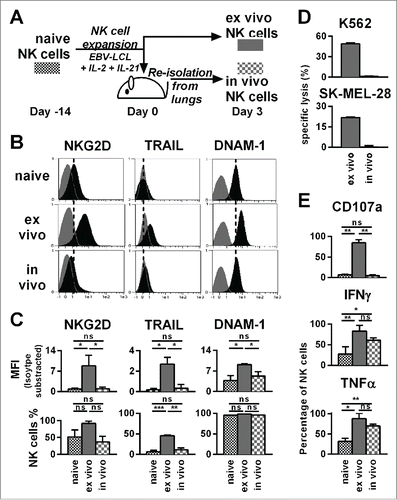
Next, we tested the functional activity of NK cells that were re-isolated from mice following their adoptive transfer. As expected from the low expression of TRAIL, DNAM-1, and NKG2D, NK cells retrieved from mice 3 d after adoptive transfer had low cytotoxicity against SK-MEL-28 and K562 target cells, compared to NK cells expanded from the same donors that were maintained in ex vivo cell culture (). In line with the reduced cytotoxicity, re-isolated NK cells had a diminished potential for degranulation compared to expanded NK cells that were maintained in ex vivo cell culture. Strikingly, the ability of ex vivo activated NK cells to produce IFNγ and TNF-α was retained after the in vivo transfer and remained at significantly higher levels than that observed with naive NK cells. Of note, similar data obtained with NK cells isolated from lungs as shown in were obtained with NK cells isolated from blood (data not shown). Taken together, although the increased potential for degranulation and direct killing of tumor cells might be short lived in vivo, our data suggested that NK cells generated by the optimized protocol had sustained competence for cytokine production in vivo.
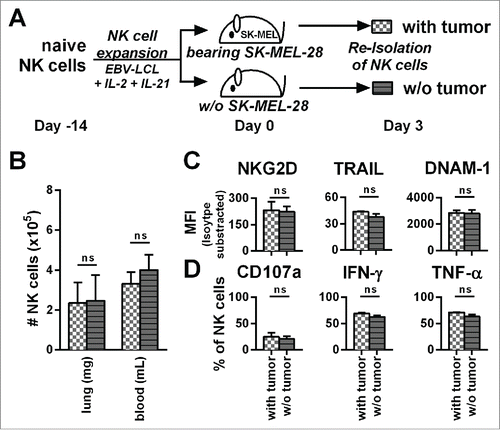
The number, phenotype, and function of transferred NK cells is not affected by SK-MEL-28 in vivo
Expanded NK cells significantly changed their function and phenotype after transfer into tumor-bearing mice (), which could result from the presence of tumor in our in vivo model. To address this point, we analyzed NK cells after adoptive transfer into tumor-bearing mice in comparison to NK cells that were injected into mice without established tumors (PBS control) (). Three days after adoptive NK cell transfer, the numbers of human NK cells were comparable in the blood and the lungs of the mice, independent of the presence or absence of tumor (). In addition, the presence of tumor affected neither NK cell expression of NKG2D, TRAIL, and DNAM-1 () nor their degranulation, nor their production of IFNγ and TNF-α in response to PMA/Iono (). In conclusion, number, phenotype, or function of the transferred NK cells were similar regardless of the presence of tumor in vivo.
Figure 5. The presence of tumor does not influence the phenotype, potential for degranulation, and cytokine production of adoptively transferred NK cells. (A) A scheme is shown for evaluation of expanded NK cells in vivo. Mice were irradiated and received human SK-MEL-28 melanoma cells (with tumor) or PBS (w/o tumor) by intravenous (i.v.) injection. Three days later after establishment of tumor, all mice were treated (i.v.) with human NK cells expanded with the optimized protocol. Three days after transfer of the NK cells, cells from the blood and lungs of the mice were recovered. (B) NK cells from the blood and lungs were enumerated and NK cell numbers per g of lung and per mL of blood are shown. (C) NK cells from mouse lungs as described in A were analyzed for the surface markers NKG2D, TRAIL, and DNAM-1 by flow cytometry and shown are the mean fluorescence intensity (MFI) corrected by isotype subtraction. (D) NK cells as described in A were stimulated with PMA/Iono and analyzed by flow cytometry for degranulation and production of IFNγ and TNF-α. Means and standard deviations for NK cells from three independent donors are shown with cells from 3–5 mice that were pooled per condition for each donor. Statistical significance was tested by paired Student's t-test.
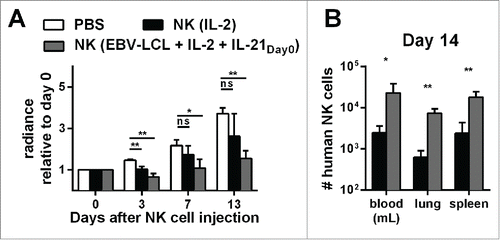
NK cells generated with the optimized expansion protocol display high antitumor activity and persistence in vivo
Finally, we compared the in vivo antitumor activity of NK cells that were generated with the optimized expansion protocol to NK cells that were activated long-term with IL-2 alone without feeder cells. For both protocols, we applied a high number of 30 × 106 NK cells per mouse. Assuming an average weight of 30 g per mouse, the dose corresponded to 109 NK cells per kg. Both protocols were suitable to obtain this high NK cell dose for use in this mouse model, even though expansion with IL-2 without feeder cells resulted in a minor NK cell expansion of 2- to 10-fold, whereas the optimized expansion protocol yielded a mean 2,900-fold NK cell expansion after 2 weeks. However, it is important to clarify that only the optimized expansion method would be able to provide this dose of NK cells for humans receiving this type of therapy. As shown before, the optimized expansion protocol led to NK cells that were able to control tumor growth also at later time points. In contrast, even though injection of conventional IL-2 activated NK cells resulted in a similar antitumor effect shortly after the transfer, it was less effective at later time points (). Importantly, despite infusing identical numbers of IL-2 activated and optimally expanded NK cells, we found a striking difference in the numbers of NK cells that could be isolated from the mice 2 weeks after the transfer. On day 14 after injection, the numbers of NK cells in blood, lungs, and spleen were around 10 times lower for IL-2 activated NK cells compared to NK cells that have been expanded with the optimized expansion method (). These data indicate that NK cells that were generated with the optimized protocol exhibited enhanced in vivo persistence and significantly controlled tumor growth at later time points.
Figure 6. Expansion of NK cells using the optimized protocol (IL-21 day0, IL-2 and irradiated EBV-LCL) results in NK cells with better in vivo persistence and antitumor activity compared to IL-2-expanded NK cells. (A) Mice-bearing SK-MEL-28 cells expressing luciferase either received intravenous (i.v.) PBS (white bars) or human NK cells. The injected NK cells were previously expanded ex vivo for 14 d, either with IL-2 (black bars) or with IL-2, irradiated EBV-LCL and IL-21 supplemented at day 0 (gray bars). In total, three independent NK cell donors were used in different experiments and the bioluminescence (radiance) of each mouse at day 3, 7, and 13 was measured and analyzed relative to the bioluminescence at day 0. For each donor, 2–8 mice were used per group, the mean and standard deviation of all donors are shown at different time points. (B) Tumor-bearing mice were treated as described in A and human NK cells were re-isolated from blood, lungs, and spleen of the mice 14 d after NK cell injection. NK cells were enumerated using flow cytometry and mean and standard deviation of NK cell numbers are shown for one representative donor using four mice per group. Statistical significance was tested by Student's t-test in all experiments.
In summary, we developed a highly efficient method to expand NK cells ex vivo that allows large numbers of NK cells to be obtained for use in clinical trials. Besides the increased cell numbers, these expanded NK cells exhibited pronounced antitumor activity in vitro. Most importantly, treatment of tumor-bearing mice with expanded NK cells resulted in control of tumor growth, which was dependent on the dose and persistence of the applied NK cells, indicating the therapeutic value of infusing large numbers of expanded NK cells in humans with cancers.
Discussion
The ability to expand ex vivo NK cells to large numbers for adoptive transfer in humans remains a major challenge for NK cell-based immunotherapy. Co-cultivation of NK cells with clinical-grade irradiated EBV-LCL feeder cells is an attractive NK cell expansion strategy, but is restricted to a relatively limited period of 2–4 weeks when expansion occurs. Here, we demonstrate for the first time that the expansion of NK cells from peripheral blood by use of clinical-grade irradiated EBV-LCL feeder cells can be greatly increased by one single initial addition of IL-21 into the culture medium resulting in a long-lasting highly efficient proliferation of NK cells with potent in vitro and in vivo antitumor activity.
Repeated stimulation with K562 feeder cells-bearing membrane bound IL-21 has been reported to facilitate a long-term expansion of NK cells, allowing around 108-fold NK cell expansion after 6 weeks.Citation9 In our study, we reached a much higher 1011-fold NK cell expansion after 6 weeks by combining repeated stimulation with irradiated EBV-LCL feeder cells with IL-21 addition at the initiation of culture. We observed that it was sufficient to supplement soluble IL-21 only at start of cultivation to achieve high expansion of NK cells using stimulation with irradiated EBV-LCL. Our data are in concordance with a publication showing exposure time of soluble IL-21 is critical for the yield of NK cells during co-culture with K562 feeder cells-expressing membrane bound IL-15.Citation16 Importantly, IL-21 has been shown to cause apoptosis of NK cells, which is enhanced when this cytokine is membrane bound compared to when it is in its soluble state.Citation17 This implies that stimulation of NK cells using feeder cells expressing membrane bound IL-21 may have disadvantages compared to the approach we utilized, where IL-21 was incorporated only briefly into the media at the start of cell culture. The combination of repeated stimulation with clinical-grade, irradiated EBV-LCL feeder cells and the short-term presence of soluble IL-21 in the medium allows to expand NK cells with a very high efficiency, which is, to the best of our knowledge, not reached by other methods so far.
Unlike other effector cells, NK cells can be applied not only in an autologous but also allogeneic setting, and receptor-ligand mismatches of donor and recipient may support a better NK cell versus tumor effect without inducing unwanted toxicities.Citation18 Thus, at some stage, with this optimized expansion method enabling the production of activated NK cells to an industrial scale, NK cell products may become a universally applicable off-the-shelf product. Until now, this has only been possible by using continuously growing NK cell lines, like NK-92, that have proven applicability in mouse studies and in pilot trials with cancer patients.Citation19,20 However, due to safety concerns, NK cell lines require proper inactivation by irradiation prior to infusion in the patient, preventing their ability to proliferate in vivo which could potentially compromise their therapeutic potential. To avoid this major drawback of NK cell lines, it would be advantageous to use primary NK cells and expand them to very high cell numbers using our optimized method to manufacture off-the-shelf NK cell products that are suitable for use in protocols treating a variety of different cancer patients.
Importantly, cultivation of NK cells ex vivo not only increases the NK cell numbers but also serves the important role of enhancing their antitumor activity prior to adoptive transfer.Citation21,22 In a previous publication, we showed that NK cells expanded with IL-2 and EBV-LCLs and NK cells activated with IL-2 alone showed a comparable profile for gene expression, phenotype and function in vitro after a cultivation period of 14 d.Citation7 Similarly, our optimized expansion method and activation with IL-2 alone both yielded NK cells with improved in vitro functionality, compared to naive NK cells. Nevertheless, NK cells expanded with our optimized method showed more pronounced antitumor activity in a mouse model and superior persistence in vivo compared to conventional IL-2 activated NK cells. To maximize this antitumor effect, we needed to inject up to 30 × 106 NK cells per mouse, corresponding to 109 NK cells/kg, which is in the upper range of considered doses for studies in humans, but which may not be obtainable with conventional NK cell expansion protocols.Citation8 Our NK cell expansion method is capable of providing these high numbers of clinical-grade NK cells. Importantly, the results of our xenograft model indeed support the rationale for transferring preferably high NK cell doses to maximize their therapeutic effect.
To maintain sustained activation and expansion of NK cells in vivo, injection of low dose IL-2 has frequently been utilized in clinical studies.Citation23-25 However, despite treatment with low dose IL-2 in our animal model, we did not detect noticeable in vivo expansion, nor did we observe maintenance of phenotypic markers of activation on transferred NK cells. In this context, a suppressive effect of the tumor on the transferred NK cells can be excluded in our model as the number, phenotype, and function of the transferred NK cells was not affected by the presence of the tumor in vivo. Possibly, the great quantity of transferred NK cells caused a very high demand for IL-2 that could not be obtained by the low dose of administered IL-2, explaining the observed decline of NK cells over time in vivo. Of note, it was published recently that, in contrast to IL-15, IL-2 was inefficient to promote in vivo NK cell expansion in a similar xenograft model.Citation26 Therefore, IL-15 may be a better cytokine to improve NK cell persistence, which also has the added benefit of avoiding unfavorable proliferation of regulatory T cells which occurs commonly with IL-2.Citation27,28
Although extending the lifespan of adoptively transferred effector cells seems desirable,Citation29 the limited in vivo persistence and the short life span of transferred NK cells could have benefits compared to T cells. Adoptive therapy using T cells expressing chimeric antigen receptors (CARs) currently represents a breakthrough for cancer immunotherapy, but the longevity of T cells may cause severe and potentially life-long side effects, such as B cell aplasia upon infusion of anti-CD20 CAR T cells, which could be avoided by the use of short-lived CAR-expressing NK cells, potentially making them better controlled, potentially superior “car drivers.”Citation30
Furthermore, repeated administration of ex vivo-expanded NK cells could be sufficient for inducing a long-lasting antitumor response, since it has been shown in mice that repeated adoptive transfer of NK cells can trigger tumor-specific endogenous memory T cell responses as antigen-presenting cells or through cross-talk with dendritic cells.Citation31 In this context, our optimized expansion protocol would enable permanent availability of NK cells allowing for repeated transfer of NK effector cells multiple times whenever required during the treatment.
Expanded NK cells had an increased potential to produce IFNγ and TNF-α that was retained after adoptive transfer. However, the role of NK cell-derived cytokines for the therapeutic effect in our in vivo model remains unclear, since the used tumor cells did not directly stimulate NK cells cytokine secretion in ex vivo assays. Nevertheless, NK cell cytokine secretion may be induced by other immune cells or soluble factors in vivo that are lacking in our xenograft model. In combination, IFNγ and TNF-α can induce permanent growth arrest in numerous human cancers,Citation32 and IFNγ and TNF-α are essential for destroying established tumors in mice by eradication of tumor-associated stroma cells.Citation33 Accordingly, in a therapeutic mouse model of RMA-S lymphoma, the production of IFNγ by transferred NK cells was essential for their tumor growth control.Citation34
In conclusion, we developed an optimized method for ex vivo expansion of primary human NK cells with outstanding cell yields. These expanded NK cells possess potent antitumor activity in vitro and in vivo. Therefore, the method has the potential to produce large numbers of highly functional antitumor NK effector cells from a single donor, suited for the use in clinical trials exploring adoptive NK cell immunotherapy for patients with a variety of different neoplastic diseases.
Material and methods
Cells, cell lines, and mice
Primary NK cells were obtained from healthy donor buffy coats (Klinikum Dortmund or DRK-Blutspendedienst). The EBV-LCL (SMI-EBV-LCL) line was kindly provided by Dr. Richard W. Childs (National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD, USA). K562, SK-MEL-28, UKRV-MEL-02, and Daudi cell lines were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium (Biowest) supplemented with 10% fetal bovine serum (Biochrom) and 2 mmol/L L-glutamine (PAA). SK-MEL-28 melanoma cells expressing the green click beetle-luciferase CBGr99 (SK-MEL-28-luc) were generated by retroviral transduction using the pCMV-eGFP-2A-CBGr99 plasmid (DKFZ, Heidelberg), containing the expression cassette from the pCMV-eGFP-2A-CBGr99 plasmid (pGL-2A, DKFZ, Heidelberg) and pBabe-puro as backbone.
NOD-scid IL-2Rgammanull (NSG) mice were bred at the DKFZ animal facility. Mice were housed under specific pathogen-free conditions and in accordance with all standards of animal care. All animal experiments were approved by the “Regierungspräsidium Karlsruhe.”
Antibodies and flow cytometric analysis
All antibodies were obtained from Miltenyi: CD56 (REAfinity antibody [REA]196) was conjugated with allophycocyanin (APC)-Vio770. CD3 (BW264/56) was conjugated with VioBlue. CD45 (5B1) was conjugated with VioGreen. Interferon(IFN)γ (45–15) and CD314 (BAT221) were conjugated with fluorescein isothiocyanate (FITC). Tumor necrosis factor (TNF)-α (cA2) and CD253 (RIK-2.1) were conjugated with phycoerythrin (PE). CD226 (DX11), and CD107a (H4A3) were conjugated with APC. To analyze the cell proliferation by dye dilution, NK cells were labeled with CellTrace Violet (Life Technologies) prior to cultivation. Cells were stained according to the product manual and analyzed by use of the MACSQuant Analyzer 10 or BD FACSCanto™. Dead cells were excluded from the analysis by means of propidium iodide staining or by using Zombie Aqua™ Fixable Viability Kit (Biolegend). Mouse immunoglobulin (Ig)G1, IgG2a, or REAs conjugated with the respective dyes were used as isotype controls.
Isolation of NK cells from buffy coats
For NK cell isolation from buffy coats, peripheral blood mononuclear cell (PBMC) preparation was performed by standard density-gradient centrifugation by means of Pancoll (PAN-Biotec). NK cell isolation kit human (Miltenyi) was applied for untouched enrichment of NK cells from PBMC. This protocol resulted typically in >95 % CD56+/CD3− NK cells.
Expansion of NK cells
NK cells were cultivated in TexMACS Research Medium (Miltenyi) supplemented with 5% human serum type AB (Life Technologies) and 500 U/mL of IL-2 (Proleukin S, Novartis), either together with 100 Gy irradiated EBV-LCL at a ratio of 1:20 and a starting concentration of 5.25 × 105 total cells/mL or without feeder cells at a seeding density of 5 × 105 cells/mL. Under some conditions, human IL-21 (premium grade, Miltenyi) was added to the medium as stated. The NK cell density was checked during cultivation by staining and counting viable CD3−/CD56+ cells by means of the MACSQuant Analyzer 10. Seven days after start of cultivation fresh medium was added to double the culture volume and every second to fifth day thereafter fresh medium was added to dilute the cell density to 5–8 × 105 NK cells/mL. Expanded NK cells were re-stimulated at later time points in some experiments by co-culturing the expanded NK cells again with irradiated EBV-LCL at a ratio of 1:20 at a cell concentration of 5.25 × 105 total cells/mL. For practical reasons, only a fraction of the cells was kept in culture over longer time and the NK cell expansion fold was determined by dividing the theoretical NK cell number at the time point of interest by the NK cell number at start of cultivation.
Analysis of cytokine production and degranulation by flow cytometry
Cytokine production and degranulation were analyzed by flow cytometry. NK cells (105/mL) were seeded in 96-well round-bottom plates in RPMI1640 supplemented with CD107a-APC and GolgiStop (BD Biosciences) according to the user manuals. NK cells were stimulated with 50 ng/mL PMA (Sigma-Aldrich) and 0.5 µM Iono (Sigma-Aldrich) or left untreated as a control. After 2 h, the cells were fixed, permeabilized and stained for IFNγ, TNF-α, and CD56 (Inside Stain Kit, Miltenyi).
Multiplex bead-array assay
To quantify cytokines in culture supernatants, a multiplex bead-array assay was used. NK cells (5 × 105/mL) were cultured in RPMI1640 with either 50 ng/mL PMA and 0.5 µM Iono or with SK-MEL-28 cells (5 × 105/mL). After 24 h, cell-free supernatants were taken and stored at −20°C until analysis. Analysis were performed using MACSplex cytokine 12-kit, human (Miltenyi) according to the product manual.
Cytotoxicity assay
To test cytotoxicity, a standard 51Cr- release assay was performed. Target cells were labeled with 51Cr, seeded at 3 × 104 cells/mL in 96-well round-bottom plates and co-incubated for 4 h with medium or NK effector cells or medium containing 10% Triton x-100 (Sigma-Aldrich). The percentage of specific lysis was calculated from counts for 51Cr release as follows: ([NK cells – medium]/[Triton x-100 – medium] × 100)
Xenograft mouse model
To evaluate for NK cell antitumor activity in vivo, tumors with human origin were established in NSG mice. NSG mice (8 weeks old) were irradiated (3.5 Gy) to support optimal engraftment of xenogenic cells. Three to five hours after irradiation, the mice received 5 × 105 luciferase transfected SK-MEL-28 melanoma cells (SK-MEL-28-luc) through tail vein injection. Three days later, after successful engraftment of the human tumor cells, NK cells or PBS were injected via tail vein. In addition, mice received 10,000 units IL-2 per mice by intraperitoneal (i.p.) injection when NK cells or PBS was injected and 1, 2, 3, 5, 7, and 11 d thereafter. To estimate the tumor burden, 4.5 mg StayBrite D-Luciferin (BioVision) was injected i.p. in anesthetized mice and the luciferase activity of engrafted tumor cells was measured using the IVIS imaging system-100 or IVIS lumina series III (Perkin-Elmer). All animal experiments were approved by the “Regierungspräsidium Karlsruhe.”
Re-isolation of human NK cells from mouse blood, lung, and spleen
NSG mice were sacrificed at day 3, 7, or 14 after injection of NK cells or PBS and single-cell suspensions were prepared from blood and lungs. Blood cells were obtained after standard red blood cell lysis using buffered ammonium chloride potassium phosphate solution (ACK buffer) for 10 min at room temperature. Lung and spleen tissues were incubated for 30 min in digestion buffer containing 0.5 mg/mL of each DNAse I and Hyaluronidase (Sigma-Aldrich) followed by density gradient centrifugation using Lympholyte-M (Cedarlane). Re-isolated NK cells were enumerated by means of the MACSQuant Analyzer 10 and gating on human markers for CD45+/ CD56+/CD3− cells.
Statistics and data analysis
Statistical comparisons were performed by means of unpaired or paired two-tailed Student's t-test as indicated. p < 0.05, p < 0.005, p < 0.0005, or p < 0.00005 is considered statistically significant and is indicated with one, two, three, or four asterisks, while “ns” displays no significance. Data analysis was performed using following software: GraphPad Prism 6.02, Microsoft Excel 2007, MACSQuantify 2.5 and 2.6, FlowJo 9.8, Living Image 2.5.
Disclosure of potential conflicts of interest
MG and VH are employees of Miltenyi Biotec. All authors declare no conflicts of interest.
Acknowledgments
We thank Maria Berg, Annette Arnold, Maria Delso Vallejo, Sabine Müller, Andrea Guter, Jens Pahl, Margareta Correia, Jing Ni, Thomas Walle, Eva-Maria Ewen, Julia Pollmann, Katharina Bauer, the DKFZ animal facility, and in particular Annette Seibel, Steffen Schmitt, Barbara Kahn, Viktor Umansky, Stefan Eichmüller, Jürgen Schmitz and Stefan Miltenyi for support, critical feedback, or/and technical assistance in enabling this work. We thank Günter Küblbeck for kindly providing the pCMV-eGFP-2A-CBGr99 plasmid.
References
- Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol 2013; 10:230-52; PMID:23604045; http://dx.doi.org/10.1038/cmi.2013.10
- Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, Pui C-H, Leung W. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol 2010; 28:955-9; PMID:20085940; http://dx.doi.org/10.1200/JCO.2009.24.4590
- Klingemann HG. Cellular therapy of cancer with natural killer cells-where do we stand? Cytotherapy 2013; 15:1185-94; PMID:23768925; http://dx.doi.org/10.1016/j.jcyt.2013.03.011
- Koehl U, Sörensen J, Esser R, Zimmermann S, Grüttner HP, Tonn T, Seidl C, Seifried E, Klingebiel T, Schwabe D. IL-2 activated NK cell immunotherapy of three children after haploidentical stem cell transplantation. Blood Cells Mol Dis 2004; 33:261-6; PMID:15528141; http://dx.doi.org/10.1016/j.bcmd.2004.08.013
- Klingemann HG, Martinson J. Ex vivo expansion of natural killer cells for clinical applications. Cytotherapy 2004; 6:15-22; PMID:14985163; http://dx.doi.org/10.1080/14653240310004548
- Berg M, Lundqvist A, McCoy P Jr, Samsel L, Fan Y, Tawab A, Childs R. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy 2009; 11:341-55; PMID:19308771; http://dx.doi.org/10.1080/14653240902807034
- Granzin M, Soltenborn S, Müller S, Kollet J, Berg M, Cerwenka A, Childs RW, Huppert V. Fully automated expansion and activation of clinical-grade natural killer cells for adoptive immunotherapy. Cytotherapy 2015; 17:621-32; PMID:25881519; http://dx.doi.org/10.1016/j.jcyt.2015.03.611
- Childs RW, Berg M. Bringing natural killer cells to the clinic: ex vivo manipulation. Hematology Am Soc Hematol Educ Program 2013; 2013:234-46; PMID:24319186; http://dx.doi.org/10.1182/asheducation-2013.1.234
- Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, Singh H, Hurton L, Maiti SN, Huls MH et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS One 2012; 7:e30264; PMID:22279576; http://dx.doi.org/10.1371/journal.pone.0030264
- Parrish-novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, Johnston J, Madden K, Xu W, West J et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature 2000; 408:57-63; PMID:11081504; http://dx.doi.org/10.1038/35040504
- Davis MR, Zhu Z, Hansen DM, Bai Q, Fang Y. The role of IL-21 in immunity and cancer. Cancer Lett 2015; 358:107-14; PMID:25575696; http://dx.doi.org/10.1016/j.canlet.2014.12.047
- Kasaian MT, Whitters MJ, Carter LL, Lowe LD, Jussif JM, Deng B, Johnson KA, Witek JS, Senices M, Konz RF et al. IL-21 limits NK cell responses and promotes antigen-specific T cell activation: A mediator of the transition from innate to adaptive immunity. Immunity 2002; 16:559-69; PMID:11970879; http://dx.doi.org/10.1016/S1074-7613(02)00295-9
- Brady J, Hayakawa Y, Smyth MJ, Nutt SL. IL-21 induces the functional maturation of murine NK cells. J Immunol 2004; 172:2048-58; PMID:14764669; http://dx.doi.org/10.4049/jimmunol.172.4.2048
- Wendt K, Wilk E, Buyny S, Schmidt RE, Jacobs R. Interleukin-21 differentially affects human natural killer cell subsets. Immunology 2007; 122:486-95; PMID:17635612; http://dx.doi.org/10.1111/j.1365-2567.2007.02675.x
- Strbo N, de Armas L, Liu H, Kolber MA, Lichtenheld M, Pahwa S. IL-21 augments natural killer effector functions in chronically HIV-infected individuals. AIDS 2008; 22:1551-60; PMID:18670213; http://dx.doi.org/10.1097/QAD.0b013e3283089367
- Lim DP, Jang YY, Kim S, Koh SS, Lee JJ, Kim JS, Thi Phan MT, Shin DJ, Shin MG, Lee SH et al. Effect of exposure to interleukin-21 at various time points on human natural killer cell culture. Cytotherapy 2014; 16:1419-30; PMID:24950680; http://dx.doi.org/10.1016/j.jcyt.2014.04.008
- Li Q, Ye L-J, Ren H-L, Huyan T, Li J, Shi J-L, Huang Q-S. Multiple effects of IL-21 on human NK cells in ex vivo expansion. Immunobiology 2015; 220:876-88; PMID:25758713; http://dx.doi.org/10.1016/j.imbio.2015.01.009
- Leung W. Infusions of allogeneic natural killer cells as cancer therapy. Clin Cancer Res 2014; 20:3390-400; PMID:24987108; http://dx.doi.org/10.1158/1078-0432.CCR-13-1766
- Tam YK, Miyagawa B, Ho VC, Klingemann HG. Immunotherapy of malignant melanoma in a SCID mouse model using the highly cytotoxic natural killer cell line NK-92. J Hematother 1999; 8:281-90; PMID:10417052; http://dx.doi.org/10.1089/106161299320316
- Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, Klingemann H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy 2008; 10:625-32; PMID:18836917; http://dx.doi.org/10.1080/14653240802301872
- Childs RW, Carlsten M. Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: the force awakens. Nat Rev Drug Discov 2015; 14:487-98; PMID:26000725; http://dx.doi.org/10.1038/nrd4506
- Ni J, Miller M, Stojanovic A, Cerwenka A. Toward the next generation of NK cell-based adoptive cancer immunotherapy. Oncoimmunology 2013; 2:e23811; PMID:23734329; http://dx.doi.org/10.4161/onci.23811
- Szmania S, Lapteva N, Garg T, Greenway A, Lingo J, Nair B, Stone K, Woods E, Khan J, Stivers J et al. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J Immunother 2015; 38:24-36; PMID:25415285; http://dx.doi.org/10.1097/CJI.0000000000000059
- Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, McKenna D, Le C, Defor TE, Burns LJ et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005; 105:3051-7; PMID:15632206; http://dx.doi.org/10.1182/blood-2004-07-2974
- Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, Jonson AL, Panoskaltsis-Mortari A, Cursinger J, McKenna D et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011; 13:98-107; PMID:20849361; http://dx.doi.org/10.3109/14653249.2010.515582
- Miller JS, Rooney CM, Curtsinger J, McElmurry R, McCullar V, Verneris MR, Lapteva N, McKenna D, Wagner JE, Blazar BR et al. Expansion and homing of adoptively transferred human natural killer cells in immunodeficient mice varies with product preparation and in vivo cytokine administration: implications for clinical therapy. Biol Blood Marrow Transplant 2014; 20:1252-7; PMID:24816582; http://dx.doi.org/10.1016/j.bbmt.2014.05.004
- Zhang H, Chua KS, Guimond M, Kapoor V, Brown MV, Fleisher TA, Long LM, Bernstein D, Hill BJ, Douek DC et al. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+CD25+ regulatory T cells. Nat Med 2005; 11:1238-43; PMID:16227988; http://dx.doi.org/10.1038/nm1312
- Bachanova V, Burns LJ, McKenna DH, Curtsinger J, Panoskaltsis-Mortari A, Lindgren BR, Cooley S, Weisdorf D, Miller JS. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol Immunother 2010; 59:1739-44; PMID:20680271; http://dx.doi.org/10.1007/s00262-010-0896-z
- Nayar S, Dasgupta P, Galustian C. Extending the lifespan and efficacies of immune cells used in adoptive transfer for cancer immunotherapies–A review. Oncoimmunology 2015; 4:e1002720; PMID:26155387; http://dx.doi.org/10.1080/2162402X.2014.1002720
- Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology 2014; 3:e28147; PMID:25340009; http://dx.doi.org/10.4161/onci.28147
- Salagianni M, Baxevanis CN, Papamichail M, Perez SA. New insights into the role of NK cells in cancer immunotherapy. Oncoimmunology 2012; 1:205-7; PMID:22720243; http://dx.doi.org/10.4161/onci.1.2.18398
- Braumüller H, Wieder T, Brenner E, Aßmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013; 494:361-5; PMID:23376950; http://dx.doi.org/10.1038/nature11824
- Zhang B, Karrison T, Rowley DA. Schreiber H. IFN-γ- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J Clin Invest 2008; 118:1398-404; PMID:18317595; http://dx.doi.org/10.1172/JCI33522
- Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med 2012; 209:2351-65; PMID:23209317; http://dx.doi.org/10.1084/jem.20120944