
ABSTRACT
There is an ever increasing amount of evidence to support the hypothesis that complement C1q, the first component of the classical complement pathway, is involved in the regulation of cancer growth, in addition to its role in fighting infections. It has been demonstrated that C1q is expressed in the microenvironment of various types of human tumors, including breast adenocarcinomas. This study compares carcinogenesis progression in C1q deficient (neuT-C1KO) and C1q competent neuT mice in order to investigate the role of C1q in mammary carcinogenesis. Significantly accelerated autochthonous neu+ carcinoma progression was paralleled by accelerated spontaneous lung metastases occurrence in C1q deficient mice. Surprisingly, this effect was not caused by differences in the tumor-infiltrating cells or in the activation of the complement classical pathway, since neuT-C1KO mice did not display a reduction in C3 fragment deposition at the tumor site. By contrast, a significant higher number of intratumor blood vessels and a decrease in the activation of the tumor suppressor WW domain containing oxidoreductase (WWOX) were observed in tumors from neuT-C1KO as compare with neuT mice. In parallel, an increase in Her2/neu expression was observed on the membrane of tumor cells. Taken together, our findings suggest that C1q plays a direct role both on halting tumor angiogenesis and on inducing apoptosis in mammary cancer cells by coordinating the signal transduction pathways linked to WWOX and, furthermore, highlight the role of C1q in mammary tumor immune surveillance regardless of complement system activation.
Abbreviations
| BALB-C1KO | = | BALB/c mice deficient for the C1qA |
| BALB-C3KO | = | BALB/c mice deficient for C3 |
| C1KO | = | C1q deficient |
| C1qR | = | C1q receptor |
| CR3 | = | complement receptor 3 |
| EMT | = | epithelial-to-mesenchymal transition |
| KLRK1 or NKG2D | = | killer cell lectin-like receptor subfamily K, member 1 |
| MDCS | = | myeloid-derived suppressor cells |
| neuT | = | rat Her2/neu transgenic |
| neuT-BKO mice | = | neuT mice deficient in antibody production |
| neuT-C1KO mice | = | neuT mice deficient for the C1qA molecule |
| neuT-C3KO mice | = | neuT mice deficient for the C3 molecule |
| NK | = | natural killer |
| pWWOX | = | phospho WWOX |
| Treg | = | T regulatory |
| WWOX | = | WW domain containing oxidoreductase |
Introduction
It has now been clearly demonstrated that the survival and progression of a primary tumor depend on three major events: immune evasion, angiogenesis, and metastasis. These complex biological processes can be promoted and halted by a number of factors, including complement cascade components.Citation1 There is sufficient evidence to demonstrate the presence of complement deposition on tumor tissues from patients in a variety of cancer types.Citation2,3 Nevertheless, the role of the complement system in tumor growth and metastatic spread has not yet been sufficiently addressed, and contradictory findings are still being reported.Citation4,5
C1, the first component of the classical complement pathway, is a multimolecular complex composed of C1q, C1r, and C1s molecules that associate in a calcium-dependent macromolecular complex. C1q, the ligand recognition subcomponent of the complex, is composed by six heterotrimeric structures, made of A (C1qA), B (C1qB), and C (C1qC) chains, with a collagen-like and a C-terminal globular region; the latter is the moiety responsible for ligand binding.Citation6 When C1q binds to IgM or clustered IgG on immunocomplexes, or to surface-bound pentraxins, other complement cascade components are sequentially activated leading to the generation of effector molecules which are able to limit infection by pathogens and play an essential homeostatic role in the clearance of damaged self-antigens, immune complexes, altered self, and apoptotic cells.Citation7
Besides this role in the activation of the classical complement pathway through the recognition of immune complexes,Citation8 it is now recognized that C1q can carry out functions that are unrelated to complement activation. Recent studies have demonstrated its involvement in several physiological processes via binding with its specific receptors.Citation9 Among the processes induced by the non-classical functions of the C1q molecule, we find: the modulation of various immune cells,Citation10 the regulation of cell migration (chemotaxis), adhesion, survival and differentiation,Citation11 coagulation,Citation12 angiogenesisCitation13 and embryonic development, including neurological synapse function.Citation14
There is mounting evidence to support the idea that C1q and its receptors are also involved in the regulation of cancer. The recently postulated ability of C1q to induce apoptosis in prostate cancer cells by coordinating signal transduction pathways linked to the tumor suppressor WW domain containing oxidoreductase (WWOX),Citation15 has highlighted its important role as a humoral factor which is needed to directly block cancer cell proliferation, without the involvement of antitumor antibodies and the consequent activation of the classical complement pathway. The direct antitumor effect played by C1q is also highlighted by the fact that it is significantly downregulated in benign prostatic hyperplasia and prostate cancer despite being expressed in normal prostate tissues.Citation15 Furthermore, recent data demonstrate that C1q receptors (C1qR) expression is upregulated on almost all types of malignant cells.Citation9 Interestingly, two of the best-known C1qR,Citation16 cC1qR and gC1qR, have opposite functions; while cC1qR is detrimental to tumor growth because it is a pro-phagocytic signal, gC1qR promotes tumor cell growth, angiogenesis, and metastases formation.Citation9 Moreover, tumor cells not only express gC1qR on their membrane but they can also secrete it into the tumor microenvironment, protecting cancer cells from C1q-induced apoptosis by impounding the C1 molecule. Soluble gC1qR, has also a role to play, by activating the complement system, in the generation of potent vasoactive peptides which are able to enhance vascular permeability, thus facilitating tumor cell escape and consequent metastatization.Citation9
A growing number of papers in recent years have linked the expression of the complement cascade components and regulators to the prognosis of breast cancer patients.Citation17,18 CD59, a membrane complement regulatory protein, has recently been found to be overexpressed in breast cancer where it facilitates tumor cell escape from complement surveillance.Citation19 Significantly differential complement cascade pathway expression was observed in luminal A, luminal B, basal-like, and Her2+ mammary cancers.Citation20 The complement factor C1q, which was found to be downregulated, appears to play a noteworthy and important role in this context.Citation18 However, the mechanisms that underlie the effects exerted by C1q on breast cancer progression have not yet been sufficiently clarified, meaning that deeper understanding of the molecular interactions between tumors and C1q may lead to the identification of additional pathways and targets which can be exploited for combined therapy.
The genetic predisposition of rat Her2/neu transgenic (neuT) mice to developing lethal mammary carcinomas, characterized by well-defined gene expression signature,Citation21 progression and long-lasting interaction with the host microenvironment, make these mice an appealing model for the evaluation of the role of immunosurveillance.Citation22,23 We have previously demonstrated that there is a deposition of C3 on tumor cells and vessels during Her2/neu-driven mammary carcinogenesis in neuT mice and that natural antibodies which are directed against antigens expressed by tumor cells arise concomitantly.Citation24 These observations suggest that complement activation through the classical pathway may play a key role in tumor immunosurveillance. To verify this hypothesis, the present work sees us generate neuT mice that are deficient for the C1qA molecule (neuT-C1KO mice) and then evaluate the progression of carcinogenesis in their mammary glands in comparison to neuT mice. Surprisingly, our data demonstrate that C1q can act as tumor-inhibiting factor, regardless of complement activation.
Results
Autochthonous Her2/neu+ carcinoma progression is accelerated in C1q deficient mice
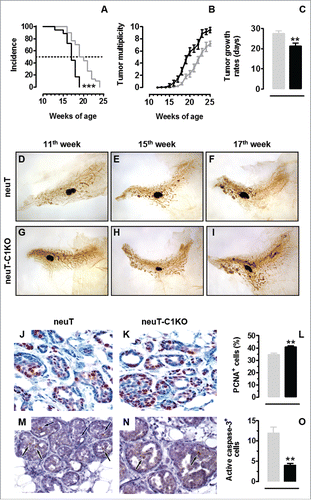
Significantly accelerated tumor incidence was observed when mammary carcinogenesis was evaluated in neuT-C1KO mice, with 100% of mice already bearing a palpable tumor at week 19 of age, when 50% of neuT mice were still free from palpable tumors (; p < 0.0001, Log-rank Mantel-Cox test). The lack of the C1q molecule in neuT mice also impacts on tumor multiplicity and tumor growth. Indeed, from the 17th week of age (; p values ranging from 0.04 to <0.0001, Student's t-test), a significant increase in tumor multiplicity was observed in neuT-C1KO mice, as compared with neuT mice, and tumors grew significantly more quickly (; p = 0.001, Student's t-test). A histological analysis of mammary glands from 11-, 15-, and 17-week-old mice confirmed that progression from hyperplastic lesions to in situ and diffuse carcinomas was accelerated in neuT-C1KO mice (). By week 11, the hyperplastic lesions appeared to be more numerous than those in neuT mice (). At week 15, neuT-C1KO mice displayed multifocal lesions, which were larger and evenly more spread throughout the mammary gland than those observed in neuT mice (). By week 17, these lesions converged into multiple, large nodules similar to in situ carcinomas that were more diffused and expanded than in neuT mice (). Nevertheless, carcinomas progressing in neuT-C1KO and neuT mice displayed a similar grade of differentiation (). However, a significantly higher number of PCNA+ tumor cells was found in carcinomas from neuT-C1KO as compared with neuT mice suggesting an increasing of tumor cell proliferation in C1q deficient neuT mice (; p = 0.001, Student's t-test). In addition, even if very few apoptotic cells were detectable in the mammary tumors from both neuT and neuT-C1KO mice, a significant decrease in the number of active Caspase-3+ cells was observed in mice lacking C1q (; p = 0.002, Student's t-test), suggesting a decreased apoptosis in the absence of C1q molecule.
Figure 1. C1q deficiency is responsible for accelerated tumor growth in neuT mice. Tumor incidence (A) and multiplicity (B) of mammary carcinomas in neuT (n = 30, gray line) and neuT-C1KO (n = 18, black line) mice. Earlier incidence (***p < 0.0001, Log-rank Mantel-Cox test) and higher tumor multiplicity (starting from the 17th week of age, p values ranging from 0.04 to <0.0001, Student's t-test) were found in neuT-C1KO as compared with neuT mice. (C) Time required for a 2 mm mean diameter tumor to reach an 8 mm threshold. Tumors that arose in neuT-C1KO (black bar) mice grew significantly faster than those growing in neuT (gray bar) mice (**p = 0.001, two-tailed Student's t-test). (D–I) Representative whole mount images of the fourth (inguinal) mammary glands of 11- (D, G), 15- (E–H), 17- (F, I) week-old neuT (D–F) and neuT-C1KO (G–I) mice. The central oval black shadows are the intra-mammary lymph nodes. Magnification × 6.3. (J, K) Histological and immunohistochemical staining for the PCNA of mammary tumor lesions in neuT (J) and neuT-C1KO (K) mice. Magnification × 400. PCNA+ tumor cell quantification (L) in neuT (gray bar) and in neuT-C1KO (black bar) mice (**p = 0.001, two-tailed Student's t-test). (M, N) Histological and immunohistochemical staining for the active caspase-3 in mammary tumor lesions of neuT (M) and neuT-C1KO (N) mice. Black arrows indicate apoptotic tumor cells. Magnification × 400. Active caspase-3+ tumor cell quantification (O) in neuT (gray bar) and in neuT-C1KO (black bar) mice (**p = 0.002, two-tailed Student's t-test).
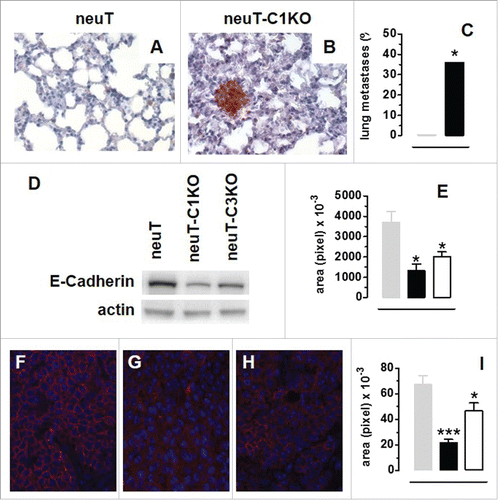
The accelerated tumor onset observed in neuT-C1KO mice was paralleled by a significantly accelerated spontaneous lung metastasis development. Indeed, at week 17, when neuT mice were completely free from lung metastases, 36% of neuT-C1KO mice displayed Her2/neu+ nodules in their lungs ( ; p = 0.05, Chi-square test). This accelerated metastatization suggests that the epithelial-to-mesenchymal transition (EMT) occurs earlier in neuT-C1KO tumors. In order to investigate this issue, we evaluated the neuT and neuT-C1KO tumor expression of E-Cadherin, whose functional loss or downregulation is considered a hallmark of EMT.Citation25 Moreover, since a link a between C3 overexpression, C3a generation and EMT has been recently reported,Citation26 we also evaluated the expression of E-Cadherin in the tumors from neuT mice deficient for the C3 molecule (neuT-C3KO mice). Western blotting showed significantly lower E-cadherin expression in neuT-C1KO and neuT-C3KO as compared with neuT tumors (; p < 0.05, Student's t-test). We used fluorescence microscopy () to confirm the immunoblot findings and determine the localization of E-cadherin in tumor cells. While tumor cells from neuT mice displayed E-cadherin expression at contact sites between cells (), tumor cells from neuT-C1KO () and neuT-C3KO () mice provided almost negative staining and fluorescent signal quantification showed significantly lower E-cadherin expression in neuT-C1KO and neuT-C3KO than in neuT mice (; p < 0.0001, p = 0.04, Student's t-test, respectively).
Figure 2. C1q deficiency is associated with anticipated metastatic spread and epithelial-to-mesenchymal transition in neuT tumors. Histological and immunohistochemical staining for Her2/neu of lungs from 17-week-old neuT (A) and neuT-C1KO (B) mice reveal earlier metastatic infiltration in neuT-C1KO mice. Magnification × 400. (C) Percentage of neuT (n = 19, gray bar) and neuT-C1KO (n = 14, black bar) mice (*p = 0.05 Chi-square test) bearing lung metastatic lesions at 17 weeks of age. (D–I) Decreased expression of E-Cadherin in neuT-C1KO and neuT-C3KO vs. neuT tumors. (D) E-Cadherin (upper panel) and actin (lower panel) protein levels as measured using the immunoblotting of whole cell lysates from 6–8 mm mean diameter carcinomas. A representative blot from three independent experiments is shown. (E) Quantification of E-Cadherin protein expression in neuT (gray bar), in neuT-C1KO (black bar) and neuT-C3KO (white bar) tumors (*p < 0.05, two-tailed Student's t-test). (F–H) Representative microscopy images of tumor sections from neuT (F), neuT-C1KO (G), and neuT-C3KO (H) mice (n = 3 per group) labeled with anti-E-Cadherin antibody (red) and DAPI (blue, labeling nuclei). Magnification × 400. (I) E-Cadherin protein was quantified in neuT (gray bar), neuT-C1KO (black bar) (***p < 0.0001, two-tailed Student's t-test) and neuT-C3KO (white bar) tumors (*p = 0.04, two-tailed Student's t-test). Results are represented as means ± SEM.
Accelerated carcinogenesis in neuT-C1KO mice is independent of the absence of the classical complement activation pathway
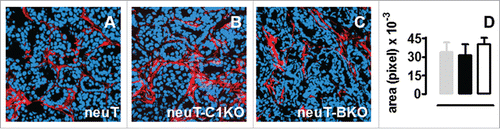
The accelerated pace of tumor progression in neuT-C1KO mice points toward a key role for the complement system in hampering Her2/neu autochthonous mammary tumor progression, as previously demonstrated in neuT-C3KO mice.Citation24 The contribution of C1q to tumor immunosurveillance should mainly be due to the triggering of the classical pathway of complement activation that occurs after binding with natural antitumor antibodies present in the sera of tumor-bearing mice.Citation24 If this were the case, C3 fragments deposition in the mammary glands would be drastically lower in neuT-C1KO mice than in neuT. Nevertheless, the confocal immunofluorescence microscopy analysis of tumors after staining with anti-C3b/iC3b/C3c antibodies showed that comparable levels of C3 cleavage products are present in neuT and neuT-C1KO tumors (). Moreover, as previously observed in neuT mice,Citation24 C3 cleavage products were clearly evident both in the tumor vasculature and stroma of mammary tumors developing in neuT-C1KO mice (Fig. S1). A further investigation was therefore performed on neuT mice that were deficient in antibody production (neuT-BKO mice).Citation27 C3 deposition in neuT-BKO tumors was similar to that observed in neuT and neuT-C1KO mice (), confirming that complement activation is independent of the classical pathway in neuT mice.
Figure 3. The dispensable role of the classical complement activation pathway in neuT tumor immunosurveillance. C3 fragment deposition at the tumor site is not altered in the absence of C1q or antibodies. (A–C) Confocal microscopy images representative of frozen tumor sections from mammary glands of 17-week-old neuT (A), neuT-C1KO (B), and neuT-BKO (C) mice labeled with anti-C3b/iC3b/C3c antibody (red) and TO-PRO®-3 iodide (blue). Magnification × 400. C3 fragment deposition was quantified (D) in neuT (gray bar), neuT-C1KO (black bar), and neuT-BKO (white bar) mice (n = 10 each group). No differences in C3 fragments deposition were found (p = ns, two-tailed Student's t-test). Results are represented as means ± SEM from 3 × 400 microscopic fields per sample.
C1q deficiency is associated with decreased activation of the oncosuppressor WWOX and increased Her2/neu expression
The influence of C1q on tumor progression does not appear to be due to complement cascade activation and so we decided to determine whether it acts on the tumor cell phenotype directly. Recent studies have reported that C1q protein may induce the apoptosis of cancer cells during the initial hyperplasia and cancerous stages of cancer progression by activating the tumor suppressor WWOX pathway.Citation15 In order to study whether a correlation between C1q deficiency and WWOX activation exists in neuT tumors, immunofluorescence staining of tumors from neuT and neuT-C1KO mice was performed. While clear positivity for the phospho (p) WWOX protein was observed in neuT tumor cells (), it was found to be significantly reduced in neuT-C1KO mice (; p = 0.01, Student's t-test). Interestingly, a similar reduction in pWWOX was also observed in tumors from neuT-C3KO mice (; p = 0.02, Student's t-test), despite the C1qA gene not being knocked out in these mice. However, significantly lower levels of C1q fragment deposition were observed at the tumor site in neuT-C3KO than in neuT mice (; p = 0.009, Student's t-test). The confocal immunofluorescence microscopy analysis of tumors after staining with anti-C1q and CD31 antibodies showed that C1q was deposited mainly in the tumor vasculature in both neuT and neuT-C3KO mice (Fig. S2).
Figure 4. Decrease of pWWOX and increase of Her2/neu expression in neuT-C1KO tumors. Confocal microscopy images of frozen tumor sections from neuT (A, E, I), neuT-C1KO (B, F, J), and neuT-C3KO (C, G, K) mice (n = 7 per group) labeled with anti-pWWOX (red, A–C), anti-C1q (red, E–G), and anti-Her2/neu (red, I–K) antibodies. Nuclei were stained with TO-PRO®-3 iodide (blue). Magnification ×100. pWWOX (D), C1q (H), and Her2/neu (L) protein quantification was performed in neuT (gray bar), neuT-C1KO (black bar), and neuT-C3KO (white bar) mice (*p = 0.02 for pWWOX; *p = 0.04 and **p = 0.009 for C1q; *p ≤ 0.04 for Her2/neu; two-tailed Student's t-test). Results are represented as means ± SEM from 3 × 400 microscopic fields per sample.
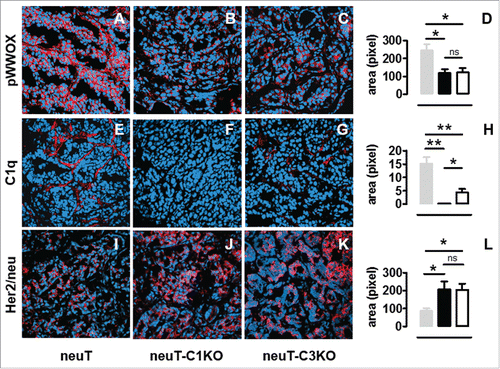
Since neuT tumors are strictly addicted to Her2/neu,Citation28 we evaluated its expression using confocal immunofluorescence microscopy. As compared with tumors in neuT mice (), Her2/neu protein expression was significantly increased in neuT-C1KO tumors (; p = 0.05, Student's t-test). This enhanced expression was similar to what observed in neuT-C3KO ( ; p = 0.02, Student's t-test) tumors. Her2/neu appeared to be expressed mainly at the cell membrane of neuT tumor cells and its expression appeared to be higher and broader in both complement-deficient strains.
C1q deficiency affects tumor vessel density but not the frequency of tumor-infiltrating leukocytes
Our data indicate that the more aggressive phenotype of neuT-C1KO, as compared with neuT tumors (), may be a result of the reduced activation of tumor suppressor WWOX and the consequent survival of tumor cell clones that present stronger Her2/neu oncoprotein expression. To evaluate whether C1q deficiency also impacts on the tumor microenvironment, we evaluated the vessel density () and the frequency of tumor-infiltrating immune cells () in neuT, neuT-C1KO, and neuT-C3KO tumors of equivalent size.
Figure 5. C1q deficiency affects intratumoral vessel density but does not modify tumor-infiltrating leukocyte recruitment. (A–C) Representative images of immunohistochemical staining for endothelial cell markers (CD31 and CD105, red) to visualize blood vessels in mouse tumors of equal volume developed in neuT (A) neuT-C1KO (B) and neuT-C3KO (C) mice. Magnification × 200. Quantification of the number (D) of vessels in neuT (gray bar; n = 3), neuT-C1KO (black bar; n = 5), and neuT-C3KO (white bar; n = 5) carcinomas (*p = 0.04; two-tailed Student's t-test). Results are represented as means ± SEM from 5 × 200 microscopic fields per sample. (E–G) Representative confocal microscopy images of tumors from neuT (E) neuT-C1KO (F) and neuT-C3KO (G) mice stained with anti-CD31 antibodies (green). Magnification × 400. Quantification of the vessel area (H) in neuT (gray bar; n = 3), neuT-C1KO (black bar; n = 5), and neuT-C3KO (white bar; n = 5) carcinomas. (***p < 0.0001; two-tailed Student's t-test). Results are represented as means ± SEM from 5 × 200 microscopic fields. (I–M) Flow cytometry analysis of infiltrating leukocytes in 6–8 mm mean diameter tumors from neuT (n = 5; gray bars), neuT-C1KO (n = 7; black bars) and neuT-C3KO (n = 6; white bars) mice. (I) CD3+ leukocytes were gated and CD3+ CD4+ CD25+ FoxP3+ were identified as Tregs (**p = 0.005; two-tailed Student's t-test). (L) CD45+ leukocytes were gated and CD3+ CD4+ cells were identified as CD4+ T, CD3+ CD8+ as CD8 T, CD3+ γδ+ as γδ T and CD3− CD49b+ as NK (**p = 0.005; two-tailed Student's t-test). (M) CD45+ CD11b+ leukocytes were gated and F4/80+ cells were identified as macrophages (MΦ), whereas GR-1+ cells were identified as myeloid-derived suppressor cells (MDSC). Bars represent the percentage of positive cells ± SEM.
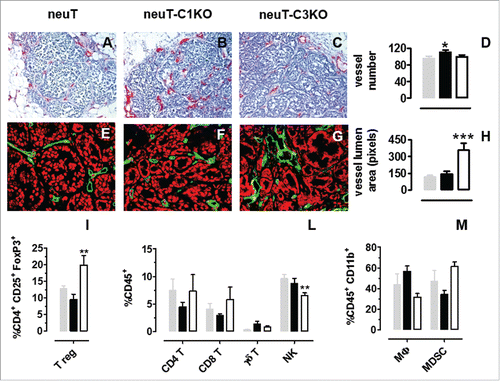
Immunohistochemical staining for endothelial cell markers () displays a statistically higher number of intratumor vessels in neuT-C1KO, as compared with neuT and neuT-C3KO mice (; p = 0.04, Student's t-test). On the other hand, the assessment of vessel diameter indicates that, although higher in number, tumor-associated blood vessels in neuT-C1KO tumors are similar in dimension when compared with those of neuT mice ( ). By contrast, as previously reported,Citation24 significantly larger intratumor vessels are evident in carcinomas developing in the absence of C3 (; p <0.0001 Student's t-test).
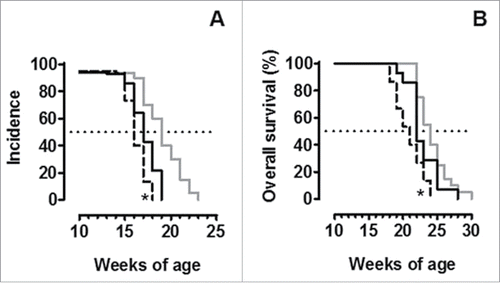
The frequency of tumor-infiltrating immune cells was evaluated using flow cytometry. Tumors of neuT and neuT-C1KO mice displayed no statistically significant differences in the frequency of T regulatory (Treg) cells, CD4+, CD8+, and γδ T cells, natural killer (NK) cells, macrophages and myeloid-derived suppressor cells (MDSC) ( ). These data suggest that the C1q molecule is not involved in regulating leukocyte recruitment at the tumor site. This is in contrast to observations in C3 deficient mice which instead show a significant increase in infiltrating Treg cells (; p = 0.005, Student's t-test), as previously shown,Citation24 and a significant reduction in NK cells (; p = 0.005, Student's t-test). These differences, together with those in vessel dimension and complement activation, justify the fact that neuT-C1KO tumors are less aggressive than those of neuT-C3KO mice (; p = 0.05 and p = 0.02, Log-rank Mantel-Cox test) despite showing comparable levels of pWWOX and Her2/neu expression ( ).
Figure 6. NeuT-C1KO tumors are less aggressive than those of neuT-C3KO mice. Tumor incidence of mammary carcinomas (A) and overall mice survival (B) in neuT (n = 20, continuous gray line), neuT-C1KO (n = 14, continuous black line) and neuT-C3KO (n = 15, dotted black line) mice. NeuT-C3KO mice displayed earlier tumor incidence (*p = 0.05, Log-rank Mantel-Cox test) and lower overall survival (*p = 0.02, Log-rank Mantel-Cox test) than neuT-C1KO mice.
Discussion
Several studies have suggested that the complement system plays a critical role in cancer growth and spread. It has been demonstrated that cancer cells establish a balance between complement activation and inhibition.Citation29 However, controversial and conflicting data on the complement system's tumor-promoting,Citation30,31 and inhibiting activities,Citation24 have been published,Citation2 and the mechanisms of complement-specific activities in the tumor microenvironment are still unclear and demands further study.
The use of genetically engineered mice, which are predestined to develop fatal tumors and are knocked out for one of the key complement cascade molecules, enabled us to test the role of complement activation in cancer growth and development and will possibly lead to the design of complement-related anticancer strategies.
We have recently exploited the genetic predestination of neuT female mice to mammary carcinogenesis to assess the weight of C3 complement-mediatedCitation24 immune surveillance in the development of autochthonous carcinomas. The data obtained demonstrated that the loss of C3 activation, whose fragments were found to specifically accumulate in vessels and stroma in and around the incipient cancers, was responsible for the dramatic increase in the aggressiveness of neu+ mammary tumors. Concomitantly, as had already been demonstrated in cancer patients,Citation32,33,34 spontaneous antitumor antibodies increased in neuT mice during the course of tumor progression.Citation24 Taken together, these observations suggest that the complement system can take on a tumor-inhibiting role that is played out through the activation of the classical pathway.
The involvement of classical complement pathway activation in tumor cell killing was further investigated by generating neuT-C1KO mice. An accelerated carcinoma progression was evident in these mice. The comparable level of C3 fragment deposition observed in the mammary glands of neuT-C1KO mice and neuT-BKO mice, which are unable to produce antibodies, highlights the fact that tumor cell killing is not related to complement classical pathway activation in neuT mice. By contrast, the decreased activation of the oncosuppressor WWOX and the overexpression of the neuT protein on the mammary tumor cells found in neuT-C1KO mice point to the key and direct role that C1q plays in tumor cell elimination. Indeed, recent data have highlighted the existence of non-canonical functions that are exerted by C1q on several types of cells, including cancer cells. Irrespectively of the beneficial or harmful impact that the complement system has on tumor growth, the C1q molecule's contribution to tumor progression and metastasization has been demonstrated regardless of complement activation, both in prostatic cancer cellsCitation15,35 and very recently in melanoma.Citation31
These results provide a new perspective on our previously published data which demonstrated that the neuT upregulation on tumor cells observed in neuT-C3KO tumors was not only directly dependent on lack of C3 activation. In particular, the extremely aggressive phenotype displayed by neuT-C3KO tumors can also be caused by the impaired activation of WWOX, which is a result of the unexpected lack of C1q deposition on the tumor site of neuT-C3KO mice. A relationship between C1q-induced expression and hypoxia has been demonstrated in neurons.Citation36 Although the mechanisms by which C1q mRNA expression is upregulated by hypoxia remain to be investigated, it has been demonstrated that cultured cells, which did not express C1q before hypoxia, did express C1q mRNA and protein during and after hypoxia, respectively.Citation36 It has recently been demonstrated, by both immunohistochemistry and western blotting experiments, that neuT tumor cells express a low but detectable level of HIF1 protein.Citation37 Similar amounts of HIF1 protein were also observed in neuT-C1KO tumors, while it was almost absent in neuT-C3KO tumors (Figs. S3A–C). These data, together with the previously reported increased blood vessel permeability and reduced necrotic areas within neuT-C3KO neoplasms, as compared with neuT lesions, suggest that neuT-C3KO tumors display a lower hypoxic phenotype than that of both neuT and neuT-C1KO tumors.Citation24 It can be speculated thus, speculate that the unexpected lack of the C1q protein observed in neuT-C3KO tumors may be due to the lack of hypoxic-induced C1q mRNA transcription. Indeed, using the LASAGNA-Search toolCitation38 (http://biogrid.engr.uconn.edu/lasagna_search/) to perform binding sites searches on mouse C1q promoter, we confirmed the presence of binding sites specific for the transcriptional factors RelA,Citation39 Meis-1a,Citation40 AhRCitation41, and Arnt,Citation42 known to be present on human C1q promoter and linked to hypoxia (GeneCards®, Human Gene Data base; http://www.genecards.org/cgi-bin/carddisp.pl?gene = C1QAandkeywords = c1qa).
Contradictory effects of complement in modulating vascularization and angiogenesis are described in the literature.Citation30,31,43 Our immunohistochemistry data showed a marked deposition of C1q on vascular endothelium and stroma of neuT tumors. On the other hand, the increased tumor vascularization observed in neuT-C1KO mice suggests that C1q could be considered an inhibitor of tumor angiogenesis, at least in our model of autochthonous Her2/neu-driven mammary cancer that display a process of angiogenesisCitation44 not superimposable to that observed in transplantable tumors.Citation31 Further investigations are warranted to define the underlying mechanisms of this C1q-mediated inhibition of tumor vasculature.
The hypothesis that C1q may have an effect in the induction of an immunosuppressive tumor microenvironment, was excluded by an analysis of tumor-infiltrating lymphocytes. Contrary to results in tumors from neuT-C3KO mice,Citation24 no differences in the percentages of MDSC and Treg cells were found. The significant reduction in NK cells in tumors from neuT-C3KO mice, as compare with both neuT and neuT-C1KO tumors, is certainly worthy of note. The lack of a C1q-induced immunosuppressive tumor phenotype, together with the presence of a significant amount of NK cells, can help explain why neuT-C1KO tumors are less aggressive than their neuT-C3KO analogs. Indeed, NK cells can lyse tumor cells after the recruitment of complement receptor 3 (CR3) by iC3b, especially when MHC class I molecules are poorly expressed.Citation45 NeuT+ tumor cells are susceptible to the activity of NK cells, as an inverse correlation exists between the expression levels of neuT protein and those of MHC class I molecules as well as other components of the antigen-processing machinery.Citation46 Moreover, signaling through the Her2/Her3 pathway in breast tumor cell lines has been shown to enhance their recognition by NK and T cells thanks to the killer cell lectin-like receptor subfamily K, member 1 (KLRK1, or NKG2D).Citation47
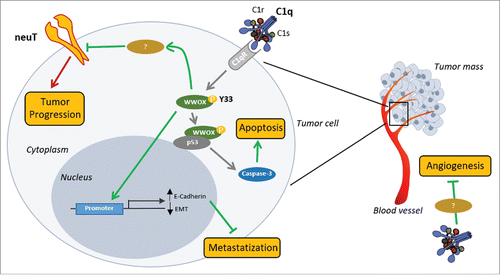
In conclusion, our data demonstrate that the C1q protein protects against the development of Her2/neu+ mammary carcinomas, at least in our preclinical BALB-neuT female mouse model. The tumor-inhibiting C1q-mediated effects do not appear to be associated with the classical pathway of complement, but with its direct role on endothelial and tumor cells. Based on our results and on data from the literature, we propose the following mechanism of C1q direct antitumoral role in neuT mammary carcinogenesis. The C1q binding with its receptor leads to the phosphorylation and consequent activation of WWOX, which is a known oncosuppressor required to block cancer cell proliferation.Citation15 Indeed, activated WWOX may induce the apoptosis of mammary tumor cancer cells, probably by interacting with p53,Citation48 and inhibit the EMT processes through the expression of E-Cadherin. Conversely, the lack of C1q prevents WWOX activation leading to tumor cell growth and metastases formation. As far as the mechanisms linking the absence of C1q and the increased levels of neuT protein expression on mammary tumor cells are concerned, our hypothesis is that WWOX activation may be involved in neuT post-translational negative regulation (). By contrast, WWOX-dependent transcriptional regulation of neuT gene is unlikely since this transgene is under the control of the MMTV promoter and not of the endogenous one.
Figure 7. Proposed mechanism of C1q influence on neuT tumor progression. C1q component of C1 complex (C1q, C1s, and C1r) appears to act directly both on tumor vasculature (on the right) and on tumor cells (on the left). Deposition of C1q on vascular endothelium inhibits tumor angiogenesis through a still undefined mechanism. C1q binding with its receptor(s) (C1qR) on tumor cells leads to the phosphorylation of tyrosine 33 (Y33) on WWOX. Activated WWOX in turn inhibits the EMT processes, through directly inducing the expression of E-Cadherin, and induces Caspase-3-mediated apoptosis, probably by engaging p53. We hypothesize that activated WWOX may be also involved in neuT post-translational negative regulation further contributing to tumor inhibition. Green lines: antitumor activities; red lines: pro-tumor activities.
However, as expected for a system with various distinct activities, this protective role in mammary cancer does not appear to be applicable to every tumor type. Indeed, recent findings have highlighted C1q's role as a cancer-promoting factor in a transplantable model of melanoma.Citation31 However, numerous variables, including the genetic background of mice used, the type of tumors and especially the use of transplantable instead of autochthonous tumors, may all influence how the complement system affects tumor progression, culminating in very different biological outcomes. Moreover, considering the opposite function exerted by the two known C1qR, we can speculate that the tumor inhibiting- or promoting- effect mediated by the C1q protein may results from different degrees of cC1qR or gC1qR expression in the tumor.
Nevertheless, it is important to note that C1q can exert its functions directly on tumor cells, independently of complement activation pathways, both in breast cancer and in melanoma. Defining the role of C1q in different tumor types may lead to the development of new drugs in the clinic, thanks to its role as a major contributor to the immunosurveillance and control of cancer progression as well as tumor growth and dissemination.
Materials and methods
Mice
BALB/c mice that were deficient for the C1qA (BALB-C1KO) and C3 (BALB-C3KO) complement component were provided by Prof. Marina Botto (Imperial College, London, UK).Citation49 NeuT male miceCitation28 from Biogem (Ariano Irpino, Italy) were crossed with BALB-C1KO and with BALB-C3KO females in order to obtain neuT+ C1+/− and neuT+ C3+/− heterozygous male mice, respectively. These heterozygous male mice were then crossed with BALB-C1KO and BALB-C3KO females; the progeny was genotyped in order to identify neuT+ C1−/− (neuT-C1KO) and neuT+ C3−/− (neuT-C3KO) female mice that were then used in the experiments. NeuT-BKO mice were generated by crossing neuT mice with BALB/c mice KO for the immunoglobulin μ chain gene, as previously described.Citation27 The mammary glands of all neuT mice were inspected and palpated twice a week for tumor appearance. Individual neoplastic masses were measured with calipers in two perpendicular diameters and the average value was recorded. Progressively growing masses > 1 mm in mean diameter were regarded as tumors. Neoplastic growth was monitored until the first tumor that exceeded a mean diameter of 10 mm was found, at which point mice were euthanized for ethical reasons. Tumor multiplicity was calculated as the cumulative number of incident individual tumors/total number of mice and is reported as mean ± SEM. All mice were maintained at the Molecular Biotechnology Center, University of Torino, in specific pathogen free conditions (Allentown Caging Equipment, Allentown Inc., Allentown, NJ) and treated in conformity with current European guidelines and policies. The Bioethical Committee of the University of Torino approved the experimental plan.
Morphological analyses
The whole mount preparation of mammary glands was performed by removing the mouse skin and fixing it overnight in 10% buffered formalin. Mammary fat pads were scored into quarters and gently scraped from the skin. These were immersed in acetone overnight and then rehydrated and stained in ferric hematoxylin (Sigma-Aldrich), dehydrated in increasing concentrations of alcohol, cleared with histo-lemon, and stored in methyl salicylate (Sigma-Aldrich). Digital pictures were taken using a Nikon Coolpix 995 (Nital, Medley, FL) mounted on a stereoscopic microscope (MZ6; Leica Microsystems, Milano, Italy) and analyzed as previously described in detail.Citation50
Histology, immunohistochemistry, and immunofluorescence
Lung samples from 14 neuT and 19 neuT-C1KO 17 week-old mice were fixed in formalin and embedded in paraffin. To optimize the detection of microscopic metastases and ensure systematic uniform and random sampling, lungs were processed as previously described.Citation24 For the evaluation of spontaneous metastases of all the experimental groups, sections were stained with hematoxylin/eosin and with anti-Her2 immunohistochemistry. Metastases were counted independently by two pathologists in a blind fashion.
Mammary glands were fixed in formalin and embedded in paraffin or fixed in paraformaldehyde 1% and frozen in a cryo-embedding medium (OCT, Bioptica) for histological and immunohistochemical analyses. Six to 8 mm mammary tumors were fixed in paraformaldehyde 1% and frozen in a cryo-embedding medium (OCT, Bioptica).
Sections were incubated with the mouse monoclonal anti-PCNA antibody (Dako Corporation, M0879), rabbit anti-active caspase-3 (R&D systems, AF835), rabbit anti-human Her-2 (Dako Corporation, A0485), rabbit anti-mouse E-Cadherin (Cell Signaling, 24E10), rabbit anti-phospho-WWOX (pTyrCitation33) (Sigma-Aldrich, SAB4504681), rat anti-mouse C1q (Abcam, ab11861), rat anti-mouse C3b/iC3b/C3c mAb (HyCult biotech, 2/11), rat anti-CD105 (BD PharMingen, 550546), and rat anti-CD31 (BD PharMingen, 550274). For immunohistochemical staining, sections were then incubated with the appropriate biotinylated secondary antibody (Jackson Immunoresearch Laboratories). Immunocomplexes were detected using NeutrAvidin™ Alkaline Phosphatase Conjugated (Thermo Scientific-Pierce Biotechnology) and Vulcan Fast Red (Biocare Medical) or Streptavidin Peroxidase (Thermo Scientific) and DAB Chromogen System (DakoCorporation). For immunofluorescence analysis, sections were then incubated with the appropriate Alexa 488 and 546 labeled secondary antibodies (all from Molecular Probes); nuclei were stained with DAPI (Sigma-Aldrich) or TO-PRO®−3 iodide (Thermo-Fisher Scientific). Images were acquired on a Zeiss ApoTome fluorescence microscope (Axiovert 200M, Zeiss, Jena, Germany) and captured using a CCD cool digital camera (Zeiss) or on a Zeiss LSM 510 META confocal microscopy.
The percentage of PCNA+ tumor cells was evaluated counting positive and negative cells on the digital images of 10 neuT and 10 neuT-C1KO tumors (3 × 400 microscopic fields per tumor) by two pathologists, independently and in a blind fashion.
The number of activated Caspase 3+ tumor cells was evaluated counting positive cells on the digital images of 10 neuT and 10 neuT-C1KO tumors (3 × 200 microscopic fields per tumor) by two pathologists, independently and in a blind fashion.
The intensity of E-Cadherin expression was evaluated and recorded using the Image J software and by analyzing ROI with similar cell numbers (3 × 630 microscopic fields per tumor, three tumors per group each from different mice).
The quantification of C3 fragment deposition, pWWOX, C1q, and Her2/neu protein was performed by image analysis with Adobe Photoshop. Positive red fluorescent pixels were selected using the magic wand tool and quantified in the histogram window in images from 10 tumors per group (3 × 400 microscopic fields per sample) for C3 and C1q quantification and seven tumors per group (3 × 400 microscopic fields per sample) for pWWOX and Her2/neu protein quantification. Results are represented as means ± SEM.
The number and the lumen area of CD31+/CD105+ vessels were evaluated on the digital images of 3–5 tumors per group (5 × 200 microscopic fields per tumor) by two pathologists, independently and in a blind fashion. Vessels area (in pixels) was evaluated with Adobe Photoshop by selecting vessels with the lasso tool and reporting the number of pixels indicated in the histogram window.
Protein preparation and immunoblotting
Total protein extracts were obtained from neuT, neuT-C1KO, and neuT-C3KO mammary tumors. Briefly flash frozen specimens were dissociated using an IKA-Ultra-Turrax® T8 homogenizer (IKA-Werke, Staufen Germany) in a buffer containing 10 mM Tris, 5 mM EDTA, 50 mM NaCl, 30 mM Sodium pyrophosphate decahydrate (Na4O7P2–10H2O), 50 mM Sodium Fluoride (NaF), 1 mM Sodium orthovanadate (Na3VO4), 1% Triton X (Adjust the pH 7.6), 1 mM Phenylmethanesulfonyl fluoride100 (all from Sigma-Aldrich), and a cocktail of protein inhibitors (Sigma-Aldrich, P8340). Samples were then centrifuged twice at 12,000 rpm at 4°C for 5 min. 40 μg of total proteins, as determined by BCA Protein Assay (Pierce, Thermo Fisher), were separated by SDS-PAGE and electroblotted onto polyvinylidene fluoride membranes (BioRad). Membranes were blocked in 5% BSA (Sigma Aldrich) Tris-buffered saline (TBS)-Tween buffer (137 mM NaCl, 20 mM Tris/HCl, pH 7.6, 0.05% Tween-20) for 1 h at RT and then incubated with appropriate primary (anti e-Cadherin, Cell Signaling, 3195; anti β-actin, Santa Cruz Biotechnology, sc-69879) and appropriate secondary antibodies (goat anti-rabbit, Sigma_Aldrich, A0545 and goat anti mouse Sigma_Aldrich, A4416, respectively) in 3% BSA TBS-Tween buffer overnight at 4°C and for 1 h at room temperature, respectively, and visualized using enhanced chemiluminescence (ECL Plus, Thermo Scientific Pierce). Protein modulations were normalized on the actin loading control and expressed as Adjusted Volume Intensity/mm2 (background subtraction) using Quantity ONE software (Biorad, Milano, Italy).
Cytometric identification of tumor-infiltrating leukocytes
For the infiltrating-cell phenotypic analyses, fresh primary tumor specimens of 6–8 mm mean diameter from neuT (n = 5), neuT-C1KO (n = 7), and neuT-C3KO (n = 6) mice were finely minced with scissors and then digested by incubation with 1 mg/mL collagenase IV (Sigma Aldrich) in RPMI-1640 (Life Technologies) at 37°C for 1 h in an orbital shaker. After washing in PBS supplemented with 2% fetal bovine serum (GIBCO), the cell suspension was incubated in an erylise buffer (155 mM NH4Cl, 15.8 mM Na2CO3, 1 mM EDTA, pH 7.3) for 10 min at RT. After washing in RPMI-1640 supplemented with 10% FBS, the cell suspension was passed through a 70−µm pore cell strainer, centrifuged at 1,400 rpm for 10 min and re-suspended in an erylise buffer. Tumor-infiltrating leukocytes were collected, washed, re-suspended in PBS, treated with Fc receptor blocker (anti CD16/CD32; 01245B; BD Bioscences), and stained with the following antibodies: anti-mouse CD45 VioGreen (Miltenyi Biotec, 130097), anti-mouse CD3 FITC (Miltenyi Biotec, 130–092962), anti-mouse CD4 APC-Vio770 (Miltenyi Biotec, 130–102–179), anti-mouse CD8 VioBlue (Miltenyi Biotec, 130–094–360), anti-mouse γδ PE/Cy7 (BioLegend, 118124), anti-mouse CD49b PE (Miltenyi Biotec, 130–091–816), anti-mouse F4/80 APC (Miltenyi Biotec, 130–102–379), anti-mouse CD11b FITC (Miltenyi Biotec, 130–081–201), and anti-mouse GR-1 PE (Miltenyi Biotec, 130–102–426). To detect FoxP3+ T regulatory cells, samples were permeabilized with the FoxP3 anti-mouse staining kit (eBioscience) and stained with the anti-mouse/rat-Foxp3-FITC antibody (eBioscience, FJK-16s;). Samples were acquired and analyzed on a CyAn ADP flow cytometer using the Summit 4.3 software (Beckman Coulter, Milano, Italy).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1253653_supplementary_data.zip
Download Zip (1.4 MB)Funding
This work was supported by grants from the Italian Association for Cancer Research (IG 11675 and IG 16724), Fondazione Ricerca Molinette Onlus, the University of Torino, and the Fondazione CRT, Torino, Italy, within the “Richieste ordinarie 2015” Call. F.R. and L.C. were supported with fellowships from “Fondazione Italiana per la Ricerca sul Cancro” and from “Fondazione Umberto Veronesi,” respectively. We thank Dr Dale Lawson for his revision and editing of the manuscript.
References
- Mamidi S, Höne S, Kirschfink M. The complement system in cancer: Ambivalence between tumour destruction and promotion. Immunobiology 2015; 222(1):45-54; PMID:26686908; http://dx.doi.org/10.1016/j.imbio.2015.11.008
- Stover C. Dual role of complement in tumour growth and metastasis (Review). Int J Mol Med 2010; 25:307-13; PMID:20127033; http://dx.doi.org/10.3892/ijmm_00000346
- Pio R, Corrales L, Lambris JD. The role of complement in tumor growth. Adv Exp Med Biol 2014; 772:229-62. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4379038&tool=pmcentrez&rendertype=abstract; PMID:24272362; http://dx.doi.org/10.1007/978-1-4614-5915-6_11
- Markiewski MM, Lambris JD. Is complement good or bad for cancer patients? A new perspective on an old dilemma. Trends Immunol 2009; 30:286-92. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid =2704572&tool=pmcentrez&rendertype=abstract; PMID:19428302; http://dx.doi.org/10.1016/j.it.2009.04.002
- Cooper PD. Complement and cancer: activation of the alternative pathway as a theoretical base for immunotherapy. Adv Immun Cancer Ther 1985; 1:125-66; PMID:3916662; http://dx.doi.org/10.1007/978-1-4612-5068-5_4
- Gaboriaud C, Frachet P, Thielens NM, Arlaud GJ. The human C1q globular domain: Structure and recognition of non-immune self ligands. Front Immunol 2012; 2:92; PMID:22566881; http://dx.doi.org/10.3389/fimmu.2011.00092
- Kishore U, Ghai R, Greenhough TJ, Shrive AK, Bonifati DM, Gadjeva MG, Waters P, Kojouharova MS, Chakraborty T, Agrawal A. Structural and functional anatomy of the globular domain of complement protein C1q. Immunol Lett 2004; 95:113-28. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3818097&tool=pmcentrez&rendertype=abstract; PMID:15388251; http://dx.doi.org/10.1016/j.imlet.2004.06.015
- Gaboriaud C, Juanhuix J, Gruez A, Lacroix M, Darnault C, Pignol D, Verger D, Fontecilla-Camps JC, Arlaud GJ. The crystal structure of the globular head of complement protein C1q provides a basis for its versatile recognition properties. J Biol Chem 2003; 278:46974-82; PMID:12960167; http://dx.doi.org/10.1074/jbc.M307764200
- Peerschke EIB, Ghebrehiwet B. cC1qR/CR and gC1qR/p33: observations in cancer. Mol Immunol 2014; 61:100-9; PMID:25044096; http://dx.doi.org/10.1016/j.molimm.2014.06.011
- Clarke EV, Tenner AJ. Complement modulation of T cell immune responses during homeostasis and disease. J Leukoc Biol 2014; 96:745-56. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4197570&tool=pmcentrez&rendertype=abstract; PMID:25210145; http://dx.doi.org/10.1189/jlb.3MR0214-109R
- Nayak A, Ferluga J, Tsolaki AG, Kishore U. The non-classical functions of the classical complement pathway recognition subcomponent C1q. Immunol Lett 2010; 131:139-50; PMID:20381531; http://dx.doi.org/10.1016/j.imlet.2010.03.012
- Bossi F, Peerschke EI, Ghebrehiwet B, Tedesco F. Cross-talk between the complement and the kinin system in vascular permeability. Immunol Lett 2011; 140:7-13. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3162365&tool=pmcentrez&rendertype=abstract; PMID:21762728; http://dx.doi.org/10.1016/j.imlet.2011.06.006
- Bossi F, Tripodo C, Rizzi L, Bulla R, Agostinis C, Guarnotta C, Munaut C, Baldassarre G, Papa G, Zorzet S et al. C1q as a unique player in angiogenesis with therapeutic implication in wound healing. Proc Natl Acad Sci U S A 2014; 111:4209-14; PMID:24591625; http://dx.doi.org/10.1073/pnas.1311968111
- Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci 2012; 35:369-89; PMID:22715882; http://dx.doi.org/10.1146/annurev-neuro-061010-113810
- Hong Q, Sze C-I, Lin S-R, Lee M-H, He R-Y, Schultz L, Chang J-Y, Chen S-J, Boackle RJ, Hsu L-J et al. Complement C1q activates tumor suppressor WWOX to induce apoptosis in prostate cancer cells. PLoS One 2009; 4:e5755. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2685983&tool=pmcentrez&rendertype=abstract; PMID:19484134; http://dx.doi.org/10.1371/journal.pone.0005755
- Eggleton P, Tenner AJ, Reid KB. C1q receptors. Clin Exp Immunol 2000; 120:406-12. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1905565&tool=pmcentrez&rendertype=abstract; PMID:10844516; http://dx.doi.org/10.1046/j.1365-2249.2000.01218.x
- Winslow S, Leandersson K, Edsjö A, Larsson C. Prognostic stromal gene signatures in breast cancer. Breast Cancer Res 2015; 17:23. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4360948&tool=pmcentrez&rendertype=abstract; PMID:25848820; http://dx.doi.org/10.1186/s13058-015-0530-2
- Nakshatri H, Qi G, You J, Kerry B, Schneider B, Zon R, Buck C, Regnier F, Wang M. Intrinsic subtype-associated changes in the plasma proteome in breast cancer. Proteomics Clin Appl 2009; 3:1305-13; PMID:21136952; http://dx.doi.org/10.1002/prca.200900040
- Ouyang Q, Zhang L, Jiang Y, Ni X, Chen S, Ye F, Du Y, Huang L, Ding P, Wang N et al. The membrane complement regulatory protein CD59 promotes tumor growth and predicts poor prognosis in breast cancer. Int J Oncol 2016; 48:2015-24; PMID:26935178; http://dx.doi.org/10.3892/ijo.2016.3408
- Zhang F, Chen JY. Breast cancer subtyping from plasma proteins. BMC Med Genomics 2013; 6(Suppl 1):S6. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=355699&tool=pmcentrez&rendertype=abstract; PMID:23369492; http://dx.doi.org/10.1186/1755-8794-6-S1-S6
- Astolfi A, Landuzzi L, Nicoletti G, De Giovanni C, Croci S, Palladini A, Ferrini S, Iezzi M, Musiani P, Cavallo F et al. Gene expression analysis of immune-mediated arrest of tumorigenesis in a transgenic mouse model of HER-2/neu-positive basal-like mammary carcinoma. Am J Pathol 2005; 166:1205-16. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1602398&tool=pmcentrez&rendertype=abstract; PMID:15793299; http://dx.doi.org/10.1016/S0002-9440(10)62339-5
- Street SEA, Zerafa N, Iezzi M, Westwood JA, Stagg J, Musiani P, Smyth MJ. Host perforin reduces tumor number but does not increase survival in oncogene-driven mammary adenocarcinoma. Cancer Res 2007; 67:5454-60; PMID:17545627; http://dx.doi.org/10.1158/0008-5472.CAN-06-4084
- Macagno M, Bandini S, Stramucci L, Quaglino E, Conti L, Balmas E, Smyth MJ, Lollini P-L, Musiani P, Forni G et al. Multiple roles of perforin in hampering ERBB-2 (Her-2/neu) carcinogenesis in transgenic male mice. J Immunol 2014; 192:5434-41; PMID:24790144; http://dx.doi.org/10.4049/jimmunol.1301248
- Bandini S, Curcio C, Macagno M, Quaglino E, Arigoni M, Lanzardo S, Hysi A, Barutello G, Consolino L, Longo DL et al. Early onset and enhanced growth of autochthonous mammary carcinomas in C3-deficient Her2/neu transgenic mice. Oncoimmunology 2013; 2:e26137. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3820812&tool=pmcentrez&rendertype=abstract; PMID:24228231; http://dx.doi.org/10.4161/onci.26137
- Gunasinghe NPAD, Wells A, Thompson EW, Hugo HJ. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev 2012; 31:469-78; PMID:22729277; http://dx.doi.org/10.1007/s10555-012-9377-5
- Cho MS, Rupaimoole R, Choi H-J, Noh K, Chen J, Hu Q, Sood AK, Afshar-Kharghan V. Complement component 3 is regulated by TWIST1 and mediates epithelial-mesenchymal transition. J Immunol 2016; 196:1412-8. Available from: http://www.jimmunol.org/cgi/doi/10.4049/jimmunol.1501886; PMID:26718342; http://dx.doi.org/10.4049/jimmunol.1501886
- Barutello G, Curcio C, Spadaro M, Arigoni M, Trovato R, Bolli E, Zheng Y, Ria F, Quaglino E, Iezzi M et al. Antitumor immunization of mothers delays tumor development in cancer-prone offspring. Oncoimmunology 2015; 4:e1005500. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4485839&tool=pmcentrez&rendertype=abstract; PMID:26155401; http://dx.doi.org/10.1080/2162402X.2015.1005500
- Quaglino E, Mastini C, Forni G, Cavallo F. ErbB2 transgenic mice: A tool for investigation of the immune prevention and treatment of mammary carcinomas. Curr Protoc Immunol 2008; PMID:18729063; http://dx.doi.org/10.1002/0471142735.im2009s82
- Pio R, Ajona D, Lambris JD. Complement inhibition in cancer therapy. Semin Immunol 2013; 25:54-64; PMID:23706991; http://dx.doi.org/10.1016/j.smim.2013.04.001
- Nunez-Cruz S, Gimotty PA, Guerra MW, Connolly DC, Wu Y-Q, DeAngelis RA, Lambris JD, Coukos G, Scholler N. Genetic and pharmacologic inhibition of complement impairs endothelial cell function and ablates ovarian cancer neovascularization. Neoplasia 2012; 14:994-1004. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3514739&tool=pmcentrez&rendertype=abstract; PMID:23226093; http://dx.doi.org/10.1593/neo.121262
- Bulla R, Tripodo C, Rami D, Ling GS, Agostinis C, Guarnotta C, Zorzet S, Durigutto P, Botto M, Tedesco F. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat Commun 2016; 7:10346. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4740357&tool=pmcentrez&rendertype=abstract; PMID:26831747; http://dx.doi.org/10.1038/ncomms10346
- Vollmers HP, Brändlein S. Natural antibodies and cancer. N Biotechnol 2009; 25:294-8; PMID:19442595; http://dx.doi.org/10.1016/j.nbt.2009.03.016
- Hamaï A, Duperrier-Amouriaux K, Pignon P, Raimbaud I, Memeo L, Colarossi C, Canzonieri V, Perin T, Classe J-M, Campone M et al. Antibody responses to NY-ESO-1 in primary breast cancer identify a subtype target for immunotherapy. PLoS One 2011; 6:e21129. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3117860&tool=pmcentrez&rendertype=abstract; PMID:21747904; http://dx.doi.org/10.1371/journal.pone.0021129
- Lu H, Ladd J, Feng Z, Wu M, Goodell V, Pitteri SJ, Li CI, Prentice R, Hanash SM, Disis ML. Evaluation of known oncoantibodies, HER2, p53, and cyclin B1, in prediagnostic breast cancer sera. Cancer Prev Res (Phila) 2012; 5:1036-43. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3790582&tool=pmcentrez&rendertype=abstract; PMID:22715141; http://dx.doi.org/10.1158/1940-6207.CAPR-11-0558
- Kouser L, Madhukaran SP, Shastri A, Saraon A, Ferluga J, Al-Mozaini M, Kishore U. Emerging and novel functions of complement protein C1q. Front Immunol 2015; 6:317; PMID:26175731; http://dx.doi.org/10.3389/fimmu.2015.00317
- Tohgi H, Utsugisawa K, Nagane Y. Hypoxia-induced expression of C1q, a subcomponent of the complement system, in cultured rat PC12 cells. Neurosci Lett 2000; 291:151-4; PMID:10984629; http://dx.doi.org/10.1016/S0304-3940(00)01399-9
- Msaki A, Pastò A, Curtarello M, Arigoni M, Barutello G, Calogero RA, Macagno M, Cavallo F, Amadori A, Indraccolo S. A hypoxic signature marks tumors formed by disseminated tumor cells in the BALB-neuT mammary cancer model. Oncotarget 2016; 7:33081-95; PMID:27105499; http://dx.doi.org/10.18632/oncotarget.8859
- Lee C, Huang C-H. LASAGNA-Search: an integrated web tool for transcription factor binding site search and visualization. Biotechniques 2013; 54:141-53; PMID:23599922; http://dx.doi.org/10.2144/000113999
- Culver C, Sundqvist A, Mudie S, Melvin A, Xirodimas D, Rocha S. Mechanism of hypoxia-induced NF-kappaB. Mol Cell Biol 2010; 30:4901-21; PMID:20696840; http://dx.doi.org/10.1128/MCB.00409-10
- Roychoudhury J, Clark JP, Gracia-Maldonado G, Unnisa Z, Wunderlich M, Link KA, Dasgupta N, Aronow B, Huang G, Mulloy JC et al. MEIS1 regulates an HLF-oxidative stress axis in MLL-fusion gene leukemia. Blood 2015; 125:2544-52; PMID:25740828; http://dx.doi.org/10.1182/blood-2014-09-599258
- Henke N, Ferreirós N, Geisslinger G, Ding MG, Essler S, Fuhrmann DC, Geis T, Namgaladze D, Dehne N, Brüne B. Loss of HIF-1α in macrophages attenuates AhR/ARNT-mediated tumorigenesis in a PAH-driven tumor model. Oncotarget 2016; PMID:27015123; http://dx.doi.org/10.18632/oncotarget.8297
- Scott C, Bonner J, Min D, Boughton P, Stokes R, Cha KM, Walters SN, Maslowski K, Sierro F, Grey ST et al. Reduction of ARNT in myeloid cells causes immune suppression and delayed wound healing. Am J Physiol Cell Physiol 2014; 307:C349-57; PMID:24990649; http://dx.doi.org/10.1152/ajpcell.00306.2013
- Langer HF, Chung KJ, Orlova VV, Choi EY, Kaul S, Kruhlak MJ, Alatsatianos M, DeAngelis RA, Roche PA, Magotti P et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood 2010; 116:4395-403; PMID:20625009; http://dx.doi.org/10.1182/blood-2010-01-261503
- Cifaldi L, Quaglino E, Di Carlo E, Musiani P, Spadaro M, Lollini PL, Wolf S, Boggio K, Forni G, Cavallo F. A Light, nontoxic interleukin 12 protocol inhibits HER-2 / neu mammary carcinogenesis in BALB / C transgenic mice with established hyperplasia. Cancer Res 2001; 61:2809-12; PMID:11306448
- Vĕtvicka V, Hanikýrová M, Vĕtvicková J, Ross GD. Regulation of CR3 (CD11b/CD18)-dependent natural killer (NK) cell cytotoxicity by tumour target cell MHC class I molecules. Clin Exp Immunol 1999; 115:229-35. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1905157&tool=pmcentrez&rendertype=abstract; PMID:9933447; http://dx.doi.org/10.1046/j.1365-2249.1999.00800.x
- Inoue M, Mimura K, Izawa S, Shiraishi K, Inoue A, Shiba S, Watanabe M, Maruyama T, Kawaguchi Y, Inoue S et al. Expression of MHC Class I on breast cancer cells correlates inversely with HER2 expression. Oncoimmunology 2012; 1:1104-10. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3494624&tool=pmcentrez&rendertype=abstract; PMID:23170258; http://dx.doi.org/10.4161/onci.21056
- Okita R, Mougiakakos D, Ando T, Mao Y, Sarhan D, Wennerberg E, Seliger B, Lundqvist A, Mimura K, Kiessling R. HER2/HER3 signaling regulates NK cell-mediated cytotoxicity via MHC class I chain-related molecule A and B expression in human breast cancer cell lines. J Immunol 2012; 188:2136-45; PMID:22301547; http://dx.doi.org/10.4049/jimmunol.1102237
- Chang NS, Doherty J, Ensign A, Lewis J, Heath J, Schultz L, Chen ST, Oppermann U. Molecular mechanisms underlying WOX1 activation during apoptotic and stress responses. Biochem Pharmacol 2003; 66(8):1347-54; PMID:14555208; http://dx.doi.org/10.1016/S0006-2952(03)00500-8
- Trendelenburg M, Fossati-Jimack L, Cortes-Hernandez J, Turnberg D, Lewis M, Izui S, Cook HT, Botto M. The role of complement in cryoglobulin-induced immune complex glomerulonephritis. J Immunol 2005; 175:6909-14; PMID:16272350; http://dx.doi.org/10.4049/jimmunol.175.10.6909
- Iezzi M, Quaglino E, Amici A, Lollini P-L, Forni G, Cavallo F. DNA vaccination against oncoantigens: A promise. Oncoimmunology 2012; 1:316-25. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3382874&tool=pmcentrez&rendertype=abstract; PMID:22737607; http://dx.doi.org/10.4161/onci.19127