
ABSTRACT
Cancer cells modulate the recruitment and function of inflammatory cells to create an immunosuppressive microenvironment that favors tumor growth and metastasis. However, the tumor-derived regulatory programs that promote intratumoral immunosuppression remain poorly defined. Here, we show in a KrasLA1/+p53R172HΔg/+-based mouse model that bone morphogenetic protein-4 (BMP4) augments the expression of the T cell co-inhibitory receptor ligand PD-L1 in the mesenchymal subset of lung cancer cells, leading to profound CD8+ T cell-mediated immunosuppression, producing tumor growth and metastasis. We previously reported in this model that BMP4 functions as a pro-tumorigenic factor regulated by miR-200 via GATA4/6. Thus, BMP4‐mediated immunosuppression is part of a larger miR‐200‐directed gene expression program in tumors that promotes tumor progression, which could have important implications for cancer treatment.
Introduction
A key regulator of tumor growth and metastasis is intratumoral inflammation, which exerts anti and pro-tumor effects in an immune cell-type-specific manner.Citation1-5 The types of inflammatory cells that infiltrate tumors correlate with patient prognosis.Citation6-9 Although tumor cells control the abundance and function of tumor-infiltrating immune cells to create a milieu that supports tumor growth and dissemination,Citation10-14 the specific genes expressed in tumor cells that create an immunosuppressive microenvironment have not been fully defined.
To study the mechanistic basis of lung cancer growth and metastasis, we have extensively used genetically-engineered mouse models expressing the two most common genetic alterations found in human lung adenocarcinoma, an activated Kras allele and mutant p53.Citation15-19 We have previously shown from gene expression profiling studies of primary lung tumors and metastases from K-rasLA1/+p53R172HΔg/+ (KP) mice that the metastatic signature is prognostic for patient outcome.Citation20 From this model, we derived an extensive panel of KP cell lines from primary and metastatic tumors and have defined their tumorigenic and metastatic capabilities in immunocompetent, syngeneic animals,Citation13,15,18,19 which recapitulate the widely metastatic behavior of human tumors to the major organs (e.g., liver, kidneys, and bone). We have demonstrated that orthotopic and subcutaneous syngeneic models display the same phenotypic behavior.Citation13,15-19,21 The series of studies with these models revealed pronounced differences between the syngeneic tumor models defined primarily by their epithelial or mesenchymal status, which is dynamically regulated by the expression of the microRNA-200 (miR-200) family. MicroRNAs coordinately regulate the expression of a broad spectrum of messenger RNAs and are therefore particularly well suited to mediate the diverse biological changes required for metastasis.Citation22 Studies in immunocompetent hosts with the KP syngeneic tumors with defined (high or low) metastatic capacity revealed that the miR-200 family expression is suppressed in highly metastatic tumor cells, while ectopic miR-200 expression in these cells abrogates invasion and metastasis, reverses epithelial-to-mesenchymal transition (EMT), and confers transcriptional features of poorly metastatic cells.Citation19 miR-200 directly targets the EMT-inducing transcription factor zinc-finger E-box-binding homeobox 1 (ZEB1). In turn, ZEB1 can directly repress the transcription of both miR-200 loci. In cancer cells, the double-negative feedback loop between miR-200 and ZEB1 is a key regulatory axis that coordinately controls the expression of many downstream genes involved in migration, invasion, and metastasis to distant sites.Citation23,24
Strikingly, in a recent study,Citation13 we used the genetically engineered KP model, the syngeneic KP models, and the Lewis lung cancer model to identify intratumoral immune cell features unique to metastasis-prone lung adenocarcinomas and found that CD8+ T cell abundance, proliferation, and activity were reduced in metastatic spontaneous lung adenocarcinomas and syngeneic tumors owing to the suppression of miR-200. This is the first report that links miR-200/ZEB1-regulated EMT to antitumor immune surveillance. Although hundreds of genes regulated by miR-200 have been identified, the precise contribution of these newly identified factors to tumor immunity remains elusive.
Among the factors that we have previously shown to be upregulated at the gene and protein level upon EMT in the KP models is bone morphogenetic protein 4 (BMP4).Citation19,25,26 We further recently described the direct regulation of BMP4 by miR-200 via the transcription factors GATA4/6, and demonstrated its pro-tumorigenic effect in our syngeneic murine lung cancer models.Citation26 Interestingly, BMP4 is a well-established factor critical to proper embryologic development of the lung and plays opposing roles in tumorigenesis and metastasis depending on cellular context.Citation26-29 These findings prompted us to further study the miR-200 target BMP4 to better understand how it might impact on the tumor microenvironment and tumor immunity.
Herein, we build upon our prior findings for a role of BMP4 in lung adenocarcinoma and provide evidence that it stimulates tumor cells to express the T cell co-inhibitory molecule PD-L1, thereby inducing CD8+ T cell dysfunction and an immunosuppressive tumor microenvironment that promotes growth and metastasis. Our work reveals that BMP4 controls the function of the intratumoral CD8+ T cells through a novel pathway involving the BMP4/STAT3/PD-L1/CD8+ T-cell axis. In parallel, tumor cell BMP4 expression produces elevated levels of intratumoral myeloid-derived suppressor cells (MDSCs) and the immunosuppressive CD4+ regulatory T cells (Tregs). Given the overall effects of BMP4 to reprogram the tumor cell signaling and the tumor microenvironment, we also demonstrate that tumors driven by BMP4 signaling require combination treatment with anti-PD-L1 and anti-CTLA4 for optimal therapeutic response.
Results
BMP4 promotes tumor growth and metastasis in miR-200-repressed tumors by altering the immune cell infiltrate and cytokine composition of the microenvironment
We previously reported that miR-200 repression in tumor cells promotes metastasis by inducing intratumoral CD8+ T cell dysfunction.Citation13 To identify tumor-derived negative regulators of CD8+ tumor-infiltrating lymphocytes (TILs), we mined transcriptional profiles of high- and low-metastatic KP cancer cells and found 31 genes related to chemokines and cytokines enriched among the 1,488 probe sets (data deposited in the Gene Expression Omnibus of the National Center for Biotechnology Information; http://www.ncbi.nlm.nih.gov/geo; accession number GSE14458). As we recently published, BMP4 was one of several cytokines expressed at low levels in poorly metastatic epithelial cancer cells (with high expression of the miR-200 family) and markedly higher levels in highly metastatic mesenchymal cancer cells.Citation19,26 Given that BMP4 belongs to the TGF-β superfamily, and since TGF-β1 potently inhibits CD8+ T cell function,Citation30,31 we posited this to be a potential mechanism for intratumoral T cell suppression, although BMP4 has not been reported to directly regulate CD8+ T cell function.Citation32 Furthermore, patients with fibrodysplasia ossificans, an autosomal dominant genetic disorder associated with ectopic expression of BMP4, have abnormal T and B lymphocyte functions.Citation33 To examine the effects of BMP4 expression on CD8+ TILs in tumors, we initially tested the KP cancer cells 344SQ (BMP4high) and 393P (BMP4low) implanted orthotopically into the lung. As we have previously published with these models, the orthotopic tumors grew and metastasized to regional lymph nodes and distant organs (Figs. S1A and B). Immune profiling of the tumors showed that the tumor cells with high BMP4 levels have fewer tumor-infiltrating CD8+ T cells (Fig. S1C). These findings are consistent with the immunosuppressive phenotype reported in our prior work with the spontaneous, orthotopic, and subcutaneous models.Citation13,15-19,26
Given the phenotypic similarities between the orthotopic and subcutaneous models, and owing to both the greater ability to carefully monitor primary tumor size over time and the greater/more rapid morbidity of orthotopic lung tumor growth in the animals, we utilized the subcutaneous models for the subsequent work outlined here. To specifically test the effect of tumor cell BMP4 expression, we utilized 393P cell line with ectopic expression of BMP4 (393P_BMP4), which has higher levels of expression (Fig. S2A) than control cells, and implanted them into immune-competent syngeneic mice. The overall tumor burden and number of lung metastases were higher in the 393P_BMP4 tumor-bearing mice than in the 393P_Vector tumor-bearing mice (). FACS analysis of the primary tumors demonstrated that ectopic BMP4 expression significantly reduced the number of CD8+ TILs, increased the percentage of exhausted CD8+ T cells and CD4+ Tregs, and substantially altered the ratio of CD8+/CD4+ Treg cells (). To further confirm the role of BMP4 in producing this immune phenotype, we used one of the previously generated cell lines of the highly metastatic mesenchymal 344SQ cells with short hairpin RNA to knock down BMP4 (344SQ-shBMP4) (Fig. S2B and and referenceCitation26). In addition, to further confirm functional BMP4 targeting, the changes of established downstream BMP4 targets ID1, ID2, and ID3 were confirmed by qRT-PCR (Fig. S3). Relative to control transfectants (scrambled shRNA), 344SQ-shBMP4 tumors were smaller, generated fewer metastases (), and contained more proliferative CD8+ T cells, as well as fewer exhausted CD8+ T cells and CD4+ Tregs (). Importantly, the dramatic differential metastatic phenotype of tumors with high or low levels of BMP4 was retained even when tumors were grown to similar sizes (Fig. S4). To test the direct effect of tumor cell BMP4 expression on T cells, BMP4-knockdown and control cells were co-cultured with murine splenocytes in vitro, and we observed fewer CD8+ T cells in the splenocyte/344SQ-scr co-cultures, and this suppression was partially abrogated by BMP4 depletion ().
Figure 1. BMP4 promotes tumor growth and metastasis via an immunosuppressive microenvironment. (A) 1 × 106 393P_vector and 393P_BMP4 tumor cells were subcutaneously injected into 129/Sv mice. Mice were sacrificed, and tumor weight and lung metastatic nodules were measured 6 weeks post-cell injection. The summary of tumor mass and lung metastatic nodules is shown in the left panel (n = 5 animals/group). The tumor photos are shown on the right top. H- and E-stained lung tissue is shown in the right bottom, indicating the metastatic lesions. Scale bar, 2 mm. (B) 1 × 106 344SQ-scr and 344SQ-shBMP4 tumor cells were subcutaneously injected into 129/Sv mice (n = 10/group). Mice were sacrificed, and tumor weight and lung metastatic nodules were measured 6 weeks post-cell injection. The summary of tumor mass and lung metastatic nodules is shown in the panel. (C) Intratumoral Ki67+CD8+ T cells in 344SQ-scr or 344SQ-shBMP4 primary tumors 2 weeks post-subcutaneous injection of cancer cells into 129/Sv mice. Representative histograms are shown on the left, and mean Ki67+ populations of gated CD8+ T cells in total T cells are shown on the right (n = 5). (D) Fluorescence-activated cell sorting analysis of CD4+TIL and CD8+TIL frequency from 393P_vector, 393P_BMP4, 344SQ-scr, or 344SQ-shBMP4 (n = 5) primary tumors. Analysis was done 2 weeks post-cancer cell injection. The representative plots are shown on the left, and the aggregate data is shown on the right. (E) 2 × 106 indicated cells were subcutaneously injected into 129/Sv mice (four mice each group). Tumors were harvested and tumor lysates prepared 2 weeks post-cancer cell injection. ELISA assays were conducted twice and the data were pooled. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (F and G) Fluorescence-activated cell sorting analysis of (F) T cell dysfunction markers PD1 and TIM3 expression on CD8+ T cells, and (G) Foxp3+ regulatory T cells from 393P_vector, 393P_BMP4, 344SQ-scr, or 344SQ-shBMP4 (n = 5) primary tumors. Analysis was done 2 weeks post-cancer cell injection. The representative plots are shown on the left, and the statistical summary is shown on the right. (H) The CD8+/Treg ratio was calculated by dividing the total number of CD8+ TILs infiltrating the tumor by the total number of CD4+Foxp3+ T-cell infiltrate. Percent of CD8+ T-cell infiltrate was calculated as a percent of total CD3+ T-cell infiltrate. The percent of Treg infiltrate was calculated as a percent of total CD4+ T cells in the tumor fraction. (I) CD8+ T cell numbers were measured in splenocytes cultured with or without cancer cells for 5 d in the presence of anti-CD3 stimulation (5 μg/mL) and Interleukin 2 (1 μg/mL). Results are means with standard deviations for triplicate samples.
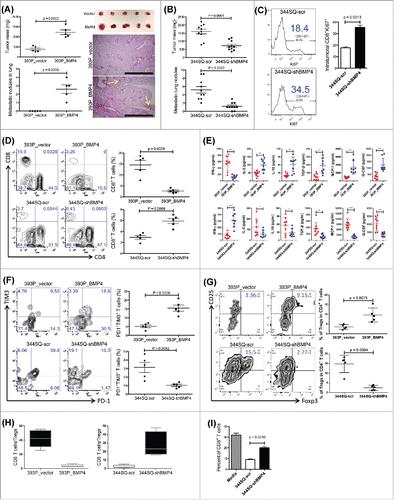
Figure 2. CD8+ T cell response plays a dominant role in BMP4-mediated tumor growth and metastasis. (A) Mice were pretreated with anti-CD8+ antibody (2.43; BioXCell; 400 μg, intraperitoneally) one week before tumor cell injection. 200 μg of anti-CD8+ antibody per mouse was injected into the mice once weekly for 5 weeks beginning on day 1 after a subcutaneous cancer cell injection (2 × 106 indicated cells per mouse; n = 5–6 mice/group). The tumor growth was measured once a week for 6 weeks. To test the CD8+ T cell depletion efficiency, spleen cells were stained with anti-CD4+ and anti-CD8+ antibodies. The tumor growth curve is shown on the top, and CD8+ T cell depletion efficiency is shown in the bottom; (B) at the endpoint, mice were necropsied to harvest primary tumors and lungs, which were weighed, and to quantify distant metastases. (C) To prepare CD8+ T cells, 129/Sv mice were challenged with 2 × 106 393P_BMP4 for 2 weeks (subcutaneously injected). CD8+ T cells were isolated from these tumors, blood, and spleens. To conduct the treatment experiment, 1 × 106 tumor cells were subcutaneously inoculated into mice 1 week before T cell transfer, then mice received cyclophosphamide at 100 mg/kg intravenously 6 h before CD8+ T cell transfer (5 × 106 per mouse, intravenously), following IL-2 (20,000 units, intraperitoneally) at 8 h after T cell transfer then every 12 h for 3 d. The tumor growth was measured once a week for 6 weeks. The tumor growth curves are shown on the right top (n = 5–6 mice/group). **p < 0.01; (D) at the endpoint, mice were necropsied to harvest primary tumors and lungs, which were weighed, and to quantify distant metastases.
We also evaluated changes in the tumor microenvironment by ELISA-based analysis of syngeneic tumor lysates, which demonstrated that tumor cell BMP4 expression elevated the levels of immune suppressive cytokines/chemokines such as IL-6, IL-10, TGF-β, MCP-1, and G-CSF, while it suppressed the antitumor immunostimulatory cytokine IFNγ in size-matched tumors (; Fig. S5A). Finally, in size-matched tumors, BMP4 expression produced an enrichment of MDSCs (Fig. S5B). These findings indicate that tumor cell expression of BMP4 creates an immunosuppressive microenvironment, with substantial changes in both the immune cell infiltrate and cytokine milieu that favor tumor growth and metastasis.
BMP4 inhibits CD8+ T cell function in a PD-L1-dependent manner to mediate tumor growth and metastasis
To determine whether CD8+ T cells were primarily responsible for the immune phenotype observed in BMP4-expressing tumors, we tested the in vivo effect of CD8+ T cells on tumor growth and metastasis in both the 393P and 344SQ syngeneic models. Tumors of 344SQ with BMP4-knockdown grew like control cells (344SQ-scr) when CD8+ T cells were depleted by an anti-CD8+ antibody (), and adoptive transfer of CD8+ T cells inhibited both growth and metastasis of 393P-BMP4-expressing tumors (). These data suggest that tumor cell BMP4 expression promotes tumor progression in a CD8+ T cell-dependent manner.
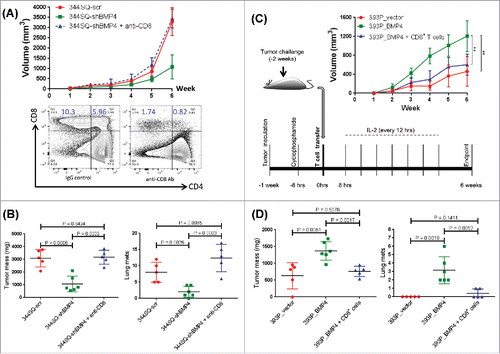
We further investigated the relationship between BMP4 expression and tumor cell EMT status by analysis of the patient samples in The Cancer Genome Atlas (TCGA) lung adenocarcinoma dataset (n = 230). We observed a significant anti-correlation between miR-200 and BMP4 expression (Fig. S6A), as well as a correlation between ZEB1 and BMP4 (Fig. S6B). Furthermore, BMP4 levels were suppressed by ectopic miR-200 expression in low-miR-200 cancer cell lines and increased by ectopic ZEB1 expression in the murine KP lung cancer cells (Fig. S6C; referenceCitation26). These data are consistent with our recent demonstration that GATA4 and GATA6 could potently drive the transcriptional activation of the BMP4 promoter and that miR-200 downregulated BMP4 via GATA4 and GATA6.Citation26 Therefore, BMP4 is regulated by the miR-200/ZEB1 axis in lung cancer. Additionally, we also recently demonstrated the regulation of tumor cell PD-L1 expression by the miR-200/ZEB1 axis, and the subsequent suppression of CD8+ T cells in the tumor microenvironment.Citation13 These findings prompted us to examine whether PD-L1 is regulated by BMP4. Indeed, the expression of PD-L1 mRNA and protein on cultured cancer cells was regulated by ectopic expression, small interfering RNA (siRNA), or short-hairpin (shRNA) knockdown of BMP4 (; Figs. S7 and S8A and B); by constitutive knockdown or expression of BMP4 in syngeneic tumors, as assessed by FACS and immunohistochemistry (IHC) analyses (); and treatment with recombinant BMP4 (rBMP4) or a pharmacologic BMP4 inhibitor (; Fig. S8C). Normally, IFNγ stimulation of cancer cells in vitro upregulates PD-L1 expression, and we observed that the level of IFNγ-induced PD-L1 upregulation was dependent on the level of BMP4 expression in cells (; Fig. S8B). These data indicate that BMP4 regulates the expression of PD-L1, even in the presence of other well-documented regulators.
Figure 3. BMP4 regulates PD-L1 expression on tumor cells and thereby causes T cell dysfunction. (A) The effect of BMP4 siRNA on PD-L1 expression in murine KP lung cancer cells. Cells were transfected with 40 nM siRNA or the control for 3 d before FACS analysis. PD-L1 expression levels were calculated by the ratio of anti-PD-L1 stained MFI/isotype stained MFI (MFI, mean fluorescence intensity). (B) The representative FACS histogram of PD-L1 (MFI, mean fluorescence intensity) expression in primary subcutaneous tumors grown in syngeneic 129/Sv mice (n = 3) injected with the indicated cell lines is shown in the upper panel. The representative PD-L1 immunohistochemical staining of each tumor type is shown in the lower panel. Samples were obtained 2 weeks post-cell injection. Scale bar, 100 μm. (C) Cell surface expression of PD-L1 analyzed by FACS 5 d post-treatment with rBMP4 at the indicated concentrations. PD-L1 expression levels were calculated by the ratio of anti-PD-L1 stained MFI/isotype stained MFI. Data are represented as mean ± SD. (D) Cells were incubated with BMP4 inhibitor LDN193189 at different concentrations (25 nM and 50 nM) for 5 d and stained with anti-PD-L1 antibody. PD-L1 expression levels were calculated by the ratio of anti-PD-L1 stained MFI/isotype stained MFI. The experiments were repeated three times. (E) Representative FACS histogram of cell surface expression of PD-L1 on different mouse lung cancer cell lines with or without IFNγ stimulation (48 h stimulation). Red line, isotype control staining; blue line, anti-PD-L1 staining. (F) FACS analysis of surface expression of PD-L1 on different cancer cells co-cultured with 129/Sv spleen cells for 2 d. Representative FACS histograms are shown. Red, control isotype antibody staining; light blue, anti-mouse PD-L1 antibody staining for cancer cells cultured without spleen cells; and pink, anti-mouse PD-L1 antibody staining for cancer cells cultured with spleen cells. (G) FACS analysis of markers of T cell dysfunction. 129/Sv spleen cells were cultured for 4 d with various cancer cells in the presence of anti-CD3 stimulation (5 μg/mL) and Interleukin 2 (1 μg/mL). 344SQ-shBMP4 cells were transfected with PD-L1 (indicated in red). The data shown in left panel (means with standard deviations) are percentages of gated CD8+ T cells that were PD1+TIM3+ and are representative of at least three independent experiments. *p < 0.05. PD-L1 transfection efficiency was measured by FACS and the representative histograms are shown in right panel.
We again utilized a functional in vitro assay system where KP tumor cell lines were co-cultured with syngeneic mouse splenocytes to examine whether BMP4-regulated PD-L1 expression on cancer cells is sufficient to induce CD8+ T cell exhaustion. Upon interaction with splenocytes, KP tumor cells differentially upregulated PD-L1 expression, which was dependent upon the level of BMP4 expression (). The PD1+TIM3+ subpopulation of exhausted CD3+CD8+ T cells was significantly lower when PD-L1 expression on cancer cells was inhibited by BMP4 knockdown (). Conversely, the number of exhausted T cells was substantially increased when PD-L1 expression on cancer cells was upregulated by BMP4, similar to ectopic PD-L1 expression (). Thus, the mechanism of cancer cells inducing CD8+ T cell exhaustion through a BMP4/PD-L1-dependent pathway was faithfully recapitulated in the in vitro co-culture assay system. Additionally, in vivo data from three different tumor cell models revealed that pharmacologic blockade of BMP4 inhibits tumor growth (Fig. S8D). Accordingly, BMP4/PD-L1-dependent signaling is sufficient to induce CD8+ T cell dysfunction and thereby promote tumor progression.
BMP4 regulates PD-L1 through STAT3
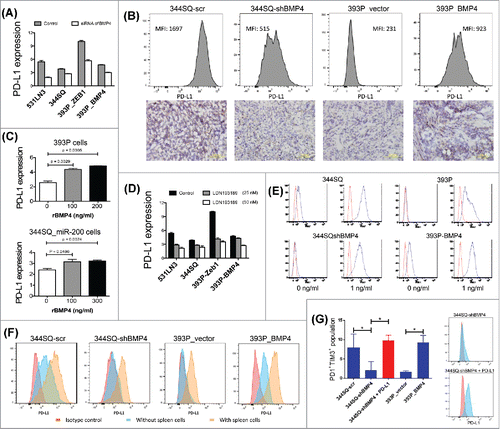
To identify the putative mediator(s) of the BMP4 effect on PD-L1 expression, we first examined the canonical SMADs because our previous study demonstrated the effect of BMP4 on SMAD1/5 signaling.Citation26 However, when K02288 and DHM1 were used to selectively inhibit SMAD1/5 activation in the BMP4high 531LN3 and 344SQ cancer cells, PD-L1 expression on these cells did not change (Fig. S9). These results suggested that an alternative pathway must be involved. Previous studies reported that BMP4 mediates STAT3 activation,Citation34 and that STAT3 regulates PD-L1 expression.Citation35 Indeed, we found that recombinant BMP4 activated STAT3 phosphorylation in two different cell line models (Fig. S10). Moreover, in reverse phase protein array (RPPA) analyses of our KP lung cancer cell line panel the mesenchymal metastatic cells (with high ZEB1 and low miR-200) showed significantly higher phospho-STAT3 (Y705) levels than the non-metastatic epithelial cells (with low ZEB1 and high miR-200),Citation13,15 both in serum free and normal media conditions, which was confirmed by Western blotting (). Additional analysis using a previously published isogenic cell line panelCitation18 revealed higher p-STAT3 levels in the epithelial 393P cells upon induction of EMT by ZEB1 expression and p-STAT3 suppression upon co-expression of miR-200 (). To test for direct interaction between STAT3 and the PD-L1 promoter, we constructed two different PD-L1 promoter luciferase reporters (PMT-1 and -2) and tested the luciferase activity upon co-transfection with a STAT3 expression vector. STAT3 expression enhanced PD-L1 promoter activity from both constructs (). In addition, the expression of PD-L1 in a panel of lung cancer cells with either high-basal expression or high-level expression due to ectopic ZEB1 or BMP4 was repressed by siRNA against STAT3 or use of a pharmacologic STAT3 inhibitor (; Fig. S11A), and PD-L1 expression strongly correlated with activated STAT3 levels (Figs. S11B and C). Thus, we conclude that BMP4 induces immunosuppression through increased expression of PD-L1 on tumor cells, a well-known immune checkpoint pathway that produces an exhausted CD8+ T cell phenotype.Citation36-39
Figure 4. BMP4high metastatic cells have high levels of activated STAT3 & STAT3 directly regulates PD-L1. (A) A panel of KP cell lines was grown in vitro under full serum (10%) conditions or washed and grown in the absence of serum for 24 h prior to harvest of protein lysates, which were then analyzed by RPPA. The samples were dichotomized into epithelial or mesenchymal groups based upon miR-200/ZEB1 expression and dot plots show the activated pY705-STAT3 levels for each group and growth condition. (B) The same panel of cell lines was used for Western blot of the indicated EMT markers, total STAT3, and activated STAT3, versus β-actin as a loading control. (C) An isogenic EMT panel of cell lines constructed from the 393P parental line was used for Western blotting of the total and activated STAT3 levels. (D) STAT3 activates PD-L1 in mouse lung cancer cells. The effects were assessed by relative luciferase activity. PD-L1 (CD274) promoters (PMT-1: −1,187 ∼ +801 bp, PMT-2: −1,701 ∼ +216 bp from the transcription start site) were isolated from a mouse BAC clone (RPCI-23 434F3, BACPAC Resources Center at the Children's Hospital Oakland Research Institute) by PCR and ligated into the pGL3-basic vector (Promega). 344SQ cells were seeded on 24-well plates (1 × 105 cells per well) one day before transfection. Cells were co-transfected with 500 ng pGL3-basic or PD-L1 promoters and 250 ng STAT3 expression vector (pIREShyg3/STAT3). The vector expression efficiency of STAT3 shown in left was measured by Western blotting. pRL-TK (50 ng, Promega) was co-transfected as an internal control. After 48 h of transfection, luciferase activity was measured with Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's protocol. Values are normalized to those of pGL3-basic vector (mean ± SD, n = 3). p-values are indicated (two-tailed Student's t-test). (E) Cells seeded in six-well plate were transfected with siRNA against STAT3 (50 nM) as well as their scrambled controls, respectively. The cells were harvested 3 d after transfection to examine PD-L1 expression. MFI of PD-L1 expression was calculated with FlowJo software. PD-L1 expression levels were calculated by the ratio of anti-PD-L1 stained MFI/isotype stained MFI. The knockdown efficiency of siRNA of STAT3 was measured by qPCR. The experiments were repeated three times. (F) Cells seeded in six-well plate were treated with STAT3 inhibitor VIII at the indicated concentrations for 3 d. PD-L1 expression was analyzed by flow cytometry. PD-L1 expression levels were calculated by the ratio of anti-PD-L1 stained MFI/isotype stained MFI.
BMP4-mediated tumor progression is targetable by the combination of PD-L1 and CTLA-4 blockade
Given the clear effect of BMP4 on tumor progression via PD-L1 expression on tumor cells, we tested the therapeutic efficacy of dosing animals with an anti-PD-L1 antibody and observed a statistically significant reduction of both primary tumor growth and metastasis that was dependent on BMP4 expression (; Fig. S12). This therapeutic effect was consistent with our prior observations for the parental 393P and 344SQ models.Citation13 Given the high levels of CD4+ Tregs found in the tumors expressing BMP4 (), we directly tested their role by depletion with an anti-CD25 antibody (PC61). However, we did not observe significant inhibition of tumor growth or metastasis upon Treg depletion alone in three different tumor models expressing high levels of BMP4 (Fig. S13), again highlighting the effect of direct PD-L1-mediated suppression of CD8+ T cells.
Figure 5. BMP4-mediated tumor progression is targetable by anti-PD-L1 alone or in combination with anti-CTLA-4. (A, B) The indicated antibody or an isotype-matched IgG control was injected into 129/Sv mice (intraperitoneally) once a week for 5 weeks beginning on day 1 after the subcutaneous indicated tumor cell injection (2 × 106 cells per mouse). Dosing per injection was 200 μg of anti-PD-L1, 150 μg of anti-CTLA-4. Tumors were measured once a week for 6 weeks. The tumor growth curves are shown. ns, no significant difference, *p < 0.05, ****p < 0.0001. (C, D) 2 × 106 indicated cells were subcutaneously injected into 129/Sv mice (four mice each group). Tumors were harvested and CTLA-4 expression on tumor-infiltrating CD4+ T cells was analyzed by FACS 2 weeks post-cancer cell injection. (E, F) Fluorescence-activated cell sorting analysis of CD4+TIL and CD8+TIL frequency from the endpoint primary tumors. The representative plots are shown on the left, and the statistical summary is shown on the right. The data from 344SQ tumors are shown in (E); the data from 393P_BMP4 tumors are shown in (F).
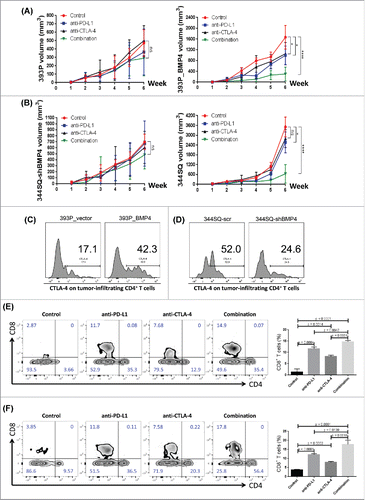
Owing to the activation of parallel, non-redundant immune checkpoint pathways in tumors, therapies against individual pathways are often insufficient to produce a clinical benefit for patients. Consequently, strategies have evolved to combine PD-1/PD-L1 axis blockade with targeting of a second immune checkpoint pathway, CTLA-4. CTLA-4 is an immune checkpoint molecule found on activated CD4+ and CD8+ T cells.Citation40,41 We noted that CTLA-4 is highly expressed on CD4+ T cells in BMP4high tumors (). We therefore evaluated whether the observed therapeutic efficacy of anti-PD-L1 antibody against BMP4-expressing tumors could be enhanced with co-administration of an anti-CTLA-4 antibody. After subcutaneous implantation of the syngeneic 344SQ or 393P_BMP4 cells into immunocompetent animals, the anti-CTLA4 and/or anti-PD-L1 antibodies or an isotype-matched control were injected intraperitoneally once a week for 5 weeks beginning on day 1 after the indicated tumor cell implantation. Although anti-PD-L1 or anti-CTLA-4 monotherapy delayed tumor growth and inhibited lung metastasis in two different BMP-4-dependent models, co-administration of the combination produced a significant enhancement of the therapeutic response of the primary tumors and number of metastases (; Fig. S12). In this setting, the combination of PD-L1 and CTLA-4 blockade increased activated CD8+ T cell infiltration ∼5–7-fold, more than either PD-L1 or CTLA-4 blockade alone (). Together, these data reveal a unique potential for anti-PD-L1 in combination with anti-CTLA-4 immunotherapy to enhance the therapeutic effect against BMP4-expressing tumors.
Discussion
Immune cells that infiltrate tumors engage in an extensive and dynamic crosstalk with cancer cells, playing an important role during tumor progression.Citation3,13,14,42,43 EMT is a critically important pathway during embryogenesis that is frequently reactivated during tumor progression. An increasing number of studies have examined the EMT state and intratumoral factors that lead to the development of an immunosuppressive tumor microenvironment,Citation13,43,44 suggesting that multiple parallel pathways are activated that may suppress an effective antitumor immune response. Although we have previously reported that PD-L1 is a direct target of miR-200, which contributes to immunosuppression in the primary tumor tissue,Citation13 the direct miR-200-PD-L1 axis likely does not entirely account for the immunosuppressive effects observed. In the current study, we have identified another molecular network involving BMP4, STAT3, and PD-L1 operating in lung cancer cells that affects the function of tumor-infiltrating CD8+ T cells.
BMP4 in cancer is an emerging field of research and potential clinical applications. Its role in the process of tumor growth, migration, invasion, EMT, stem cell properties, distant metastases, and tumor recurrence has been widely implicated.Citation26,27,45-48 Several earlier reports suggest that BMP4 has dual functionality that harbors both tumor-suppressive and tumor-promoting features. In glioblastoma, BMP4 causes the depletion of tumor-initiating cells and thus inhibits tumorigenesis.Citation49 BMP4 has previously been shown to inhibit the expression of G-CSF from breast tumor cells, thus blocking MDSC activity.Citation28 However, we observed an enrichment of MDSCs, along with high levels of G-CSF and multiple other chemokines/cytokines in BMP4high lung tumors. In pancreatic cancer, loss of ATM alters TGF-β superfamily signaling as shown by a perturbed BMP4/SMAD1/5/8 and NODAL/SMAD2/3 signaling axis. Elevated BMP4 signaling acts as a switch in the maintenance of acinar cell integrity and induces trans-differentiation of acinar cells to metaplastic ductal cells.Citation27 Similarly, other published work shows that the BMP4/SMAD1/MMP2 axis drives EMT in pancreatic ductal adenocarcinoma (PDAC).Citation50 Therefore, whether BMP4 induces a pro- or antitumorigenic response appears to be context/tissue-dependent, behaving very differently in distinct cancer types. It is well accepted that EMT has a critical impact on immunosuppression during tumor progression.Citation13,14,43,51 Consistent with this notion, BMP4, as a downstream mediator of EMT, forges a strong immunosuppressive microenvironment upon loss of miR-200 expression in tumors.
Tumors have multiple redundant pathways that confer the ability to evade destruction by the host immune system. Evidence shows that functional tumor cell killing by tumor-infiltrating T cells can be inhibited by both the CTLA-4 and PD-1 co-inhibitory signals, and that tumor cells frequently express PD-L1, which can negatively signal T cells through both the PD-1 and B7-1 molecules on their surface.Citation40,52 Our data show that BMP4 can upregulate PD-L1 expression on cancer cells, suggesting BMP4 as another potential target for cancer treatment. Indeed, when we treated mice with a BMP4 inhibitor, the tumor growth was significantly reduced. Although PD-L1 blockade led to significant inhibition of BMP4-mediated tumor growth and lung metastasis, the anti-PD-L1 monotherapy did not entirely suppress the tumors over time. Since it is well known that anti-CTLA-4 reduces CD4+ regulatory T cells within tumors,Citation40,41 we treated mice with the combination of anti-PD-L1 and anti-CTLA-4 in a BMP4-mediated lung cancer setting. The data show that the combination with anti-CTLA-4 enhances the antitumor efficacy of anti-PD-L1, demonstrating an additive effect. The significant therapeutic response suggests that BMP4/STAT3/PD-L1-induced intratumoral immunosuppression is targetable, thus having important clinical implications for cancer treatment. Each tumor has its own unique cell autonomous signaling requirements, anatomic compartments, and microenvironments, along with differences between different tumor types.Citation53-55 In this context, our work initiates studies on the relationship between BMP4 and CD8+ T cell functions in tumor, shedding light on identifying potential therapeutic targets. We further found that BMP4 enhances immunosuppression through upregulation of several cytokines/chemokines, including IL-6, IL-10, TGF-β, MCP1, and G-CSF as well as downregulation of IFNγ. It is not clear which signaling pathways underlie the impact of BMP4 on these factors, and we are actively investigating these connections.
Our previous work has shown that BMP4 functions as a pro-tumorigenic factor through enhancement of cancer cell migration and invasion in a murine lung cancer model, and that miR-200 downregulates BMP4 via direct targeting of the GATA4 and GATA6 transcription factors that stimulate BMP4 transcription.Citation26 Results from the present work reveal that BMP4 also produces an immunosuppressive tumor microenvironment in a CD8+ T cell-dependent manner with the accumulation of tumor-infiltrating regulatory T cells and suppressive cytokines. Therefore, the findings obtained from the previous and present studies establish that miR-200/ZEB1/GATA and BMP4/STAT3/PD-L1 form a critical metastasis-promoting network that drives metastasis in lung cancer. Better understanding this network may ultimately lead to discovery of novel therapeutic targets for lung cancer.
Materials and methods
Plasmids, viruses, cell lines, mice, reagents, and treatments
pLenti4.1-miR200 vector was obtained from Dr Goodall and used as described previously.Citation19,24 Mouse ZEB1 open reading frame (ORF) fragment was digested with EcoR I and then subcloned into the pcDNA3.1/His C vector (Invitrogen) to generate the ZEB1 expression plasmid. BMP4 ORF fragment was digested with BamH I and then subcloned into the pBABEhygro vector to generate the BMP4-overexpression plasmid. shRNA specific to mouse PD-L1 and scrambled controls were cloned in pGFP-V-RS vector (Origene) to generate the shPDL1 knockdown constructs, respectively. Mouse PD-L1 expression vector (pUNO1-mCD274) was purchased from Invivogen.
The 393P, 393LN, 412P, 713P, 307P, 531P1, 344P, 344SQ, 344LN, 531LN1, 531LN2, and 531LN3 cell lines were derived from K-rasLA1/+p53R172HΔg/+ mice as previously described, and are named based upon the mouse from which they were derived and the site of the tumor, with “P” representing a pulmonary tumor, “LN” a lymph node metastasis, and “SQ” a subcutaneous metastasis.Citation15,18,19 344SQ_miR200, 531LN2_miR200, 393P_ZEB1, and 393P_ZEB1_miR-200 stable cell lines were generated by pLenti4.1-miR200 lentivirus transductionCitation15,18 Murine BMP4 shRNAs (#TG516759) were purchased (Origene), cloned into the pLKO.1 vector (Addgene) according to the manufacturer protocol (http://www.addgene.org/tools/protocols/plko/), and introduced into 344SQ or 531LN3 cells by viral infection. After antibiotics selection for more than 2 weeks, stable transfectants were established and previously characterized.Citation26 Splenocytes were freshly isolated from 129/Sv mice. Six 8-week-old 129/Sv male mice were purchased from the Charles Rivers Laboratory (Massachusetts, USA). Specific pathogen-free mice were maintained in facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care International. All animal procedures were reviewed and approved by the MD Anderson Cancer Center Animal Care and Use Committee.
Recombinant IFNγ (Pepro Tech), VIII (Sigma-Aldrich), and rBMP4 (LifeSpan BioSciences) were used for in vitro studies. LDN193189 (Selleckchem), IL-2 (Miltenyi Biotec), and Cyclophosphamide (Sigma-Aldrich) were used for in vivo studies. siRNAs against BMP4 and STAT3 were purchased from OriGene.
Tumor models
To establish orthotopic mouse model of lung cancer, KP-derived cancer cells 344SQ (BMP4high) and 393P (BMP4low) were orthotopically injected into 129/Sv murine lung (1 × 106 cells per mouse). Six weeks post-injection, mice were necropsied to quantify primary tumors and distant metastases. To examine intratumoral lymphocyte populations, 2 × 106 cancer cells in 100 µL were subcutaneously injected into the flanks of 129/Sv mice. If not otherwise specified, tumors were harvested and analyzed 2 weeks after injection. For studying the growth rate of the primary tumor mass and formation of distant lung metastases, 1 × 106 cancer cells in 100 µL of phosphate-buffered saline (PBS) were injected subcutaneously into the mouse flank. Mice were monitored regularly and euthanized 5–6 weeks after the tumor cell injections. At end points, mice were sacrificed to examine the primary and metastatic tumors.
Co-culture assay
The murine KP cancer cells were seeded at 5 × 106 cells in 150 × 10 mm dishes for 16 h. 1.5 × 106 splenocytes were then added directly to each dish. Splenocyte only cultures were used as control. Cells were cultured in complete RPMI 1640 medium supplemented with 10% FBS, normocin (Gibco) in the presence of anti-CD3 stimulation (5 μg/mL) and interleukin 2 (1 μg/mL). Co-cultured splenocytes were transferred to new dishes seeded with cancer cells (5 × 106 total cells per 150 × 10 mm dish for plating) every other day. In some studies, 344SQ-shBMP4 cell lines were transiently transfected with pUNO1-mCD274 plasmid 2 d prior to the co-culture assay. Co-cultures were harvested 4 d later and separated into non-adherent and adherent fractions. The non-adherent cells were primarily splenocytes, while the adherent cells were mainly the cancer cells. FACS staining was performed on the non-adherent cells for T cell profiles and the adherent cells for cell surface expression of PD-L1.
Antibody-mediated cell depletion
Mice were pretreated with anti-CD8+ antibody (2.43; BioXCell; 400 μg, intraperitoneally) 1 week before tumor cell injection. 200 μg of anti-CD8+ antibody per mouse was injected into the mice once weekly for 5 weeks beginning on day 1 after a subcutaneous cancer cell injection.
CD8+ T cell adoptive transfer
The protocol was modified according to the previous reportCitation56. To prepare CD8+ T cells, 129/Sv mice were challenged with 2 × 106 393P_BMP4 for 2 weeks by subcutaneous injection. CD8+ T cells were isolated from these tumors, blood, and spleens by MACS technology according to the manufacturer's instructions (Miltenyi Biotec). CD8+ T cells (5 × 106) were injected via the tail vein into 129/Sv mice 6 h after receiving cyclophosphamide.
Immunotherapy
Anti-CD25 (PC61; BioXcell; 150 μg per mouse), anti-PD-L1 (9G2; BioXcell; 200 μg per mouse), and anti-CTLA-4 (9H10; BioXcell; 150 μg per mouse) antibody or an isotype-matched IgG control were injected into 129/Sv mice intraperitoneally once a week for 5 weeks beginning on day 1 after the subcutaneous implantation of the indicated tumor cell model.
siRNA knockdown assay
Cells seeded in six-well plate were transfected with siRNA against BMP4 (40 nM), STAT3 (50 nM), and their scrambled non-targeting controls. The cells were harvested 3 d after transfection to examine the expression of the target and PD-L1.
PD-L1 promoter luciferase assay
PD-L1 promoters (PMT-1: −1,187 ∼+801 bp, PMT-2: −1,701 ∼ +216 bp from the transcription start site) were isolated from a mouse BAC clone (RPCI-23 434F3, BACPAC Resources Center at the Children's Hospital Oakland Research Institute) by PCR and ligated into the pGL3-basic vector (Promega). 344SQ cells were seeded on 24-well plates (1 × 105 cells per well) one day before transfection. Cells were co-transfected with 500 ng pGL3-basic or PD-L1 promoters and 250 ng STAT3 expression vector (pIREShyg3/STAT3). pRL-TK (50 ng, Promega) was co-transfected as an internal control. After 48 h of transfection, luciferase activity was measured with Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's protocol. Values are normalized to those of pGL3-basic vector (mean ± SD, n = 3). p-values are indicated (two-tailed Student's t-test).
RNA analysis, western blotting, and reverse phase protein array (RPPA)
Total RNA was isolated from cultured cells using Trizol reagent (Invitrogen) according to the manufacturer's instructions. cDNA was prepared from the RNA using SuperScript III first-strand synthesis system (Invitrogen) and was used for RT-PCR reactions. cDNA products were amplified using SYBR Green PCR Master Mix (Applied Biosystems) and analyzed by using ABI Prism 7500 Fast System (Applied Biosystems). Primers were designed using NCBI primer design software. The amount of each gene product was calculated using the 2−ΔΔCt method.Citation57 Relative levels of expression were normalized on the basis of L32 ribosomal RNA. Primers used for amplification are shown below:
L32 forward 5′-GTAACCCGTTGAACCCCATT-3′
reverse 5′-CCATCCAATCGGTAGTAGCG-3′
BMP4 forward 5′-GGACCTACCCTTGCAAACAA-3′
reverse 5′-GGTGGCACAAGATCACCTTT-3′
STAT3 forward 5′-CGATCCTCTCCTCCAGCATGG-3′
reverse 5′-GATAACTTCATTAGCAGAATCT-3′
ID1 forward 5′- TTGGTCTGTCGGAGCAAAGCGT-3′
reverse 5′- CGTGAGTAGCAGCCGTTCATGT-3′
ID2 forward 5′- TCACCAGAGACCTGGACAGAAC-3′
reverse 5′- TGCTATCATTCGACATAAGCTCAG-3′
ID3 forward 5′- GCGTGTCATAGACTACATCCTCG-3′
reverse 5′- GTCCTTGGAGATCACAAGTTCCG-3′
Cell lysates were prepared by lysing the samples in lysis buffer containing 10 mM Tris (pH 7.4), 1 mM EDTA, 0.5 mM EGTA, 150 mM NaCl, 1% Triton X-100, 50mM NaF, 10 mM Na4P2O7·10H2O, 1 mM PMSF, and protease inhibitors cocktail (Sigma). The amounts of protein in the lysates were estimated by using BCA protein assay reagents (Pierce). Samples (40 µg of protein) were electrophoresed in SDS-polyacrylamide gel and transferred onto PVDF membranes. The membranes were then blocked with 5% skim milk in Tris-buffered saline (TBS) containing 0.2% Tween-20 (TBS-T) and subsequently incubated with primary antibodies overnight at 4°C. Membranes were probed with the following antibodies: primary antibodies anti-BMP4 (Abcam, ab39973, 1:200 dilution), anti-ZEB1 (Santa Cruz, sc-25388, 1:500 dilution), anti-Ncad (BD Biosciencses, 610921, 1:500 dilution), anti-Vim (Cell Signaling, 3932s, 1:500 dilution), anti-STAT3 (Abcam, ab2415, 1:2,000 dilution), anti-p-STAT3 (Y 705) (Santa Cruz, sc-7988, 1:1,000 dilution), anti-Actin (Abcam, ab8227, 1:5,000 dilution), and secondary antibody labeled by horseradish peroxidase (Amersham GE Healthcare). The secondary antibody was visualized using a chemiluminescent reagent Pierce ECL kit (Thermo Scientific).
For RPPA analysis, five serial dilutions of each protein lysate were printed on nitrocellulose-coated slides using an Aushon Biosystems 2470 arrayer (Burlington, MA) and stained sequentially with primary and secondary antibodies in an autostainer (BioGenex), prior to signal detection using a signal amplification system and DAB-based colorimetric reaction. MicroVigene Software (VigeneTech) as well as an in-house R package was used to assess spot intensities and the SuperCurve method was applied to estimate protein levels in each sample. For comparisons, data were log transformed (to the base of 2) and median-centered across antibodies to correct for protein loading. All statistical analyses were performed using R packages (version 2.10.0).
Flow cytometry
Single-cell suspensions were prepared and stained according to standard protocols for flow cytometry with the following antibodies: CD45-FITC (mouse; BD Biosciences, 553080, 1:100 dilution), CD3-PerCP (mouse; BD Biosciences, 560527, 1:100 dilution), CD4-APC (mouse; BD Biosciences, 553051, 1:100 dilution), CD4-FITC (mouse; BD Biosciences, 553729, 1:50 dilution), CD8b-APC (mouse; eBioscience, 17-0083, 1:100 dilution), CD8b-PE (mouse; Bioscience, 12-0083-83, 1:200 dilution), CD11b-APC (mouse; BD Biosciences, 553312, 1:150 dilution), Ki67-PE (eBioscience, 12-5698-82, 1:100 dilution), CTLA-4/CD152-PE (mouse; BioLegend, 106306, 1:150 dilution), PD1-PE (mouse; eBioscience, 12-9985-83, 1:150 dilution), PD1-FITC (mouse; eBioscience, 11-9985-82, 1:150 dilution), PD-L1-PE (mouse; BD Biosciences, 558091, 1:200 dilution), PD-L1-APC (mouse; BioLegend, 124312, 1:200 dilution), TIM3-PE (mouse; eBioscience, 12-5870-83, 1:150 dilution), CD25-APC (mouse; eBioscience, 17-0251-82, 1:100 dilution), Gr-1-PE (mouse; BD Biosciences, 553128, 1:100 dilution), Foxp3-PE (mouse; BioLegend, 126404, 1:50 dilution), and pSTAT3-Ser727 (mouse; eBioscience, 12-2231-83, 1:100 dilution). For intracellular staining, cells were fixed and permeabilized with BD Cytofix/Cytoperm (BD Biosciences). The data were acquired on a Fortessa or Calibur platform (BD Biosciences) and analyzed with FlowJo software (version 7.6; Tree Star). For analyzing the abundance and the function of CD4+ or CD8+ TILs, single-cell suspensions were prepared from tumors and inguinal lymph nodes and stained; the staining of inguinal lymph node cells was used as the reference of lymphocyte gating, then CD3+ cells were gated, and then CD4+ or CD8+ population was analyzed.
ELISA
The indicated cancer cells were subcutaneously injected into 129/Sv mice (four mice per group). Tumors were harvested and prepared tumor lysates 2 weeks post-cancer cell injection. The antibodies (IFNγ, eBioscience, 88-7314-88; IL-6, R&D Systems, M6000B; IL-10, R&D Systems, M1000B; TGF-β, abcam, ab119557; MCP-1, eBioscience, 88-7391-86; G-CSF, R&D Systems, MCS00) were used to perform the ELISA assays. The experiments were conducted twice and the data were pooled.
Histology and immunohistochemistry
For hematoxylin and eosin (HE) staining, lung tissues were fixed in 10% PFA and embedded in paraffin. HE stained sections (4 µm) were examined to define micrometastases. For IHC, the antibodies against mouse pSTAT3-ser727 (Santa Cruz, USA; sc-8001-R; dilution 1:150) and mouse PD-L1 (abcam, USA; ab18259; dilution 1:100), and HRP-conjugated secondary antibody were used for staining. Images (× 20) were acquired with an Olympus BX41 microscope.
Human samples
The set of samples analyzed consisted of lung adenocarcinoma cases from TCGA project (http://cancergenome.nih.gov/),Citation58 which represent early-stage surgical resection specimens and were collected in accordance with TCGA Human Subjects Protection and Data Access Policies.
Statistical analyses
All statistical analyses were performed using GraphPad Prism V5.0 software. A significant difference (p < 0.05) between two groups was determined using Student's t tests. Statistical comparisons of the means of multiple groups were determined using one-way ANOVA.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1234570_s02.docx
Download MS Word (8.3 MB)Funding
This work was supported by 2P50CA070907-16A1 to L.C.; CPRIT RP150405, Uniting Against Lung Cancer/Lung Cancer Research Foundation award, Rexanna's Foundation for Fighting Lung Cancer to D.L.G.; ACS RSG LIB-117155 and 5-P50-CA70907-12 PP-3b to X.F.Q.; NRF-2014R1A1A1002340 and 2010-0027945 to Y.H.A.; CPRIT Graduate Scholar Training Grant (RP140106) to D.P. (David Peng); and NIH Cancer Center Support Grant (CA016672) to MDACC core facilities. D.L.G. and L.A.B. are R. Lee Clark Fellows of the University of Texas MD Anderson Cancer Center, supported by the Jeane F. Shelby Scholarship Fund. The work was also supported by the generous philanthropic contributions to The University of Texas MD Anderson Lung Cancer Moon Shots Program.
ORCID
Jonathon D. Roybal http://orcid.org/0000-0003-3321-2870
Yongbin Yang http://orcid.org/0000-0002-3398-9244
David Peng http://orcid.org/0000-0003-0511-1888
Stephen E. Ullrich http://orcid.org/0000-0003-4748-0913
References
- Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420:860-7; PMID:12490959; http://dx.doi.org/10.1038/nature01322
- de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer 2006; 6:24-37; PMID:16397525; http://dx.doi.org/10.1038/nrc1782
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010; 140:883-99; PMID:20303878; http://dx.doi.org/10.1016/j.cell.2010.01.025
- Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer 2009; 9:239-52; http://dx.doi.org/10.1038/nrc2618
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature 2008; 454:436-44; PMID:19279573; http://dx.doi.org/10.1038/nature07205
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313:1960-4; PMID:17008531; http://dx.doi.org/10.1126/science.1129139
- Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med 2005; 353:2654-66; PMID:16371631; http://dx.doi.org/10.1056/NEJMoa051424
- Sharma P, Shen Y, Wen S, Yamada S, Jungbluth AA, Gnjatic S, Bajorin DF, Reuter VE, Herr H, Old LJ et al. CD8 tumor-infiltrating lymphocytes are predictive of survival in muscle-invasive urothelial carcinoma. Proc Natl Acad Sci USA 2007; 104:3967-72; PMID:17360461; http://dx.doi.org/10.1073/pnas.0611618104
- Zhuang X, Xia X, Wang C, Gao F, Shan N, Zhang L, Zhang L. A high number of CD8+ T cells infiltrated in NSCLC tissues is associated with a favorable prognosis. Appl Immunohistochem Mol Morphol 2010; 18:24-8; PMID:19713832; http://dx.doi.org/10.1097/PAI.0b013e3181b6a741
- Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol 2004; 22:329-60; PMID:15032581; http://dx.doi.org/10.1146/annurev.immunol.22.012703.104803
- DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, Crowley D, Chen J, Jacks T. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 2011; 19:72-85; PMID:21251614; http://dx.doi.org/10.1016/j.ccr.2010.11.011
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 2005; 5:263-74; PMID:15776005; http://dx.doi.org/10.1038/nrc1586
- Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun 2014; 5:5241; PMID:25348003; http://dx.doi.org/10.1038/ncomms6241
- Chen L, Heymach JV, Qin FX, Gibbons DL. The mutually regulatory loop of epithelial-mesenchymal transition and immunosuppression in cancer progression. Oncoimmunol 2015; 4:e1002731; PMID:26155392; http://dx.doi.org/10.1080/2162402X.2014.1002731
- Ahn YH, Gibbons DL, Chakravarti D, Creighton CJ, Rizvi ZH, Adams HP, Pertsemlidis A, Gregory PA, Wright JA, Goodall GJ et al. ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J Clin Invest 2012; 122:3170-83; PMID:22850877; http://dx.doi.org/10.1172/JCI63608
- Yang Y, Ahn YH, Chen Y, Tan X, Guo L, Gibbons DL, Ungewiss C, Peng DH, Liu X, Lin SH et al. ZEB1 sensitizes lung adenocarcinoma to metastasis suppression by PI3K antagonism. J Clin Invest 2014; 124:2696-708; PMID:24762440; http://dx.doi.org/10.1172/JCI72171
- Yang Y, Ahn YH, Gibbons DL, Zang Y, Lin W, Thilaganathan N, Alvarez CA, Moreira DC, Creighton CJ, Gregory PA et al. The Notch ligand Jagged2 promotes lung adenocarcinoma metastasis through a miR-200-dependent pathway in mice. J Clin Invest 2011; 121:1373-85; PMID:21403400; http://dx.doi.org/10.1172/JCI42579
- Ungewiss C, Rizvi ZH, Roybal JD, Peng DH, Gold KA, Shin DH, Creighton CJ, Gibbons DL. The microRNA-200/Zeb1 axis regulates ECM-dependent beta1-integrin/FAK signaling, cancer cell invasion and metastasis through CRKL. Sci Rep 2016; 6:18652; PMID:26728244; http://dx.doi.org/10.1038/srep18652
- Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ, Thilaganathan N, Du L, Zhang Y, Pertsemlidis A et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev 2009; 23:2140-51; PMID:19759262; http://dx.doi.org/10.1101/gad.1820209
- Gibbons DL, Lin W, Creighton CJ, Zheng S, Berel D, Yang Y, Raso MG, Liu DD, Wistuba II, Lozano G et al. Expression signatures of metastatic capacity in a genetic mouse model of lung adenocarcinoma. PloS One 2009; 4:e5401; PMID:14744438; http://dx.doi.org/10.1371/journal.pone.0005401
- Saintigny P, Massarelli E, Lin S, Ahn YH, Chen Y, Goswami S, Erez B, O'Reilly MS, Liu D, Lee JJ et al. CXCR2 expression in tumor cells is a poor prognostic factor and promotes invasion and metastasis in lung adenocarcinoma. Cancer Res 2013; 73:571-82; PMID:23149918; http://dx.doi.org/10.1158/0008-5472.CAN-12-0263
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116:281-97, [pii]; PMID:14744438; http://dx.doi.org/10.1016/S0092-8674(04)00045-5
- Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res 2008; 68:7846-54; PMID:18829540; http://dx.doi.org/10.1158/0008-5472.CAN-08-1942
- Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008; 10:593-601; PMID:18376396; http://dx.doi.org/10.1038/ncb1722
- Schliekelman MJ, Gibbons DL, Faca VM, Creighton CJ, Rizvi ZH, Zhang Q, Wong CH, Wang H, Ungewiss C, Ahn YH et al. Targets of the tumor suppressor miR-200 in regulation of the epithelial-mesenchymal transition in cancer. Cancer Res 2011; 71:7670-82; PMID:21987723; http://dx.doi.org/10.1158/0008-5472.CAN-11-0964
- Kim JS, Kurie JM, Ahn YH. BMP4 depletion by miR-200 inhibits tumorigenesis and metastasis of lung adenocarcinoma cells. Mol Cancer 2015; 14:173; PMID:26395571; http://dx.doi.org/10.1186/s12943-015-0441-y
- Russell R, Perkhofer L, Liebau S, Lin Q, Lechel A, Feld FM, Hessmann E, Gaedcke J, Güthle M, Zenke M et al. Loss of ATM accelerates pancreatic cancer formation and epithelial-mesenchymal transition. Nat Commun 2015; 6:7677; PMID:26497898; http://dx.doi.org/10.1038/ncomms8677
- Cao Y, Slaney CY, Bidwell BN, Parker BS, Johnstone CN, Rautela J, Eckhardt BL, Anderson RL. BMP4 inhibits breast cancer metastasis by blocking myeloid-derived suppressor cell activity. Cancer Res 2014; 74:5091-102; PMID:25224959; http://dx.doi.org/10.1158/0008-5472.CAN-13-3171
- Weaver M, Dunn NR, Hogan BL. Bmp4 and Fgf10 play opposing roles during lung bud morphogenesis. Development 2000; 127:2695-704; PMID:10821767
- Chen ML, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, von Boehmer H, Khazaie K. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-β signals in vivo. Proc Natl Acad Sci USA 2005; 102:419-24; PMID:15623559; http://dx.doi.org/10.1073/pnas.0408197102
- Zhang Q, Yang XJ, Kundu SD, Pins M, Javonovic B, Meyer R, Kim SJ, Greenberg NM, Kuzel T, Meagher R et al. Blockade of transforming growth factor-{β} signaling in tumor-reactive CD8(+) T cells activates the antitumor immune response cycle. Mol Cancer Ther 2006; 5:1733-43; PMID:16891459; http://dx.doi.org/10.1158/1535-7163.MCT-06-0109
- Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-β signalling inhibitors for cancer therapy. Nat Rev Drug Discov 2004; 3:1011-22; PMID:15573100; http://dx.doi.org/10.1038/nrd1580
- Kaplan FS, Le Merrer M, Glaser DL, Pignolo RJ, Goldsby RE, Kitterman JA, Groppe J, Shore EM. Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol 2008; 22:191-205; PMID:18328989; http://dx.doi.org/10.1016/j.berh.2007.11.007
- Rajan P, Panchision DM, Newell LF, McKay RD. BMPs signal alternately through a SMAD or FRAP-STAT pathway to regulate fate choice in CNS stem cells. J Cell Biol 2003; 161:911-21; PMID:12796477; http://dx.doi.org/10.1083/jcb.200211021
- Wolfle SJ, Strebovsky J, Bartz H, Sähr A, Arnold C, Kaiser C, Dalpke AH, Heeg K. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur J Immunol 2011; 41:413-24; PMID:21268011; http://dx.doi.org/10.1002/eji.201040979
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006; 439:682-7; PMID:16382236; http://dx.doi.org/10.1038/nature04444
- Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 2013; 13:227-42; PMID:23470321; http://dx.doi.org/10.1038/nri3405
- Zinselmeyer BH, Heydari S, Sacristán C, Nayak D, Cammer M, Herz J, Cheng X, Davis SJ, Dustin ML, McGavern DB. PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J Exp Med 2013; 210:757-74; PMID:23530125; http://dx.doi.org/10.1084/jem.20121416
- Horne-Debets JM, Faleiro R, Karunarathne DS, Liu XQ, Lineburg KE, Poh CM, Grotenbreg GM, Hill GR, MacDonald KP, Good MF et al. PD-1 dependent exhaustion of CD8(+) T cells drives chronic malaria. Cell Rep 2013; 5:1204-13; PMID:24316071; http://dx.doi.org/10.1016/j.celrep.2013.11.002
- Curran MA, Montalvo W, Yagita H, Allison J. P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. P Natl Acad Sci USA 2010; 107:4275-80; PMID:20160101; http://dx.doi.org/10.1073/pnas.0915174107
- Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015; 520:373-7; PMID:25754329; http://dx.doi.org/10.1038/nature14292
- Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 2009; 30:1073-81; PMID:19468060; http://dx.doi.org/10.1093/carcin/bgp127
- Lou Y, Diao L, Cuentas ER, Denning WL, Chen L, Fan YH, Byers LA, Wang J, Papadimitrakopoulou VA, Behrens C et al. Epithelial-mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma. Clin Cancer Res 2016; 22:3630-42; PMID:26851185; http://dx.doi.org/10.1158/1078-0432.CCR-15-1434
- Mak MP, Tong P, Diao L, Cardnell RJ, Gibbons DL, William WN, Skoulidis F, Parra ER, Rodriguez-Canales J, Wistuba II et al. A patient-derived, PAN-cancer EMT signature identifies global molecular alterations and immune target enrichment following epithelial-to-mesenchymal transition. Clin Cancer Res 2016; 22:609-20; PMID:26420858; http://dx.doi.org/10.1158/1078-0432.CCR-15-0876
- Lee YC, Cheng CJ, Bilen MA, Lu JF, Satcher RL, Yu-Lee LY, Gallick GE, Maity SN, Lin SH. BMP4 promotes prostate tumor growth in bone through osteogenesis. Cancer Res 2011; 71:5194-203; PMID:21670081; http://dx.doi.org/10.1158/0008-5472.CAN-10-4374
- Kallioniemi A. Bone morphogenetic protein 4-a fascinating regulator of cancer cell behavior. Cancer Genet 2012; 205:267-77; PMID:22749032; http://dx.doi.org/10.1016/j.cancergen.2012.05.009
- Alarmo EL, Huhtala H, Korhonen T, Pylkkänen L, Holli K, Kuukasjärvi T, Parkkila S, Kallioniemi A. Bone morphogenetic protein 4 expression in multiple normal and tumor tissues reveals its importance beyond development. Mod Pathol 2013; 26:10-21; PMID:22899288; http://dx.doi.org/10.1038/modpathol.2012.128
- Richter A, Valdimarsdottir L, Hrafnkelsdottir HE, Runarsson JF, Omarsdottir AR, Ward-van Oostwaard D, Mummery C, Valdimarsdottir G. BMP4 promotes EMT and mesodermal commitment in human embryonic stem cells via SLUG and MSX2. Stem Cells 2014; 32:636-48; PMID:24549638; http://dx.doi.org/10.1002/stem.1592
- Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006; 444:761-5; PMID:17151667; http://dx.doi.org/10.1038/nature05349
- Gordon KJ, Kirkbride KC, How T, Blobe GC. Bone morphogenetic proteins induce pancreatic cancer cell invasiveness through a Smad1-dependent mechanism that involves matrix metalloproteinase-2. Carcinogenesis 2009; 30:238-48; PMID:19056927; http://dx.doi.org/10.1093/carcin/bgn274
- Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol 2015; 25:675-86; PMID:26437589; http://dx.doi.org/10.1016/j.tcb.2015.07.012
- Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol 2007; 19:813-24; PMID:17606980; http://dx.doi.org/10.1093/intimm/dxm057
- Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, Hieber M, Arbeiter A, Klein S, Kong B et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 2013; 23:406-20; PMID:23453624; http://dx.doi.org/10.1016/j.ccr.2013.01.023
- Di Piazza M, Nowell CS, Koch U, Durham AD, Radtke F. Loss of cutaneous TSLP-dependent immune responses skews the balance of inflammation from tumor protective to tumor promoting. Cancer Cell 2012; 22:479-93; PMID:23079658; http://dx.doi.org/10.1016/j.ccr.2012.08.016
- Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012; 22:425-37; PMID:23079654; http://dx.doi.org/10.1016/j.ccr.2012.08.024
- Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986; 233:1318-21; PMID:3489291; http://dx.doi.org/10.1126/science.3489291
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001; 29:e45; PMID:11328886; http://dx.doi.org/10.1093/nar/29.9.e45
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014; 511:543-50; PMID:25079552; http://dx.doi.org/10.1038/nature13385