
ABSTRACT
While RNA-pulsed dendritic cell (DC) vaccines have shown promise, the advancement of cellular therapeutics is fraught with developmental challenges. To circumvent the challenges of cellular immunotherapeutics, we developed clinically translatable nanoliposomes that can be combined with tumor-derived RNA to generate personalized tumor RNA-nanoparticles (NPs) with considerable scale-up capacity. RNA-NPs bypass MHC restriction, are amenable to central distribution, and can provide near immediate immune induction.
We screened commercially available nanoliposomal preparations and identified the cationic lipid 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) as an efficient mRNA courier to antigen-presenting cells (APCs). When administered intravenously, RNA-NPs mediate systemic activation of APCs in reticuloendothelial organs such as the spleen, liver, and bone marrow. RNA-NPs increase percent expression of MHC class I/II, B7 co-stimulatory molecules, and maturation markers on APCs (all vital for T-cell activation). RNA-NPs also increase activation markers on tumor APCs and elicit potent expansion of antigen-specific T-cells superior to peptide vaccines formulated in complete Freund's adjuvant. We demonstrate that both model antigen-encoding and physiologically-relevant tumor-derived RNA-NPs expand potent antitumor T-cell immunity. RNA-NPs were shown to induce antitumor efficacy in a vaccine model and functioned as a suitable alternative to DCs in a stringent cellular immunotherapy model for a radiation/temozolomide resistant invasive murine high-grade glioma.
Although cancer vaccines have suffered from weak immunogenicity, we have advanced a RNA-NP formulation that systemically activates host APCs precipitating activated T-cell frequencies necessary to engender antitumor efficacy. RNA-NPs can thus be harnessed as a more feasible and effective immunotherapy to re-program host-immunity.
Introduction
The promise of RNA-pulsed dendritic cell (DC) vaccines was recently demonstrated in a randomized clinical trial design demonstrating a significantly prolonged survival benefit in patients with glioblastoma (GBM) receiving an optimized DC vaccine platform.Citation1 While these therapeutic approaches have shown considerable promise for GBM patients, the advancement of cellular therapeutics through the commercialization pipeline has been fraught with challenges in the development, manufacturing, and marketing of successful cancer immunotherapies.Citation1-3 To circumvent the challenges associated with DC vaccines, we developed a novel treatment platform, which leverages the use of commercially available and clinically translatable nanoparticles (NPs) that can be combined with tumor-derived RNA to peripherally activate T cells against tumor antigens.Citation4-8 RNA-nanoparticles (RNA-NPs) can be generated in a few days after tumor tissue procurement and abrogate the need for complex cellular processing. While several NP delivery strategies for vaccine development have focused on the establishment of novel NP formulations, the advancement of these platforms into clinical evaluation has been slow, often due to the significant costs and time associated with scale-up synthesis and preclinical toxicity evaluation of compositions with unknown reactivity in humans.Citation4,5 We therefore chose to evaluate readily available and clinical-grade NP formulations for complexing and protecting mRNA transcripts encoding for bioactive proteins and focused on the engineering of rapidly translatable NP complexes. We screened commercially available nanoliposomal preparations and identified the cationic lipid 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) as the most promising mRNA courier to DCs in vitro. These RNA-NPs induced in vivo gene expression and preserved RNA stability over time. We determined that i.v. injection of RNA-NPs was requisite for expansion of functional antigen-specific immunity, superior to other routes of immunization and of greater stimulatory capacity than standard peptide vaccines formulated in complete Freund's adjuvant (CFA). Intravenously administered RNA-NPs increased serum interferon-α levels and mediated systemic activation of APCs in reticuloendothelial organs precipitating sharp increases in the percentages of MHC class I/II expression and B7 co-stimulatory molecules. By simply co-encapsulating immunomodulatory RNAs encoding for bioactive cytokines, we enhanced the immunogenicity of RNA-NPs. We demonstrated that RNA-NPs mediate activity against intracranial and subcutaneous melanomas and potentiate antitumor T-cell responses in a cellular immunotherapy model against a radiation/temozolomide resistant invasive murine high-grade glioma. Antitumor efficacy elicited by RNA-NPs was abrogated through blockade of interferon-α. Although cancer vaccines have suffered from weak immunogenicity, we have advanced a RNA-nanoliposomal formulation that can reshape a host's immune profile through systemic immune activation, precipitating activated T-cell frequencies necessary to engender antitumor efficacy.Citation9,10
Results
Efficient transfection of DCs in vitro by RNA-NPs
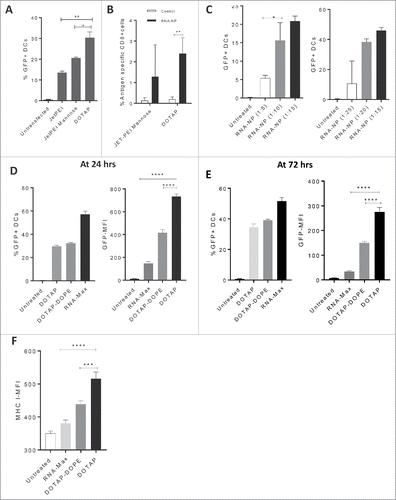
We screened several translatable NP formulations for their ability to transfect DCs in vitro with GFP mRNA. We demonstrated that the NP DOTAP is a superior formulation compared with linear polyethylenimine NPs with (JETPEI-Mannose) (*p < 0.05, unpaired t test) and without DC targeting mannose receptors (JETPEI) (**p < 0.01, unpaired t test) (); we corroborated DOTAP's superiority based on expansion of antigen-specific CD8+ immunity (). Later, we investigated the optimal ratio of RNA to DOTAP based on DC transfection efficiency in vitro and determined that ratios ranging from 1 ug of RNA to 10–20 ug of DOTAP achieved peak transfection efficiencies (). Based on these data, we selected a ratio of 1:15 ug for composition of subsequent cationic nanoliposomal formulations. We then compared the transfection efficiency of DOTAP to alternative cationic liposomal preparations embedded with pH buffers (DOTAP:DOPE) and lipofectamine RNAiMAX at 24 h () and 72 h () post-transfection. While lipofectamine RNAiMAX had an increased transfection efficiency, DOTAP had significantly increased mean fluorescent intensity (MFI) of GFP-transfected cells ( and ). We also assessed DC2.4s for expression of MHC I after co-culture with each NP formulation. Compared with DOTAP-DOPE and lipofectamine RNAiMAX, DOTAP elicited the greatest increase in MHC I expression by MFI (). Since the cationic lipid DOTAP elicited a greater GFP-MFI and MHCI-MFI by DC2.4s, we moved this NP forward for evaluation in subsequent experiments.
Figure 1. Identification of a target RNA-NP. (A) An immortalized murine bone marrow-derived DC line (DC2.4) was transfected with GFP RNA comparing the cationic liposome DOTAP to JETPEI and JETPEI-mannose, and cells were screened one day later for assessment of GFP expression via flow cytometry. (B) C57Bl/6 mice, were given a low precursor frequency of OT-Is, and vaccinated intravenously with OVA RNA or control RNA (luciferase) encapsulated in either DOTAP, or linear polyethylenimine NPs with DC targeting mannose receptors (JETPEI-Mannose); OVA-specific T cell immunity was assessed from spleens of vaccinated mice 1 week later by flow cytometry. (C) Different ratios of GFP RNA to NP DOTAP were encapsulated and assessed for %transfection efficiency of DC2.4s in vitro. Cells were grown in vitro and harvested 1 d after addition of RNA-NPs to culture media. (D) Cationic lipid preparations of DOTAP and DOTAP-DOPE were compared with RNA-iMAX lipofectamine based on percent GFP transfection efficiency (left) and GFP-MFI (right) after 24 h. (E) Cationic lipid preparations of DOTAP and DOTAP-DOPE were compared with RNA-iMAX lipofectamine based on percent GFP transfection efficiency (left) and GFP-MFI (right) after 72 h. (F) Cationic lipid preparations of DOTAP and DOTAP-DOPE were compared with RNA-iMAX lipofectamine based on MHCI MFI 24 h after transfection of DC2.4s with GFP RNA. (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, unpaired t test).
RNA-NPs form stable complexes with positive zeta potentials and nanometer size distribution, and elicit in vivo gene expression
To further characterize our formulation, we performed cryo-electron microscopy on uncomplexed NPs and RNA encapsulated NPs and determined each to have heterogeneous sizes ranging above and below 100 nm (); this was corroborated by dynamic light scatter (), showing a distribution of sizes ranging from 70 to 200 nm. To determine the zeta potential of RNA-NPs, total tumor-derived RNA was encapsulated with DOTAP and successive runs were performed on the Brookhaven ZetaPlus instrument. The average zeta potential of these runs measured at 27.28 mV.
Figure 2. RNA-NP characterization, stability, and in vivo gene expression. (A) DOTAP NPs were left uncomplexed or complexed with luciferase mRNA and size was measured using cryo-electron microscopy. (B) DOTAP NPs were complexed with total RNA derived from a glioma cell line and particle size was measured using dynamic light scattering. (C) NPs encapsulating luciferase RNA were stored over time at room temperature (blue) or 37°C (red) before transfection of HEK cells. Transfection efficiency over time (relative to transfection efficiency at hour zero) is measured using bioluminescent imaging. (D) NPs encapsulating luciferase RNA were injected locally into the peritoneum of albino C57Bl/6 mice followed by peritoneal luciferin injection 6 h post-RNA-NPs. (E) NPs encapsulating luciferase RNA were injected intravenously into three C57Bl/6 mice followed by organ harvests of spleens lymph nodes, livers, kidneys, and lungs re-suspended in a 24-well plate with PBS to which luciferin substrate was added and measured for bioluminescent activity.
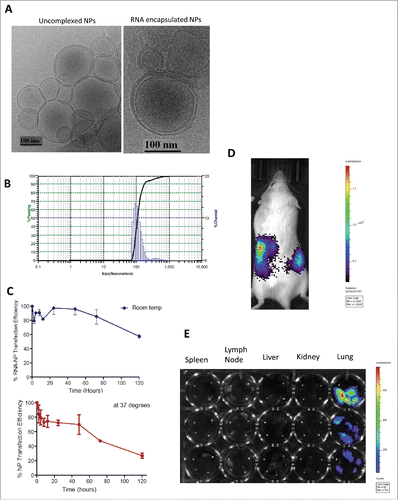
To evaluate the viability and shelf-life of our formulation, we determined the translation efficiency of luciferase RNA-NPs at different time points after RNA encapsulation. We in vitro transfected human embryonic kidney (HEK) cells with luciferase RNA-NPs and compared the transfection efficiency at these time points over the baseline transfection efficiency at time point zero. Based on bioluminescent imaging, our data demonstrates maintenance of optimal translation efficiency in vitro for nearly 60 h after RNA-NP complexation at room temperature (). We then investigated the translation efficiency of our formulation in vivo. After local intraperitoneal injection of RNA-NPs into albino C57Bl/6 mice, our formulation induced in vivo luciferase RNA expression (). We also recorded luciferase expression in mice after intra-venous injection of luciferase encoding RNA-NPs, and demonstrated a predominant uptake in the lungs ().
I.V. administration of NPs is the optimum route for expansion of functional antigen-specific CD8+ T-cell immunity
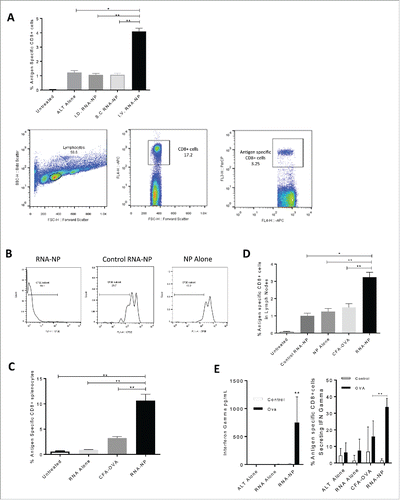
To identify the optimal route for expansion of antigen-specific immunity, RNA-NP complexes were injected i.v. into naïve immunocompetent C57Bl/6 mice that had been spiked with a low frequency of antigen-specific CD8+ T cells (OT-I) and compared with other routes of injection. Spleens were harvested after 1 week for assessment of antigen-specific CD8+ cells. I.V. RNA-NP complexes showed superior expansion of antigen-specific CD8+ cells compared with intradermal (i.d.) injections (**p < 0.01, Mann–Whitney test), subcutaneous (s.c.) injections (**p < 0.01, Mann–Whitney test) and unvaccinated animals receiving adoptive lymphocyte transfer (ALT) from OTIs (*p < 0.05, Mann–Whitney test) (). To assess proliferation of antigen-specific T cells, we labeled splenocytes from OT-I transgenic mice with carboxyfluorescein succinimidyl (CFSE) and transferred a small precursor frequency to naïve C57Bl/6 mice in conjunction with i.v. RNA-NPs. Based on CFSE dilution, we determined that RNA-NPs induce in vivo T cell (OT-I) proliferation in an antigen-dependent manner (). We then compared the immunogenicity of RNA-NPs versus peptide vaccines. Through emulsification of the class I SIINFEKL epitope from OVA peptide in CFA (a composite of mineral oil and mycobacterium that is too toxic for clinical use, but has been shown to induce potent immune responses in miceCitation15), we compared OVA peptide vaccines with RNA-NPs and found that RNA-NPs elicited a greater expansion of antigen-specific CD8+ cells in the spleens (**p < 0.01, Mann–Whitney test) () and lymph nodes (**p < 0.01, unpaired t test) () of immunocompetent mice. To determine T-cell functionality, splenocytes from mice vaccinated with RNA-NPs were harvested and re-stimulated with OVA peptide for assessment of interferon (IFN)-gamma production. Compared to peptide vaccines in CFA, C57Bl/6 mice receiving RNA-NPs had a significantly increased percentage of antigen-specific CD8+ cells secreting IFN-gamma (**p < 0.01, Mann–Whitney test) ().
Figure 3. Antigen-specific immunogenicity of RNA-NPs. (A) C57Bl/6 mice were vaccinated with a single OVA RNA-NP complex following OT-I administration before splenocytes were harvested 1 week later. Lymphocytes were gated off by forward and side scatter, followed by gating of CD8+ T cells (APC) by side scatter. OVA-specific CD8+ cells were then identified by percent Ly5.1 positive (PercP) of CD8+ (APC) T cells (*p < 0.05; **p < 0.01, Mann–Whitney test). (B) Expansion of CFSE labeled splenocytes from OT-I transgenic mice in the presence of OVA mRNA-NPs, NPs alone, or GFP mRNA NPs. (C) C57Bl/6 mice, injected with OT-I T cells, were vaccinated with a single OVA RNA-NP complex, RNA alone or CFA-OVA peptide before splenocytes were harvested 1 week later and assessed for expansion of antigen-specific CD8+ cells (*p < 0.05; **p < 0.01, Mann–Whitney test). (D) C57Bl/6 mice were injected with OT-I T cells followed by vaccination with a single OVA RNA-NP complex, control RNA-NPs, NPs alone, RNA alone, or CFA-OVA peptide before lymph nodes were harvested 1 week later and assessed for expansion of antigen-specific CD8+ cells (*p < 0.05; **p < 0.01, unpaired t test). (E) C57Bl/6 mice were vaccinated with a single OVA RNA-NP complex following OT-I administration before splenocytes were harvested 1 week later. Splenocytes were then co-cultured with OVA peptide for 1 to 2 d, before cells were spun down for assessment of functional antigen-specific T cells by intracellular staining (right); supernatants were separated and simultaneously assessed for IFN-gamma production (left) (**p < 0.01, Mann–Whitney test).
Systemic activation of CD11c+ cells via RNA-NPs
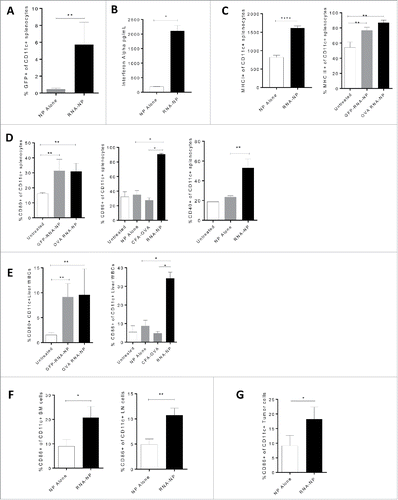
In the context of drug and gene delivery, systemic nanoliposomes have suffered from poor in vivo targeting techniques and reticuloendothelial deposition. While reticuloendothelial deposition has limited various drug and gene therapies, these areas may be ideal for nanoliposomal transfection of APCs and induction of T-cell priming.Citation11-14 We assessed systemic RNA-NP localization to reticuloendothelial sites such as the spleen. Since CD11c is a marker on DCs and APCs, we analyzed GFP expression on CD11c+ splenocytes. Compared to animals receiving NPs alone, mice receiving GFP RNA-NPs displayed a ∼5-fold increase in GFP fluorescence representing transfection of ∼5% of total CD11c+ splenocytes (*p < 0.05; **p < 0.01, Mann–Whitney test) (). C57BL/6 mice were then vaccinated with a single RNA-NP vaccine and serum was collected from mice within 24 h for assessment of IFN-α production. Compared with NPs alone, animals receiving RNA-NPs had a robust increase in serum IFN-α levels (). We then assessed effects of RNA-NPs on MHC class I and II expression. Within 24 h of vaccination, RNA-NPs induced nearly a 100% increase in MHC I expression by MFI and a robust increase in the baseline expression of MHC II on CD11c+ cells (*p < 0.05; **p < 0.01, Mann–Whitney test) (). Afterwards, we assessed effects of RNA-NPs on distinct activation markers including B7 co-stimulatory molecules and CD40. After RNA-NP injection, there was about a 2-fold change in the percent expression of CD80 on CD11c+ cells; CD86 expression was upregulated from expression on ∼30% of APCs to ∼90% of CD11c+ cells; and there was a 2-fold change in the percent expression of CD40 on CD11c+ splenocytes (*p < 0.05; **p < 0.01, Mann–Whitney test). By vaccinating mice with either GFP or OVA RNA-NPs, we demonstrated that increased expression of MHC II on CD11c+ cells on CD11c+ cells was independent of mRNA species and required RNA-NP complexes as RNA or NP alone had no effect on immune activation (). To evaluate whether elevation of activation markers was observed in other vaccine formulations, we compared expression levels of CD86 between RNA-NPs and peptide vaccines formulated in CFA (). RNA-NPs induced increased expression of CD86 on CD11c+ splenocytes, which was not observed following peptide vaccines formulated in CFA (**p < 0.01, Mann–Whitney test) (). We then evaluated RNA-NP effects on APCs in other reticuloendothelial organs. Within 24 h of RNA-NP administration, CD11c+ liver white blood cells (WBCs) displayed a ∼6- and 3-fold increase in the expression of CD80 and CD86, respectively (*p < 0.05; **p < 0.01, Mann–Whitney test) (). There was also about a 2-fold increase in the percentage of CD11c+ cells expressing CD86 in the lymph nodes and bone marrow of RNA-NP-vaccinated mice (*p < 0.05, **p < 0.01; unpaired t test) (). To demonstrate if RNA-NPs could activate CD11c cells intratumorally, we implanted C57Bl/6 mice with B16F10 melanomas and administrated RNA-NPs to tumor-bearing mice 2 weeks later. Within 24 h of administration, there was a near 2-fold increase in the percentage of CD11c+ tumor cells expressing CD86 (*p < 0.05; unpaired t test) ().
Figure 4. Systemic immune activation of RNA-NPs. (A) NPs were complexed with GFP RNA and injected i.v. into C57Bl/6 mice. Spleens were harvested 1 d after injection for assessment of CD11c+ cells expressing GFP by flow cytometry (*p < 0.05; **p < 0.01, Mann–Whitney test). (B) NPs alone or RNA-NPs were administered to C57Bl/6 mice bearing subcutaneous B16F10 melanomas and serum was collected 6 h later for assessment of IFN-α levels by ELISA (upper limit of detection for assay is (2,000 pg/dL) (*p < 0.05, Mann–Whitney test) (C) RNA-NPs were administered systemically to C57Bl/6 mice and spleens were collected within 24 h for assessment of MHC I (****p < 0.0001, unpaired t test) and MHC II (**p < 0.01, Mann–Whitney test) on CD11c+ splenocytes. (D) RNA-NPs were administered systemically to C57Bl/6 mice and spleens were collected within 24 h for assessment of CD80, CD86, and CD40 on CD11c+ splenocytes (*p < 0.05; **p < 0.01, Mann–Whitney test). (E) RNA-NPs were administered systemically to C57Bl/6 mice and livers were collected within 24 h for assessment of CD80 and CD86 on CD11c+ liver WBCs (*p < 0.05; **p < 0.01, Mann–Whitney test). (F) Intravenous RNA-NPs were administered to C57Bl/6 mice and lymph nodes and bone marrow were harvested within 24 h for assessment of %CD86 on CD11c+ cells (*p < 0.05; **p < 0.01, unpaired t test). (G) Intravenous RNA-NPs were administered to C57Bl/6 mice 2 weeks after subcutaneous implantation of B16F10 (1 × 106 /mouse); tumors were harvested within 24 h for assessment of %CD86 on CD11c+ cells (*p < 0.05, unpaired t test).
RNA-NPs mediate antitumor efficacy in tumor-bearing mice that can be enhanced through incorporation of immunomodulatory RNAs
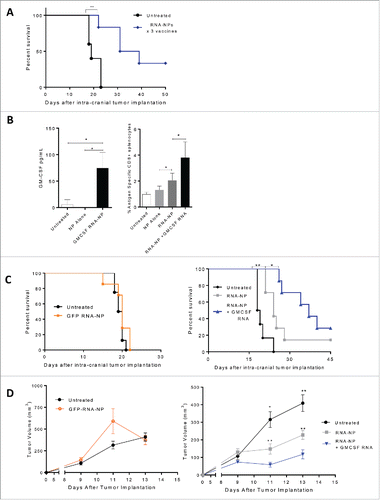
We sought to determine if RNA-NPs could generate antitumor efficacy mediated through the endogenous host T cell response (without OT-1 transfer). We implanted B16F10-OVA tumors intra-cranially into naïve C57Bl/6 mice, and 5 d after implantation, once weekly i.v. RNA-NP complexes (× 3) were administered. Compared with unvaccinated mice, RNA-NPs improved the median and overall survival in tumor-bearing animals. The median survival increased from 19 d to 35 d, with 33% long-term survivors (**p < 0.01, Gehan–Breslow–Wilcoxon test) (). Since GM-CSF has been shown to enhance the potency of immunotherapeutic strategies, and has been reported to be an adequate adjuvant for mRNA vaccines, we assessed if co-encapsulation of GM-CSF encoding RNA would enhance the antitumor immunity elicited by RNA-NPs.Citation15,16 By transfecting DC2.4 cells in vitro with GM-CSF RNA-NPs, we ensured that GM-CSF encoding RNA could elicit production of GM-CSF protein (*p < 0.05, unpaired t test) (). We then determined that co-encapsulation GM-CSF RNA significantly improved the antigen-specific immune responses elicited by RNA-NPs (*p < 0.05; **p < 0.01, Mann–Whitney test) (). In C57Bl/6 mice bearing intracranial () and subcutaneous () B16F10-OVA tumor cells, RNA-NPs co-encapsulated with GM-CSF RNA also enhanced the antitumor efficacy of our formulation (*p < 0.05; **p < 0.01, Gehan–Breslow–Wilcoxon test ; *p < 0.05; **p < 0.01, Mann–Whitney test ).
Figure 5. Antitumor efficacy from RNA-NPs and capacity for immunomodulatory modification. (A) OVA RNA-NP complexes were injected once weekly (× 3) into naïve C57Bl/6 mice starting 5 d after intracranial implantation of B16F10-OVA (6,250 cells/mouse) (**p < 0.01, Gehan–Breslow–Wilcoxon test). (B) DC2.4 cells were grown in culture and transfected with GM-CSF RNA-NPs. One day later, cells were spun down and supernatants were collected for analysis of GM-CSF production by ELISA (*p < 0.05, unpaired t test). (B) C57Bl/6 mice were injected with OT-I T cells followed by a single RNA-NP complex co-encapsulated with or without GM-CSF RNA and spleens were harvested 1 week later for analysis of %antigen-specific CD8+ cells (*p < 0.05; **p < 0.01, Mann–Whitney test). (C) C57Bl/6 mice were injected with OT-I T cells followed by vaccination with a single RNA-NP (GFP RNA-NP, OVA RNA-NP, or OVA+ GM-CSF RNA-NP) complex 5 d after intra-cranial B16F10-OVA (6,250 cells/mouse) implantation (*p < 0.05; **p < 0.01, Gehan–Breslow–Wilcoxon test). (D) C57Bl/6 mice were injected with OT-Is followed by vaccination with a single RNA-NP (GFP RNA-NP, OVA RNA-NP, or OVA+ GM-CSF RNA-NP) complex 1 d after subcutaneous B16F10-OVA tumor (1 × 106 /mouse) implantation (*p < 0.05; **p < 0.01, Mann–Whitney test).
RNA-NPs targeting physiologically relevant antigens supersede DC vaccines in cellular immunotherapy of high-grade glioma
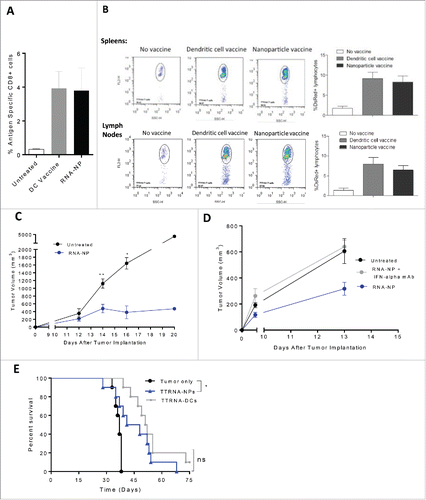
We anticipated that if RNA-NP-mediated immunity was comparable to RNA-pulsed DCs, then our formulations would supplant DC vaccines based on relative ease and capacity for enhancement. To compare antigen-specific immunity, we vaccinated C57Bl/6 mice with either RNA pulsed DCs or RNA-NPs, in conjunction with OT-I T cells, and found no significant difference in the expansion of antigen-specific CD8+ cells (). To demonstrate that immune responses generated by RNA-NPs were not restricted to high-affinity model antigens such as ovalbumin, we evaluated tumor-specific T cell expansion using physiologically relevant total tumor-derived RNA (TTRNA). We extracted TTRNA from an infiltrative high-grade murine glioma cell line derived from a genetically engineered mouse model (KR158B-luc) and vaccinated C57Bl/6 mice with TTRNA-NPs or TTRNA-pulsed DCs.Citation17 Citation18 To track each animal's immunologic response to vaccination, RNA-NPs and DC vaccines were given after administration of ex vivo activated DS red labeled tumor-specific T cells (as described in Methods). Compared with mice receiving DC vaccines, mice vaccinated with RNA-NPs had comparable expansion of tumor-specific T cells (). To demonstrate that RNA-NPs elicit efficacy in a vaccine model against physiologically relevant antigens, we implanted B16F0 melanomas into the flanks of C57Bl/6 mice; 1 d later, we administrated TTRNA (derived from B16F0) encapsulated NPs once weekly (× 3 weeks). Compared with untreated mice, mice vaccinated with TTRNA-NPs had smaller tumor volumes (). To determine if type I interferon was essential for antitumor immunity elicited by RNA-NPs, we administered INF-α blocking antibodies prior to vaccination with TTRNA-NPs in our subcutaneous murine melanoma model. In animals receiving IFN-α blocking antibodies, the antitumor efficacy elicited by TTRNA-NPs was abrogated (). To demonstrate that TTRNA-NP could also elicit antitumor efficacy in an adoptive cellular therapy model, naïve C57Bl/6 mice were implanted with a radiation resistant and temozolomide resistant infiltrative murine astrocytoma (KR158B-luc).Citation17,18 We have previously demonstrated that antitumor efficacy against this tumor is contingent on DC vaccines in a cellular immunotherapy model.Citation17 In this model, there was no significant difference in the efficacy elicited by TTRNA-NPs versus TTRNA-pulsed DCs (*p < 0.05, Gehan–Breslow–Wilcoxon test) ().Citation17,18
Figure 6. Efficacy of RNA-NPs targeting physiologically relevant antigens. (A) C57Bl/6 mice were vaccinated with a single OVA RNA-NP or DC vaccine pulsed with OVA mRNA following OT-I administration before splenocytes were harvested 1 week later for assessment of %antigen-specific CD8+ cells. (B) DS red+ tumor-specific T cells were expanded in vitro from primed spleens of mice immunized against TTRNA extracted from KR158B-luc. These T cells were adoptively transferred after 5–7 d of in vitro activation into KR158B-luc intracranial tumor-bearing C57Bl/6 mice followed by TTRNA-NPs or TTRNA-pulsed DCs. (C) C57Bl/6 mice were implanted with B16F0 melanomas (250,000 cells/mouse) in the flank and vaccinated 1 d later with weekly TTRNA-NPs × 3 (*p < 0.05; **p < 0.01, Mann–Whitney test). (D) C57Bl/6 mice were implanted with B16F0 melanomas (250,000 cells/mouse) in the flank and vaccinated 1 d later with weekly TTRNA-NPs that were pre-treated with an IFN-α-blocking antibody. (E) C57Bl/6 mice were stereotactically implanted with KR158B-luc astrocytoma cells and received a single dose of 9Gy TBI (on Day 4) followed by an i.v. injection of 5 × 104 lineage negative bone marrow-derived stem cells (within 6 h of TBI) and i.v. injection of 107 tumor-specific T lymphocytes (1 d post-TBI).Citation17 This was immediately followed by vaccination of 2.5 × 105 total tumor RNA-pulsed DCs or TTRNA-NPs. The second and third RNA-NP/DC vaccines were administered at weekly intervals (*p < 0.05, Gehan–Breslow–Wilcoxon test).
Discussion
Although ex vivo generation of TTRNA-loaded DCs holds considerable promise, the advancement of cellular vaccines is fraught with developmental challenges and manufacturing delays making it difficult to generate readily available vaccines for patients with invasive malignancies.Citation2,19 After leukopheresis collection, autologous DC manufacturing and testing typically requires several weeks. This production time provides a significant delay for patients with aggressive malignancies and may preclude candidates from trial with rapid disease progression. To circumvent the challenges of cellular therapeutics and develop an immunotherapeutic strategy that could be available expeditiously, we have utilized commercially available and clinically translatable nanoliposomes to deliver TTRNA to APCs in vivo. While TTRNA has been shown to be a rich source of antigenic material for induction of antitumor immunity, “naked” RNA is inherently unstable.Citation20 Clinically available nanoliposomes can be combined with tumor-derived RNA extracted and amplified from patient specific biopsies or in vitro transcribed mRNAs encoding for tumor-specific antigens to generate personalized tumor RNA-NPs with considerable scale-up capacity.Citation4-8 We have shown that the nanoliposome DOTAP can encapsulate RNA into ∼70–200 nm particles protecting nucleic acids from degradation while simultaneously delivering them to APCs in lymphoid organs where peripheral T cells can be activated against intracranial malignancies.Citation21
It has previously been shown that mRNA encapsulated in cationic liposomes can be utilized to induce immunity against model antigens, but there have not been head to head studies versus DCs using physiologically relevant antigens.Citation22 In the above studies, we pursued a translational line of investigation, which has enabled us to generate distinct and impactful findings: (1) we compared cationic liposomes to polyethylenamine NPs and identified DOTAP as the most promising formulation based on in vitro transfection efficiency, MHC I expression, and antigen-specific immune response in vivo; (2) we characterized a target formulation not only based on size and charge, but stability overtime; (3) we revealed intravenous delivery as the optimal route for RNA-NP administration and harnessed the natural reticuloendothelial depots of systemic nanoliposomal formulations demonstrating RNA-NP transfection of CD11c+ cells; (4) we demonstrate systemic immune activation through a surge in maturation markers and B7 co-stimulatory molecules on reticuloendothelial and intratumoral APCs; (5) we demonstrate superior immunogenicity to peptide vaccines; and (6) we demonstrated induction of antitumor immunity using physiologically relevant RNA-loaded nanoliposomes in an aggressive intracranial tumor model.
While reticuloendothelial localization is a clear impediment in the context of standard liposomal drug delivery, these areas of localization are critical and advantageous for APC transfection and T-cell priming. RNA-NPs induce systemic immune activation and increase percent expression of MHC class I/II B7 co-stimulatory molecules, and maturation markers on splenic APCs (all vital for T cell activation). We corroborated the results of Kranz et al. and Broos et al. demonstrating that RNA-NPs mediate antitumor immunity through a type I interferon dependent mechanism.Citation23,24 While other nanoliposomal formulations including DOTMA-DOPE and lipofectamine RNAiMAX may be relevant for RNA delivery, optimization and selection of the most efficient RNA-lipoplexes may require some prioritization schema based on in vivo functional activity (i.e., immune response or induction of therapeutic antitumor immunity).Citation23,24
Although mature APCs such as DCs are the intended targets of RNA-NPs, localization to myeloid-derived suppressor cells or tumor-associated macrophages may stymie the downstream effector response elicited by these formulations. However, a distinct advantage of this platform compared with other approaches is the unique ability to package RNA-NPs to deliver combinatorial therapies using a single delivery platform.Citation4,25 As such, RNA-NPs can be harnessed to embed immunomodulatory RNAs that influence regulatory cellular compartments. Depending on the immunomodulatory modifications employed and the timing of each RNA-NP administration, this versatility allows for development of RNA-NPs tailored to modulate the host innate and adaptive immune response against a desired epitope or cohort of tumor antigens.
While RNA-pulsed DC vaccines have shown promise in clinical trials in patients with GBM, they are limited by their cost, complexity, and manufacturing delays.Citation1 Alternatively, RNA-NPs can be generated in a few days after a patient's biopsy cutting down the time to generation of personalized vaccines. Nanoliposomes such as DOTAP, have been utilized for enhancing pharmacologic drug delivery in several clinical trials, are readily available in clinical-grade formulation, and can be engineered to upregulate innate and adaptive host immunity.Citation26-28 In a phase I dose-escalation study using systemic RNA-loaded liposomes targeting four tumor-associated antigens, apart from transient flu-like symptoms, RNA-lipoplexes were well tolerated.Citation24 While tumor mRNA-encapsulated liposomes need further safety interrogation, they appear to have a promising safety profile in humans and are well tolerated in mice.Citation24 Since RNA-NPs bypass MHC restriction (through encoding full-length antigens that can be processed and presented by host APC machinery), circumvent the complexity of cellular therapeutics, and are amenable to central distribution, these formulations can provide near immediate immune induction and be harnessed as a more feasible and effective therapy for patients with refractory malignancies.Citation29 Although the local and systemic milieu of invasive malignancies such as GBM is notoriously immunosuppressive, the immune modulating properties of RNA-NPs can be harnessed to re-program the quiescent immune environment toward an activated phenotype.Citation30
Materials and methods
Isolation of RNA
Total tumor-derived RNA from tumor cells (B16F0, B16F10, and KR158-luc) was isolated using commercially available RNeasy mini kits (Qiagen) based on manufacturer instructions.Citation17 To generate model RNA templates (Green Fluorescent Protein (GFP), luciferase, and OVA) for in vitro transcription (IVT), plasmids were linearized using restriction enzymes (i.e., SpeI) and purified with Qiagen PCR MiniElute kits. Linearized DNA was subsequently transcribed using the mmRNA IVT kit (Life technologies, Invitrogen) and cleaned up using RNA Maxi kits (Qiagen).
Nanoparticles
Cationic liposomes (DOTAP, DOTAP-DOPE) were acquired from Avanti, Polar Lipids Inc. (Alabaster, AL, USA). For preparation, chloroform was added to re-suspend 25–100 mg of cationic liposome. Chloroform was then evaporated off until a thin lipid layer remained. The mixture was re-suspended in 5–20 mL of PBS (Gibco, Phosphate-Buffered Saline pH 7.4, 1x) before being placed in a 50°C water bath for 2 h and vortexed intermittently. One day later, 5–20 mL of PBS was added to the mixture, vortexed and placed in a bath sonicator for 5 min before being filtered through a 0.43 μm syringe and subsequently a 0.22 μm syringe (PALL Acrodisc syringe filter with Supor membrane). The prepared NP solution was then stored at 4°C until use. Ratio of RNA to filtered DOTAP was based on pre-filtration DOTAP concentration (2.5 ug/uL). Polyethylenimine NPs (JETPE, and JETPEI mannose) were obtained from Polyplus transfection and prepared using manufacturer's instructions. Lipofectamine® RNAiMAX transfection reagent was obtained from Thermo Fisher and prepared using manufacturer's instructions.
RNA-NP complex preparation
For in vivo preparations, 25 μg of RNA were added to 375 μg of DOTAP (per mouse) in HBS or PBS buffer. For in vitro preparations, 1.67 μg of RNA were added to 25 μg of DOTAP (per 1 × 105 cells) in HBS or PBS buffer. This mixture was kept at room temperature for 15 to 20 min to allow RNA-DOTAP complexes to form. For in vivo i.v. injections, 200 μL of RNA-DOTAP was injected into the tail vein of each C57Bl/6 mouse.
Cryo-electron microscopy
Thin films were prepared by placing 3 μL of sample suspension onto holey carbon grids (C-Flats; Protochips, Inc.); these were vitrified by plunging into nitrogen cooled ethane, using a Vitrobot™ Mark IV freezing device (FEI Co.). Citation31 The frozen grids were then imaged using a 16-megapixel CCD camera (Gatan, Inc.) in a Tecnai G2 F20-TWIN Transmission Electron Microscope (FEI), which was operated at a voltage of 200 kV using low-dose conditions (∼20 e/Å2).Citation31
Dynamic light scatter and zeta-potential analysis
The mean particle diameter and size distribution were determined by using the Microtrac Nanotrac dynamic light scattering DLS. The surface charge of RNA-NP complexes was determined by analysis of the ζ-potential using the Brookhaven Zeta Plus instrument. Measurements were performed at 25°C using deionized water and repeated for each sample;10 runs of 5 cycles were performed with pH assumed to be 7 and measurements were obtained using the Smoluchowski calculation.
Mice
Four- to eight-week old C57Bl/6 mice were procured from Jackson Laboratory (Bar Harbor, ME, stock#000664).Citation17 CD8+ OVA-specific TCR (OT-I) transgenic mice on the C57Bl/6 background were purchased from Jackson Laboratories. As proposed by the committee on care of Laboratory Animal Resources Commission on Life Sciences, National Research Council, the investigators adhered to the “Guide for the Care and Use of Laboratory Animals.”Citation17 The facilities are fully accredited by the American Association for Accreditation of Laboratory Animal Care, and the Institutional Animal Care and Use Committee approved all studies.Citation17
Bioluminescent imaging
Transfected cells in vitro and animals vaccinated with luciferase RNA-NPs were imaged in vivo using an IVIS Kinetic (Perkin Elmer).Citation17 Mice were injected with 100 μL luciferin substrate and imaged 6 h after injection of RNA-NPs.Citation17 HEK cells and animal organs were assessed for bioluminescence after addition of luciferin substrate 6 h after administration of luciferase RNA-NPs.
OT-I transfer and RNA-NP vaccination
Naïve C57Bl/6 mice received WBCs harvested from the spleens of OT-I transgenic mice (C57Bl/6-Tg(TcraTcrb)1100Mjb/J)(0.5–1 × 107 cells total). WBCs from OT-Is were administered either i.v./i.p. and labeled with CFSE (Celltrace) in select experiments. Immediately following cell transfer, mice were vaccinated with RNA-NPs or with 100 µg i.d. of OVA class I peptide (SIINFEKL; American Peptide Company, Inc., CA) in 10% DMSO with an equal volume of CFA (DIFCO Laboratories, MI) (100 µL/mouse).Citation32 Fresh spleens were harvested from vaccinated mice 1 week later prior to undergoing RBC lysis. Splenocytes were then washed and co-stained with anti-CD8 APC antibody (eBioscience) and a Ly5.1 antibody (BD PharMingen) or a tetramer specific for H-2Kb-restricted SIINFEKL (MBI). Samples were analyzed using flow cytometry.
Flow cytometric analysis
Organs were harvested from mice one day after RNA-NP injection. Organs underwent RBC lysis before WBC extraction before being washed and stained for surface expression of CD11c in conjunction with CD80, CD86, MHC I/II, or CD40 (Affymetrix). GFP expression in vivo was amplified by anti-GFP Alexa-488 antibody in select experiments. After incubation, cells were fixed and analyzed with a BD Bioscience FACS Calibur.
Cytokine bead assay
To analyze memory recall responses, we harvested, lysed, and washed spleens before performing a re-stimulation assay with 1 µM SIINFEKL chicken OVA peptide or negative control peptide in complete T-cell media (RPMI 10% FBS 100 µM non-essential amino acids, 1 mM sodium pyruvate, 2 mM L-glutamine, 50 µg/mL gentamycin sulfate, 100 IU/mL penicillin/Streptomycin, and 55 µM 2-mercaptoethanol).Citation32 Cells were incubated at 37°C (5% CO2), and supernatants were collected within 24–48 h.Citation32 A 50 µL culture supernatant was mixed with 50 µL capture beads and 50 µL detection reagent for Th1/Th2 cytokine release (BD Bioscience) and incubated for 2 h at room temperature.Citation32 Cells were washed and analyzed by flow cytometric analysis per manufacturer instructions.Citation32
Monoclonal antibodies
Interferon-α monoclonal antibodies were purchased from BioXcell and 200–500 μg/dose were administered twice weekly in the peritoneum of C57Bl/6 mice. IFN-α ELISA was performed using a mouse IFN-α Platinum ELISA (eBioscience) per manufacturer's instructions.
Tumor cells
B16F0 cells were purchased from ATCC. The tumor cell lines B16F10 and B16F10-OVA were obtained as a kind gift from Dr Richard G. Vile, PhD, at Mayo Clinic.Citation33,34 KR158B-luc cells were obtained through a kind gift from Dr Tyler Jacks (Massachusetts Institute of Technology, Boston, MA).Citation17,18 This KR158 line was originally isolated from a spontaneously arising astrocytoma in an NF1;Trp53 mutant mouse that was on a C57Bl/6 background.Citation17
Tumor culture conditions
Culture media for B16F0, B16F10, and B16F10-OVA consisted of DMEM with sodium pyruvate, supplemented with 10% heat inactivated fetal bovine serum (FBS), and 5.5 mL Penicillin/ Streptomycin; cells were kept in a 37˚C incubator (5% CO2 levels).Citation32 Culture media for KR158-luc consisted of DMEM without sodium pyruvate (LifeTechnologies), supplemented with 10% heat inactivated fetal bovine serum (FBS) (LifeTechnologies), 5.5 mL Penicillin/Streptomycin (Life Technologies); cells were kept in a 37˚C incubator (5% CO2).Citation17
Tumor implantations
B16F0, B16F10, B16F10-OVA, and KR158B-luc cells were harvested with trypsin (Gibco) before washed once in serum-containing medium, and once in phosphate-buffered saline (PBS).Citation17 Trypan blue dye exclusion was utilized to re-suspend cell pellets in PBS at the appropriate concentration of viable cells.Citation17 B16F0, B16F10, and B16F10-OVA were injected subcutaneously in the left flank of C57Bl/6 mice anesthetized with isoflurane. Animals bearing subcutaneous tumors reaching human endpoints (i.e., tumor ulceration) were removed from analysis of tumor volume measurements at later time points across groups. For intracranial implantations, B16F10-OVA and KR158B-luc tumors cells were mixed with an equal volume of 10% methylcellulose in PBS before being loaded into a 250-µL syringe (Hamilton, Reno, NV) with an attached 25 gauge needle.Citation32 Intracranial implantations were performed by positioning the tip of the needle at the bregma and 2 mm to the right of the cranial midline suture and 4 mm below the surface of the cranium using a Kopf stereotactic frame (David Kopf Instruments, Tujunga, CA).Citation17
Dendritic cell vaccines
DCs were generated based on previously published methods by Flores et al.Citation17 Briefly, tibias and femurs from C57Bl/6 mice were harvested and bone marrow was flushed with RPMI (LifeTechnologies) in 10% FBS (LifeTechnologies).Citation17 Red blood cells were lysed with 10 mL of Pharmlyse (BD Bioscience) and mononuclear cells were re-suspended in complete DC media (RPMI-1640, 5% FBS, 1 M HEPES (LifeTechnologies), 50 mM, 55 mM bmercaptoethanol (LifeTechnologies), 100 mM Sodium pyruvate (LifeTechnologies), 10 mM Nonessential amino acids (LifeTechnologies), 200 mM L-glutamine (LifeTechnologies), 10 mg GM-CSF (R&D Systems), 10 mg IL- 4 (R&D Systems), 5.5 mL Penicillin/Streptomycin (LifeTechnologies) before being plated into tissue culture treated six-well plates at a concentration of 106 cells/mL in a total volume of 3 mL/well.Citation17 Non-adherent cells were discarded at day 3; at day 7, non-adherent cells were collected and re-plated onto 100 mm tissue treated culture dishes at a concentration of 106 cells/mL in a total volume of 5 mL/dish.Citation17 24 h later, resulting cells were electroporated with 25 ug of total RNA (extracted from KR158B-luc cells), or OVA mRNA (RNeasy, Qiagen).Citation17 RNA-pulsed DCs were collected the next day and suspended in PBS at a final concentration of 1.25 × 106 cells/mL; 200 uL was administered via intradermal or intraperitoneal injection.Citation17
Generation of tumor-specific T cells
Tumor-specific T cells were generated based on previously published methods by Flores et al.Citation17 Briefly, total RNA was isolated from KR158B-luc tumor before being electroporated into bone marrow-derived DCs using BTX Single Waveform Electroporation System (Harvard Apparatus).Citation17 Naıve mice or DsRed transgenic mice on a C57Bl/6 background (Jackson Laboratories, stock #006051) were then vaccinated with total tumor RNA-pulsed DCs, before having their spleens harvested 7 d later; splenocytes were expanded ex vivo using RNA-pulsed DCs and 100IU IL-2 (R&D Systems) for 7 d.Citation17 Tumor-reactive T cells were then adoptively transferred intravenously after 5–7 d of in vitro activation.Citation17
Cellular immunotherapy model for invasive high-grade Glioma
A cellular immunotherapy malignant glioma model was generated based on previously published methods by Flores et al.Citation17 Briefly, naïve C57Bl/6 mice were stereotactically implanted with 104 KR158B-luc astrocytoma cells into the right caudate nucleus on Day 0.Citation17 Mice subsequently received a single dose of 9 Gray (Gy) total body irradiation (TBI) on Day 4 before receiving haematopoietic stem cell (HSC) rescue through i.v. injection of 5 × 104 lineage negative bone marrow-derived stem cells within 6 h of TBI.Citation17 HSCs were derived from the bone marrow of C57Bl/6 mice and isolated through magnetic separation of lineage negative bone marrow stem cells (Miltenyi Biotec).Citation17 Afterwards, 107 tumor-specific T lymphocytes were intravenously administered between 16 and 24 h post-TBI and were immediately followed by vaccination with 2.5 × 105 total tumor RNA-pulsed DCs or RNA-NPs.Citation17 The second and third RNA-NP and DC vaccines were administered at weekly intervals. Mice were followed for survival and sacrificed when moribund.Citation17
Statistical analysis
Survival data from the animal studies were analyzed by the Gehan-Breslow-Wilcoxon test. Subcutaneous tumor measurements were plotted with standard errors from the mean and time-points were analyzed by Mann-Whitney non-parametric tests. Student's t test and Mann–Whitney non-parametric test were used to determine statistical significance for in vivo immunogenicity experiments. Student's t test was used to determine statistical significance for in vitro experiments. Statistical significance was set for p values less than 0.05. GraphPad Prism and Microsoft excel were used to conduct all analyses.
Disclosure of potential conflicts of interest
JHS receives honoraria from Celldex Therapeutics and funding under the Duke University Faculty Plan from license fees paid to Duke University by Celldex Therapeutics. EJS, GDL, CP, HK, JC, AG, JH, LSP, CF, and DAM declare no conflicts of interest.
Acknowledgments
The authors would like to acknowledge David Snyder for his technical assistance with intravenous injections and intracranial implantations and Weihua Xie and Jeffrey Drake for their molecular biology assistance. The authors would also like to thank Paul Chipman at the electron microscopy core at the University of Florida Interdisciplinary Center of Biotechnology Research for his assistance with cryo-electron microscopy and Gary Scheiffele for his assistance with dynamic light scatter and zeta potentials.
Funding
This work was supported by the National Institutes of Health- National Cancer Institute R01-CA195563–01 (to D.A. Mitchell), 5T32-CA009307–34, and K08CA199224 (to E.J. Sayour). Additional grants used to support this work were from Alex's Lemonade Stand (to D.A. Mitchell), The Beez Foundation (to E.J. Sayour), The Thrasher Research Fund (Early Career Award to E.J. Sayour), Hyundai Hope on Wheels (Hope Award to E.J. Sayour), St. Baldrick's Foundation (Hannah's Heroes Scholar Award to E.J. Sayour), and the American Brain Tumor Association (Discovery Award to E.J. Sayour). Additional support was provided by the Pediatric Brain Tumor Foundation of the United States (D.A. Mitchell and J.H. Sampson).
References
- Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, Congdon KL, Reap EA, Archer GE, Desjardins A et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015; 519:366-9; PMID:25762141; http://dx.doi.org/10.1038/nature14320
- Mitchell DA, Sampson JH. Toward effective immunotherapy for the treatment of malignant brain tumors. Neurotherapeutics 2009; 6:527-38; PMID:19560742; http://dx.doi.org/10.1016/j.nurt.2009.04.003
- Sonabend AM, Ogden AT, Maier LM, Anderson DE, Canoll P, Bruce JN, Anderson RC. Medulloblasoma: challenges for effective immunotherapy. J Neuro-Oncol 2012; 108:1-10; PMID:22173741; http://dx.doi.org/10.1007/s11060-011-0776-1
- Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol 2007; 2:751-60; PMID:18654426; http://dx.doi.org/10.1038/nnano.2007.387
- Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer 2005; 5:161-71; PMID:15738981; http://dx.doi.org/10.1038/nrc1566
- Nguyen DN, Mahon KP, Chikh G, Kim P, Chung H, Vicari AP, Love KT, Goldberg M, Chen S, Krieg AM et al. Lipid-derived nanoparticles for immunostimulatory RNA adjuvant delivery. Proc Natl Acad Sci U S A 2012; 109:E797-803; PMID:22421433; http://dx.doi.org/10.1073/pnas.1121423109
- Perche F, Benvegnu T, Berchel M, Lebegue L, Pichon C, Jaffres PA, Midoux P. Enhancement of dendritic cells transfection in vivo and of vaccination against B16F10 melanoma with mannosylated histidylated lipopolyplexes loaded with tumor antigen messenger RNA. Nanomedicine 2011; 7:445-53; PMID:21220051; http://dx.doi.org/10.1016/j.nano.2010.12.010
- Copland MJ, Baird MA, Rades T, McKenzie JL, Becker B, Reck F, Tyler PC, Davies NM. Liposomal delivery of antigen to human dendritic cells. Vaccine 2003; 21:883-90; PMID:12547598; http://dx.doi.org/10.1016/S0264-410X(02)00536-4
- Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Translat Med 2012; 4:127ps8; PMID:22461638; http://dx.doi.org/10.1126/scitranslmed.3003634
- Lu C, Stewart DJ, Lee JJ, Ji L, Ramesh R, Jayachandran G, Nunez MI, Wistuba II, Erasmus JJ, Hicks ME et al. Phase I clinical trial of systemically administered TUSC2(FUS1)-nanoparticles mediating functional gene transfer in humans. PloS One 2012; 7:e34833; PMID:22558101; http://dx.doi.org/10.1371/journal.pone.0034833
- Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, Pierce RH, Crispe IN. Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood 2007; 110:4077-85; PMID:17690256; http://dx.doi.org/10.1182/blood-2007-02-073841
- Klugewitz K, Blumenthal-Barby F, Schrage A, Knolle PA, Hamann A, Crispe IN. Immunomodulatory effects of the liver: deletion of activated CD4+ effector cells and suppression of IFN-gamma-producing cells after intravenous protein immunization. J Immunol 2002; 169:2407-13; PMID:12193708; http://dx.doi.org/10.4049/jimmunol.169.5.2407
- Polakos NK, Klein I, Richter MV, Zaiss DM, Giannandrea M, Crispe IN, Topham DJ. Early intrahepatic accumulation of CD8+ T cells provides a source of effectors for nonhepatic immune responses. J Immunol 2007; 179:201-10; PMID:17579039; http://dx.doi.org/10.4049/jimmunol.179.1.201
- Lorenzer C, Dirin M, Winkler AM, Baumann V, Winkler J. Going beyond the liver: Progress and challenges of targeted delivery of siRNA therapeutics. J Control Release 2015; 203:1-15; PMID:25660205; http://dx.doi.org/10.1016/j.jconrel.2015.02.003
- Carralot JP, Probst J, Hoerr I, Scheel B, Teufel R, Jung G, Rammensee HG, Pascolo S. Polarization of immunity induced by direct injection of naked sequence-stabilized mRNA vaccines. Cell Mol Life Sci 2004; 61:2418-24; PMID:15378210; http://dx.doi.org/10.1007/s00018-004-4255-0
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Eng J Med 2010; 363:411-22; PMID:20818862; http://dx.doi.org/10.1056/NEJMoa1001294
- Flores C, Pham C, Snyder D, Yang S, Sanchez-Perez L, Sayour E, Cui X, Kemeny H, Friedman H, Bigner DD et al. Novel role of hematopoietic stem cells in immunologic rejection of malignant gliomas. Oncoimmunology 2015; 4:e994374; PMID:25949916; http://dx.doi.org/10.4161/2162402X.2014.994374
- Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet 2000; 26:109-13; PMID:10973261; http://dx.doi.org/10.1038/79075
- Farrell D, Ptak K, Panaro NJ, Grodzinski P. Nanotechnology-based cancer therapeutics–promise and challenge–lessons learned through the NCI Alliance for Nanotechnology in Cancer. Pharm Res 2011; 28:273-8; PMID:20814720; http://dx.doi.org/10.1007/s11095-010-0214-7
- Weide B, Carralot JP, Reese A, Scheel B, Eigentler TK, Hoerr I, Rammensee HG, Garbe C, Pascolo S. Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. J Immunother 2008; 31:180-8; PMID:18481387; http://dx.doi.org/10.1097/CJI.0b013e31815ce501
- Templeton NS, Lasic DD, Frederik PM, Strey HH, Roberts DD, Pavlakis GN. Improved DNA: liposome complexes for increased systemic delivery and gene expression. Nat Biotechnol 1997; 15:647-52; PMID:9219267; http://dx.doi.org/10.1038/nbt0797-647
- Hess PR, Boczkowski D, Nair SK, Snyder D, Gilboa E. Vaccination with mRNAs encoding tumor-associated antigens and granulocyte-macrophage colony-stimulating factor efficiently primes CTL responses, but is insufficient to overcome tolerance to a model tumor/self antigen. Cancer Immunol Immunother 2006; 55:672-83; PMID:16133108; http://dx.doi.org/10.1007/s00262-005-0064-z
- Broos K, Van der Jeught K, Puttemans J, Goyvaerts C, Heirman C, Dewitte H, Verbeke R, Lentacker I, Thielemans K, Breckpot K. Particle-mediated intravenous delivery of antigen mRNA results in strong antigen-specific T-cell responses despite the induction of type I interferon. Mol Ther Nucleic Acids 2016; 5:e326; PMID:27327138; http://dx.doi.org/10.1038/mtna.2016.38
- Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016; 534:396-401; PMID:27281205; http://dx.doi.org/10.1038/nature18300
- McNeil SE. Nanoparticle therapeutics: a personal perspective. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2009; 1:264-71; PMID:20049796; http://dx.doi.org/10.1002/wnan.6
- Porteous DJ, Dorin JR, McLachlan G, Davidson-Smith H, Davidson H, Stevenson BJ, Carothers AD, Wallace WA, Moralee S, Hoenes C et al. Evidence for safety and efficacy of DOTAP cationic liposome mediated CFTR gene transfer to the nasal epithelium of patients with cystic fibrosis. Gene Ther 1997; 4:210-8; PMID:9135734; http://dx.doi.org/10.1038/sj.gt.3300390
- Strieth S, Dunau C, Michaelis U, Jager L, Gellrich D, Wollenberg B, Dellian M. Phase I/II clinical study on safety and antivascular effects of paclitaxel encapsulated in cationic liposomes for targeted therapy in advanced head and neck cancer. Head Neck 2013; 36:976-84; PMID:23733258; http://dx.doi.org/10.1002/hed.23397
- Fasol U, Frost A, Buchert M, Arends J, Fiedler U, Scharr D, Scheuenpflug J, Mross K. Vascular and pharmacokinetic effects of EndoTAG-1 in patients with advanced cancer and liver metastasis. Ann Oncol 2012; 23:1030-6; PMID:21693769; http://dx.doi.org/10.1093/annonc/mdr300
- Schlake T, Thess A, Fotin-Mleczek M, Kallen KJ. Developing mRNA-vaccine technologies. RNA Biol 2012; 9:1319-30; PMID:23064118; http://dx.doi.org/10.4161/rna.22269
- Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res 2013; 19:3165-75; PMID:23613317; http://dx.doi.org/10.1158/1078-0432.CCR-12-3314
- Kailasan S, Garrison J, Ilyas M, Chipman P, McKenna R, Kantola K, Söderlund-Venermo M, Kučinskaitė-Kodzė I, Žvirblienė A, Agbandje-McKenna M. Mapping Antigenic Epitopes on the Human Bocavirus Capsid. J Virol 2016; 90:4670-80; PMID:26912619; http://dx.doi.org/10.1128/JVI.02998-15
- Sanchez-Perez LA, Choi BD, Archer GE, Cui X, Flores C, Johnson LA, Schmittling RJ, Snyder D, Herndon JE 2nd, Bigner DD et al. Myeloablative temozolomide enhances CD8(+) T-cell responses to vaccine and is required for efficacy against brain tumors in mice. PloS One 2013; 8:e59082; PMID:23527092; http://dx.doi.org/10.1371/journal.pone.0059082
- Sanchez-Perez L, Kottke T, Diaz RM, Ahmed A, Thompson J, Chong H, Melcher A, Holmen S, Daniels G, Vile RG. Potent selection of antigen loss variants of B16 melanoma following inflammatory killing of melanocytes in vivo. Cancer Res 2005; 65:2009-17; PMID:15753401; http://dx.doi.org/10.1158/0008-5472.CAN-04-3216
- Daniels GA, Sanchez-Perez L, Diaz RM, Kottke T, Thompson J, Lai M, Gough M, Karim M, Bushell A, Chong H et al. A simple method to cure established tumors by inflammatory killing of normal cells. Nat Biotechnol 2004; 22:1125-32; PMID:15300260; http://dx.doi.org/10.1038/nbt1007