
ABSTRACT
High hydrostatic pressure (HHP) promotes key characteristics of immunogenic cell death (ICD), in thus far resembling immunogenic chemotherapy and ionizing irradiation. Here, we demonstrate that cancer cells succumbing to HHP induce CD4+ and CD8+ T cell-dependent protective immunity in vivo. Moreover, we show that cell death induction by HHP relies on the overproduction of reactive oxygen species (ROS), causing rapid establishment of the integrated stress response, eIF2α phosphorylation by PERK, and sequential caspase-2, −8 and −3 activation. Non-phosphorylatable eIF2α, depletion of PERK, caspase-2 or −8 compromised calreticulin exposure by cancer cells succumbing to HHP but could not inhibit death. Interestingly, the phagocytosis of HHP-treated malignant cells by dendritic cells was suppressed by the knockdown of caspase-2 in the former. Thus, caspase-2 mediates a key function in the interaction between dying cancer cells and antigen presenting cells. Our results indicate that the ROS→PERK→eIF2α→caspase-2 signaling pathway is central for the perception of HHP-driven cell death as immunogenic.
Introduction
Conventional anticancer treatments including chemotherapy and radiotherapy operate by inducing widespread cancer cell death thereby helping in “debulking” of the tumor. It has been long hypothesized that most anticancer therapies induce poorly immunogenic or even tolerogenic cancer cell death.Citation1 However, accumulating evidence indicates that in response to certain chemotherapeutics (e.g., anthracyclines, mitoxantrone, oxaliplatin, or bortezomib),Citation2,3 ionizing irradiation,Citation4 oncolytic viruses,Citation5,6 and Hypericin-based photodynamic therapy (Hyp-PDT),Citation7,8 tumor cells can undergo an immunogenic form of apoptosis called “immunogenic cell death” (ICD) inducing an effective antitumor immune response in immunocompetent mice compared with vaccination of immunodeficient mice.Citation9,10 These findings demonstrate the important role of the immune system in the efficacy of anticancer therapy.
The ICD is mediated largely by spatiotemporally defined release or exposure of “danger signals” or damage-associated molecular patterns (DAMPs) that can function as either adjuvants or danger signals for the innate immune systemCitation10 leading to the induction of host protective anticancer immunity.Citation11,12 Several DAMPs have recently been associated with ICD, of which surface exposure (ecto-) of the endoplasmic reticulum (ER)-resident chaperone calreticulin (CALR) constitutes one of the major checkpoints determining the immunogenicity of cell death.Citation12,13 Ecto-CALR is best characterized for its prominent function as an “eat me” signal for CD91 positive cells (mostly macrophages and dendritic cells) and stimulates antigen-presenting cells, particularly DCs, to efficiently engulf dying cells, process their antigens, and prime a specific immune response.Citation14
Ecto-CALR's importance for ICD is outlined by the fact that its blockade (via CALR-neutralizing antibodies), depletion of CALR with small interfering RNAs (siRNAs) or depletion/inhibition of danger signaling pathway components mediating ecto-CALR exposure, abolishes the immunogenicity of ICD in multiple tumor models.Citation4,7,11,13,15 CALR translocates from the ER lumen to the cell surface after treatment with various ICD inducers, before the cell manifests signs of programmed cell death.Citation16 Rapid, pre-apoptotic ecto-CALR is potently triggered by chemotherapeutics and physicochemical modalities such as Hyp-PDT, which induce the production of reactive oxygen species (ROS) and ER stress response (concomitant or sequential).Citation17 Of note, both ROS and ER stress “modules” are required for efficient danger signaling and ICD such that the absence of either compromises immunogenicity.Citation7,15 For instance, scavenging of ROS by antioxidants abolishes ecto-CALR induced by anthracyclinesCitation15 and Hyp-PDT.Citation18 Similarly, ER stress response also plays an important role in mediating CALR exposure. However depending on the ICD inducer, ecto-CALR mediating signaling components can be subdivided into either “core components” (i.e., signaling components shared by all ICD inducers for ecto-CALR exposure) or “private components” (i.e., signaling components specific to certain ICD inducers).Citation19 Here, in the case of chemotherapy, ER stress response consisting of the ER stress sensor, PERK (protein kinase R (PKR)-like endoplasmic reticulum kinase)-induced phosphorylation of eukaryotic translation initiation factor, eIF2α, both, playing an important role in ecto-CALR exposure.Citation15 Ecto-CALR exposure in response to chemotherapy requires downstream of ER stress, caspase-8-mediated cleavage of the ER-resident protein, BAP31, and conformational activation of Bax and Bak.Citation15 However, the Hyp-PDT pathway differs markedly, such that only PERK and Bax/Bak are required for ecto-CALR exposure.Citation7 Thus, based on these observations, although PERK and Bax/Bak represent the “core signaling components” mediating ecto-CALR for both chemotherapy and Hyp-PDT, eIF2α phosphorylation, caspase-8 and BAP31 represent the “private signaling components” only applicable to chemotherapy-induced ecto-CALR. However, in absence of analysis for other ICD inducers, it is not yet known whether such a subdivision of danger signaling components is consistently applicable to other contexts and whether additional as-yet-undiscovered “private signaling components” mediating ecto-CALR, exist.Citation10 We previously described a novel physical modality, high hydrostatic pressure (HHP), inducing ICD in a wide spectrum of primary human tumor cells and human cancer cell lines.Citation20,21 The early danger signaling pathways activated by HHP in cancer cells are completely unknown. Therefore, we decided to investigate the signaling events associated with the ICD induced by HHP treatment and compare them with known pathways triggered by immunogenic chemotherapy or Hyp-PDT.Citation7,15
Materials and methods
Mice
Female BALB/c and male C57BL/6 (B6) mice were obtained from the animal facility of the Institute of Physiology (Academy of Sciences of the Czech Republic), v.v.i. Mice were used at 9–15 weeks of age and kept in the conventional animal facility of Institute of Microbiology of ASCR, v.v.i. Mice were regularly screened for MHV and other pathogens according to FELASA. All experiments were approved by the Animal Welfare Committee at the Institute of Microbiology of ASCR, v.v.i.
Treatment of CT26 colon carcinoma and LL2 lung carcinoma in vivo
BALB/c (CT26 carcinoma) or B6 (LL2 carcinoma) mice were s.c. injected into lower left flank with 5 × 106 HHP-treated CT26 or LL2 cells in 200 µL of PBS on days 0 and 21, respectively. Control mice were injected with the same volume of PBS. Mice were then s.c. injected into lower right flank with 105 live CT26 cells or LL2 cells in 100 µL of PBS on day 31. 250 µg of depleting anti-CD4+ (clone GK1.5, BioXcell) and/or anti-CD8+ (clone 53-6.72, BioXcell) mAbs were injected i.p. and control mice were injected with the same volume (250 µL) of PBS. Mice surviving day 130 without any signs of tumor were considered as long-term survivors (LTS). Tumor size was measured every 2–4 d by caliper. A total of 10 mice per group were used in the experiments. Every experiment was repeated twice with the similar results.
Cell lines
All cell lines were purchased from American Type Culture Collection (Manassas, VA, USA). Ovarian cancer cell line OV-90 (ATCC) and mouse colon adenocarcinoma CT26 cell lines were cultured in RPMI 1640 (Gibco) supplemented with 10% heat-inactivated FBS (PAA), 2 mM GlutaMAX I CTS (Gibco) and 100 U/mL penicillin + 100 µg/mL streptomycin (Gibco). MEF-wild type (WT) and Bax−/−Bak−/−, a kind gift of Dr. G. Kroemer (INSERM U848, Institut Gustave Roussy, France). MEF cells expressing normal eIF2α (WT) or a non-phosphorylable mutant heterozygously (S51A knock-in mutation) were kindly provided by Dr. R.L. Rasor, University of Michigan. MEF and LL2 cell lines were cultured in DMEM medium (Sigma Aldrich) supplemented with 10% heat-inactivated FBS (PAA), 2 mM GlutaMAX I CTS (Gibco), and 100 U/mL penicillin + 100 µg/mL streptomycin (Gibco).
Antibodies and reagents
Antibodies against phospho-eIF2α (Ser51), eIF2α, phospho-PERK, PERK, caspase-3, caspase-8, caspase-2, CHOP, Bax, Bak (Cell Signaling Technology, Inc.), and GAPDH (GeneTex) were used. Secondary anti-rabbit and anti-mouse antibodies conjugated to horseradish peroxidase (Jackson ImmunoResearch Laboratories) were also used.
Anti-calreticulin antibodies were purchased from Enzo Life Sciences and Abcam. The chicken polyclonal antibody against calreticulin was purchased from ThermoFisher Scientific. Anti-mouse DyLight 649- and anti-rabbit Alexa Fluor 647-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratories, Molecular Probes, and Cell Signaling, respectively. The chicken IgY isotype control antibody was from GeneTex. Cell death was assessed by Annexin V-PerCP-eFluor 710 (eBioscience) and DAPI (Molecular Probes) staining. For phagocytosis, the dendritic cells and tumor cells were stained with Vybrant® DiO and Vybrant® DiD cell labeling solutions (Molecular Probes), respectively.
CellRox orange reagent (Molecular Probes) was used for the detection of ROS production. N-acetyl-L-cysteine (NAC; 5 mM) and reduced L-glutathione (GSH; 5 mM) from Sigma Aldrich were used as ROS inhibitors. NAC and GSH were prepared at 5 mM concentration in complete media followed by pH adjustment to pH 7.3–7.4. Idarubicin hydrochloride and thapsigargin were from Sigma Aldrich.
Generation of shRNA stable clones of OV-90 cells
For knockdown experiments, cells were stably transfected using Scramble sequences (SHC001V; SHC002V) or shRNA transduction particles expressing siRNA against target genes. Sequences of the shRNAs are provided in Table S1. Transfection was performed according to the manufacturer's instructions (Mission® pLKO.1-puro lentiviral particles, Sigma Aldrich). Cells were seeded at 5 × 103 in 96-well plates, and infected with a multiplicity of infection of Puromycine (1 μg/mL, Sigma Aldrich) selected infected cells.
siRNA transfection of CT26 cells
A total of 1 × 105 OV-90 or CT26 cells per well were plated in six-well plates and allowed to reach 50% confluence on the day of transfection. siRNA specific for mouse PERK (ON-TARGET plus Mouse Eif2ak3 siRNA SMART pool L-044901-00-0010), human PERK (ON-TARGET plus Human Eif2ak3 siRNA SMART pool L-004883-00-0010), human caspase-2 (ON-TARGET plus Human CASP2 siRNA SMART pool L-003465-00-0010, mouse caspase-8 (ON-TARGET plus Mouse Casp8 siRNA SMART pool L-043044-00-0010) and an unrelated control (ON-TARGET plus Non-targeting pool D-001810-10-05) were purchased from Dharmacon. Cells were transfected with siRNA at a final concentration of 25 nM using Lipofectamine® RNAiMAX Transfection Reagent (Invitrogen) according to the manufacturer's instructions. Knockdown of PERK, caspase-8, and caspase-2 was confirmed by western blotting.
Induction of cell death
Tumor cell death was induced by UV-B (312 nm), idarubicin or HHP. UV-B irradiated and idarubicin treated (37 µM for human OV-90 cells and 18.5 µM for mouse MEF and CT26 cells) tumor cells were used as a negative and positive control, respectively. Cells were treated by value of HHP 250 MPa (OV-90 cells), 175 MPa (CT26 and LL2), and 150 MPa (MEF and CT26 cells) for 10 min in the custom made service (Resato International BV, Netherlands). A total of 1×106 UV-B irradiated (7.6 J/cm2), idarubicin or HHP treated cells per well were plated in 24-well plates, cultured for 1, 2, 6, and 24 h at 37 °C under 5% CO2, and used for subsequent experiments.
Flow cytometric analysis of apoptosis and cell surface CALR expression
Briefly, 2 × 105 cells per sample were collected and washed in PBS. The cells were then incubated for 30 min with primary anti-CALR antibody diluted in PBS, followed by washing and incubation with DyLight 649- or Alexa Fluor 647-conjugated secondary antibody. For cell death assessment, the cells were washed with PBS and resuspended in an incubation buffer containing Annexin V-PerCP-eFluor 710 (eBioscience). The samples were kept in the dark and incubated for 15 min prior to the addition of DAPI (Molecular Probes) and subsequent analysis on LSRFortessa (BD Biosciences) using FlowJo software (Tree Star). The cell surface expression of CALR was analyzed only on non-permeabilized Annexin+/DAPI− cells. Annexin+/DAPI+ and Annexin−/DAPI+ cells were excluded from analysis.
Preparation of cell extracts and immunoblotting analysis
Cell extracts were prepared at the indicated time points following UV-B irradiation, idarubicin, or HHP treatment. The cells were washed with ice-cold PBS and lysed on ice in RIPA buffer (10 mM Tris pH 7.5, 150 mM NaCl, 5 mM EDTA, and 1% Triton X-100) with a protease inhibitor cocktail (Roche Diagnostics) and 1 mM PMSF (phenylmethylsulfonyl fluoride) or in sample buffer (300 mM Tris pH 6.8, 5% SDS, 50% Glycerol, 360 mM β-Mercaptoethanol, and 0.05% Bromophenol blue). Proteins were separated by 12% SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad).
The membranes were blocked in 5% nonfat dry milk in TBST buffer (50 mM Tris, 150 mM NaCl, and 0.05% Tween 20) for 1 h at room temperature and incubated with primary antibody overnight at 4 °C. Then, the membranes were washed in TBST and incubated for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibodies. Detection was performed with the enhanced chemiluminescence (ECL) detection system. Equal protein loading was ensured with a BCA assay, verified by an analysis of Ponceau-S staining of the membrane and GAPDH reprobing.
Generation of immature dendritic cells
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats of healthy donors by Ficoll-Paque PLUS gradient centrifugation (GE Healthcare) and CD14+ cells were isolated by EasySep Human CD14 positive selection kit (STEMCELL Technologies). CD14+ cells were subsequently cultured for 5 d in RPMI 1640 (Gibco) supplemented with 10% heat-inactivated FBS (PAA), 2 mM GlutaMAX I CTS (Gibco), and 100 U/mL penicillin + 100 µg/mL streptomycin (Gibco) in the presence of GM-CSF (Gentaur) at a concentration of 500 U/mL and 20 ng/mL of IL-4 (Gentaur).
Uptake of dying tumor cells by dendritic cells
For flow cytometry analysis of phagocytosis, OV-90 cells were harvested and labeled with Vybrant® DiD cell labeling solution (Molecular Probes). A fraction of tumor cells was labeled with Vybrant® DiI cell labeling solution (Molecular Probes) and used for fluorescent microscopy analysis. Stained OV-90 cells were treated by HHP, plated in 24-well plates at a concentration of 1 × 106 cells/mL and cultured for 6 h at 37 °C under 5% CO2 before use. To prepare UVB-irradiated cells, stained OV-90 cells were seeded in RPMI 1640 (Gibco) supplemented with 10% heat-inactivated FBS (PAA), 2 mM GlutaMAX I CTS (Gibco), and 100 U/mL penicillin + 100 µg/mL streptomycin (Gibco) in 24-well plates at a concentration of 1 × 106 cells/mL and subjected to a 312 nm UV-B-irradiation for 10 min to induce apoptosis. Cells were then incubated for 24 h at 37 °C with 5% CO2 before use. OV-90 cells treated by idarubicin (37 µM) were also used after 24 h of culture in the presence of chemotherapeutic agent. To determine the uptake of killed tumor cells by dendritic cells, immature dendritic cells were stained with Vybrant® DiO cell labeling solution (Invitrogen) and co-cultured with OV-90 cells at a cell ratio of 5:1 in Nunclon U-bottom 96-well plates (Nunc) for 3 h at 37 °C under 5% CO2. The cells were extensively washed prior to feeding to dendritic cells. Parallel control cultures were set up on ice to evaluate the passive transfer of dye or labeled tumor fragments to dendritic cells. The phagocytic ability of dendritic cells was evaluated by flow cytometry and fluorescent microscopy.
Immunofluorescence
Phagocytosis detection
A suspension of Vybrant® DiO labeled dendritic cells and Vybrant® DiI labeled UV-B irradiated, idarubicin or HHP treated OV-90 cells was washed in PBS and the cells were fixed with 4% paraformaldehyde (Serva) for 20 min. After washing with PBS, the cells were mounted on slides with ProLong Gold antifade reagent (Molecular Probes) using StatSpin Cytofuge (Iris Sample Processing, Westwood, MA). The slides were analyzed by a fluorescence microscope Leica DMI6000 B.
ROS detection
For the detection of ROS activity, cells were stained with CellRox orange reagent (Molecular Probes) before HHP treatment. After the treatment, cells were washed twice with culture medium and fixed in 4% paraformaldehyde for 5 min and mounted on slides.
Statistical analysis
Results are shown as mean values of at least three independent experiments and standard deviation ( ± SD) represented by bars. The significance of differences was estimated by Student's unpaired or paired two-tailed t test using GraphPad Prism 6 (GraphPad Software, Inc.). Values *p < 0.05, **p < 0.01, '***p < 0.001 represent the level of significance (p < 0.05 was considered significant).
Results
HHP-treated cancer cells are immunogenic and induce CD4+ and CD8+ T cell-dependent immunity in mice
To test whether HHP-treated cancer cells are immunogenic in vivo, we have exposed CT26 and LL2 cells to 175 MPa and used such treated cells for immunization of BALB/c and B6 mice, respectively. Mice were immunized with two doses of HHP-treated cancer cells 3 weeks apart and immunized mice were challenged with live tumor cells about 10 d after last immunization (). Both CT26 and LL2 tumor cells treated with HHP elicited protective immunity in experimental mice (and ) since immunized mice showed slower growth of tumors and shift in the survival curves ( and). The resistance to the given challenge of tumor cells was recorded only in the CT26 model as 3 out of 10 mice survived more than 150 d without any signs of tumor growth. Further, we decided to investigate if the antitumor immunity induced by HHP-treated cancer cells is T-cell-dependent and address the importance of CD4+ or CD8+ T cells populations in immunization experiments. Antitumor immunity induced by HHP-treated CT26 cancer cells is clearly T-cell-dependent as depletion of both CD4+ and CD8+ T cells almost completely eliminated the immunity elicited by HHP-treated CT26 cells ( and ). Depletion of either CD4+ or CD8+ T cells dampened the antitumor immunity to certain extend; however, CD8+ T cells seems to be more important than CD4+ T cells although both participate in antitumor immunity induced by HHP-treated cancer cells.
Figure 1. High hydrostatic pressure (HHP)-treated cancer cells elicit T-cell-dependent antitumor immunity. BALB/c mice were s.c. injected into lower left flank with 5 × 106 HHP-treated CT26 cells in 200 µL of PBS on days 0 and 21. Control mice were injected with the same volume of PBS. Mice were than s.c. injected into lower right flank with 105 live CT26 cells in 100 µL of PBS on day 31 (A). Growth of the tumors (B) and survival of mice (C) were recorded. The same experiment was performed in LL2 cell line growing in B6 mice (D, E). BALB/c mice s.c. injected with HHP-treated CT26 cells were also i.p. injected with either 250 µg of anti-CD4+ mAb, anti-CD8+ mAb or both in 250 µL of PBS on day 28 (F, G). Significant differences are shown (*p ≤ 0.05, ***p < 0.001).
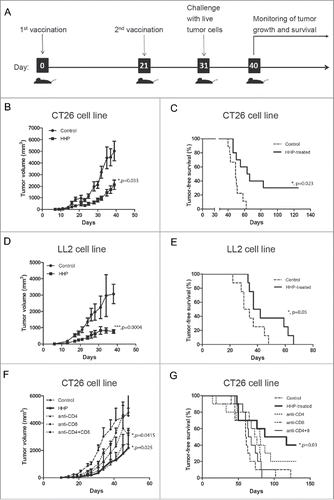
ROS generation in response to HHP treatment elicits pro-danger and pro-death signals
ROS production and ER stress have been characterized as major proximal pre-requisites for ecto-CALR induction in anthracyclines and Hyp-PDT-induced immunogenic apoptosis. HHP triggered vast ROS production in human ovarian cancer cell line (OV-90) shortly after the treatment ( and ). Moreover, N-acetyl-L-cysteine (NAC) and reduced L-glutathione (GSH), two well-established ROS scavengers, partly reduced the HHP-induced ROS production ( and ).
Figure 2. HHP induces the ROS production playing role in CALR surface exposure. Analysis of ROS production using CellRox orange reagent staining in HHP treated OV-90 cells in the presence or absence of N-acetyl-L-cysteine (NAC; 5 mM) and reduced L-glutathione (GSH; 5 mM) 1 h after treatment. Cells were analyzed by immunofluorescence (A) and flow cytometry (B). Scale bar: 20 μm. Data are presented as the mean ± SD for three independent experiments. Significant differences are shown (*p < 0.05). (C) Kinetics of the pro-caspase-2, −3, −8 cleavage and eIF2α phosphorylation in OV-90 cells pretreated with ROS scavenger NAC after 0 and 2 h after HHP application was analyzed by western blotting. The activity of eIF2α was determined by a phospho-specific antibody, and then the membranes were reprobed with antibodies against total eIF2α. Equal protein loading was demonstrated by GAPDH reprobing. Experiments were performed in triplicate. (D) CALR surface exposure in HHP treated OV-90 cells pretreated with ROS scavengers (NAC, GSH) using flow cytometry at 1 and 2 h after treatment. Data are presented as the mean ± SD for three independent experiments. Significant differences are shown (*p ≤ 0.05, **p < 0.01).
HHP-treated cancer cells seemed to undergo rapid cell death (Fig. S1A) which was largely dependent on oxidative stress since suppression of ROS production by NAC/GSH antioxidants significantly reduced cell death in this set-up (Fig. S1A). We then decided to analyze the impact of HHP-mediated ROS on ICD associated danger-signaling events, including the eIF2α phosphorylation and caspase signaling arms, which are known to regulate the surface exposure of the major immunogenic eat-me signal ecto-CALR.Citation15
Immunoblotting of OV-90 cell lysates collected immediately after HHP treatment (0 h) showed an early induction of P-eIF2α and of caspase-2 cleavage, followed by caspase-8 and −3 cleavage 2 h post-HHP. Pretreatment of OV-90 cells with the ROS scavengers, and especially with NAC, blunted the fast induction of phosphorylated eIF2α and caspase-2 processing, and later caspase-signaling events as well (). Moreover, reduction of ROS levels in HHP-treated cells, caused a partial but significant reduction of ecto-CALR () (especially 2 h post-treatment). Of note, ecto-CALR was analyzed only on DAPI− cells (i.e., residual cells that managed to survive the HHP-insult) to exclude intracellular CALR in permeabilized cells (DAPI+) by flow cytometry (Fig. S1B). These findings suggest that HHP-induced ROS production contributes to the early induction of pro-danger signals and the rapid evolution of cell death.
Integrated stress response participates in HHP-induced CALR surface exposure
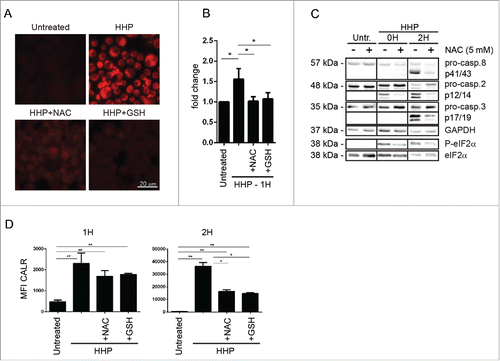
To further characterize the relevance of the early pro-danger signal activated by HHP treatment, we decided to validate and compare the results obtained in OV-90 cells in the colon carcinoma CT26 cell line. We also compared the ICD signature elicited by HHP to that of the more characterized idarubicin. We initially focused on the role of the apical PERK-P-eIF2α axis, since P-eIF2α pathway was one of the earliest signaling event observed in HHP treated cells and PERK-P-eIF2α is considered an apical danger module during ICD.Citation15
Similarly to what found in OV-90 cells, in CT26 cells HHP induced molecular signatures of ER stress and/or integrated stress response (ISR) such as rapid PERK and eIF2α phosphorylation however in absence of CHOP induction ().
Figure 3. HHP induces ER stress (PERK and eIF2α phosphorylation) involved in CALR surface exposure. (A) Kinetics of the PERK and eIF2α phosphorylation and CHOP activation in CT26 cells 1, 2, and 6 h after HHP treatment. Thapsigargin was used as a positive control. Equal protein loading was demonstrated by GAPDH reprobing. (B) Western blot analysis of eIF2α phosphorylation kinetics in OV-90 control and OV-90 shPERK cells 1, 2, and 6 h after UV-B, HHP, and idarubicin treatment. The activity of eIF2α was determined by a phospho-specific antibody, and then the membranes were reprobed with antibody against total eIF2α. Equal protein loading was demonstrated by using GAPDH as a loading control. Experiments were performed in triplicate. (C) Western blot analysis of pro-caspase-2 cleavage in non-transfected OV-90 cells, SCR, and shPERK cells. PERK knockdown was verified by western blotting. Equal protein loading was demonstrated by using GAPDH as loading control. (D) Western blot analysis of eIF2α phosphorylation and pro-caspase-2 cleavage in non-transfected OV-90 cells, SCR and shcaspase-2 cells 2 h after HHP treatment. Caspase-2 knockdown was verified by western blotting. Equal protein loading was demonstrated by using GAPDH as loading control. (E) OV-90 cells were transfected with siRNA against caspase-2 and PERK. Knockout was verified by western blotting. The activity of eIF2α was determined by a phospho-specific antibody and the membranes were reprobed with antibody against total eIF2α. Equal protein loading was demonstrated by using GAPDH as a loading control. (F) CALR surface exposure was measured in untreated, UV-B and HHP treated OV-90 shPERK and OV-90 control cells after 1, 2, and 6 h by flow cytometry. The data show the compiled results (mean ± SD) of three independent experiments. Significant differences are shown (*p < 0.05, **p < 0.01). (G) Detection of surface CALR exposure in untreated, UV-B irradiated and HHP treated CT26 control and CT26 siRNA PERK cells at 1, 2, and 6 h post-treatment using flow cytometry. The data show the compiled results (mean ± SD) of three independent experiments. Significant differences are shown (*p < 0.05, **p < 0.01). (H) CALR surface exposure in untreated, UV-B irradiated and HHP treated MEF WT and eIF2α S51A cells at 1, 2, and 6 h after treatment was measured by flow cytometry. Data are presented as the mean ± SD for three independent experiments. Significant differences are shown (*p < 0.05, **p < 0.01).
To elucidate the role of PERK phosphorylation evoked by HHP treatment, we performed a set of additional experiments with OV-90 cells with shRNA-based stable knockdown of PERK (OV-90 shPERK—we used clone 1399 in which PERK was near-to-completely depleted (Fig. S1C). These cells had slightly reduced capacity to phosphorylate eIF2α upon UV-B, HHP and idarubicin treatment (1–6 h post-treatment) (). Moreover, knockdown of PERK reduced the cleavage of pro-caspase-2 (). On the contrary, we did not observe any effect of caspase-2 knockdown on eIF2α phosphorylation (). These results were also confirmed in OV-90 in which PERK or caspase-2 was depleted by using siRNAs () and suggest that PERK activity could be upstream of pro-caspase-2 cleavage following HHP treatment.
Unlike ROS production, these molecular events (i.e., ER stress/ISR or caspase activation) did not contribute toward HHP-induced cell death per se since strategies that ablated PERK or caspases (−2, −3, −8) expression (e.g., via siRNA-mediated knockdown of gene expression or by gene deletion) or the ability of eIF2α to get phosphorylated, did not impede HHP-induced cell death (Figs. S1D–F, S2D–F, and S3A), thereby, establishing these events as bystanders to cell death in the current setting.
It has been previously documented by Panaretakis et al., that following treatment with anthracyclines, ER stress associated events acting as bystanders to cell death (i.e., activation of the PERK-eIF2α arm) mediated ecto-CALR induction.Citation15 Therefore, we decided to ascertain the importance of the above events in regulating HHP-induced ecto-CALR. For this, we used three different types of “modified” cancer cells, OV-90 shPERK cells, CT26 in which PERK was depleted by transient transfection with specific siRNA against mouse PERK (CT26 siRNA PERK) and mice embryonic fibroblasts (MEF) cells in which the wild-type (WT) eIF2α was replaced heterozygously with a non-phosphorylatable S51A mutant (MEF eIF2α S51A).
Importantly, PERK knockdown reduced the early ecto-CALR induced by HHP (especially at 1–2 h post-treatment) (). UV-B irradiation, a non-ICD inducer, did not induce CALR and there was no significant difference between UV-B treated OV-90 control and OV-90 shPERK cells (). These data were substantiated in CT26 siRNA PERK cells ().
We also found out that not only ROS and PERK, but also eIF2α plays a role in ecto-CALR exposure, as MEF cells in which WT eIF2α has been replaced by the non-phosphorylable S51A mutant (Fig. S1G) exhibited lower capacity to expose early ecto-CALR in response to HHP ().
These data together suggest that HHP elicits a high degree of oxidative stress leading to a stress response involving ROS production, PERK and eIF2α phosphorylation which tend to be involved in HHP-induced ecto-CALR.
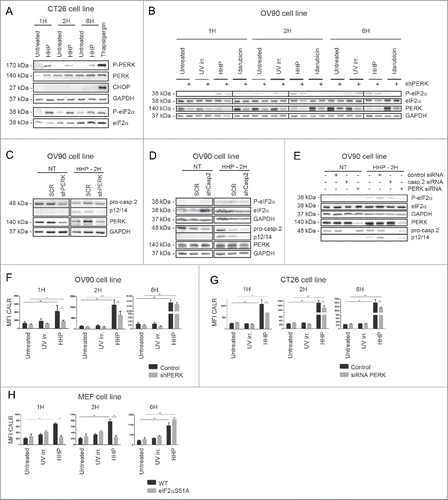
Caspase-8 contributes to CALR surface exposure induced by HHP
The pathways involved in ER stress-associated molecular mechanisms behind pre-apoptotic ecto-CALR induction are governed by a caspase-module involving an ER-proximal caspase-8 and Bax/Bak activation.Citation15 In accordance with published results, we next decided to assess the importance of caspase-8 in CALR induced by HHP; for which we conducted a set of experiments on OV-90 cells with caspase-8 knockdown (OV-90 shcaspase-8—we used clone 75 in which caspase-8 was appreciably depleted by using shRNA; Fig. S2A) and on CT26 cells transiently transfected with specific siRNA against mouse caspase-8 (CT26 siRNA caspase-8; Fig. S2B). We observed pro-caspase-8 cleavage 2 h after HHP treatment (). OV-90 shcaspase-8 expressing cells, upon HHP treatment, exhibited reduced capacity to cleave pro-caspase-3 (at 1, 2, and 6 h, post-treatment) but we observed only slight reduction in cleavage of pro-caspase-2 1 h after HHP treatment ().
Figure 4. Caspase-8 knockdown reduces CALR exposure induced by HHP. (A) The kinetics of pro-caspase-8, pro-caspase-3 and pro-caspase-2 cleavage in OV-90 control and OV-90 shcaspase-8 cells 1, 2, and 6 h after UV-B, HHP, or idarubicin treatment was analyzed by western blotting. Caspase-8 knockdown was verified by western blotting and equal protein loading was demonstrated by using GAPDH as a loading control. Experiments were performed in triplicate. (B) CALR surface exposure was measured in OV-90 control and OV-90 shcaspase-8 cells 1, 2, and 6 h after UV-B or HHP treatment by flow cytometry. Data are presented as the mean ± SD for three independent experiments. Significant differences are shown (*p < 0.05, **p < 0.01). (c) CALR surface exposure in UV-B or HHP treated CT26 control and CT26 siRNA caspase-8 cells at 1, 2, and 6 h post-treatment was analyzed by flow cytometry. The data show the compiled results (mean ± SD) of three independent experiments. Significant differences are shown (*p < 0.05, **p < 0.01). (d) CALR surface detection in UV-B or HHP treated MEF Bax−/−/Bak−/− cells at 1, 2, and 6 h post-treatment using flow cytometry. Relevant control cells (MEF WT) were included in the experiments. Data are presented as the mean ± SD for three independent experiments. Significant differences are shown (*p < 0.05, **p < 0.01).
Of note, caspase-8 knockdown led to a small but significant reduction in ecto-CALR after HHP treatment () and similar results were obtained for CT26 siRNA caspase-8 (). On the other hand we did not observe any reduction in ecto-CALR in either of these cells upon idarubicin treatment (Fig. S2E and F).
Next, we also decided to ascertain the role of Bax and Bak in ecto-CALR exposure induced by HHP; since downstream of caspase-8, Bax/Bak have been found to be crucial for anthracycline-induced ecto-CALR.Citation15,16 For addressing this, we used MEF cells with Bax and Bak double knockout phenotype (MEF Bax−/−/Bak−/−; Fig. S3B). We found that Bax/Bak deficiency did not affect CALR exposure induced by HHP (). Similarly, knockdown of caspase-3, an executioner caspase, did not block the translocation of CALR to the cell surface of HHP-treated tumor cells (Fig. S2C).
In contrast, cells lacking Bax/Bak showed compromised ability to expose CALR on the surface in response to idarubicin 24 h post-treatment (Fig. S3B). In the case of UV-B irradiation, we did not observe differences in CALR expression between relevant controls and MEF Bax−/−/Bak−/− cells ().
In conclusion, HHP causes pro-caspase-8 cleavage which to a small extent participates in ecto-CALR induction following HHP treatment. On the other hand, surprisingly, Bax and Bak were found to be dispensable for HHP-induced CALR trafficking.
Caspase-2 is involved in CALR surface exposure in HHP treated cancer cells
Recent studies have shown that cellular stress responses can induce caspase-2 activation.Citation22–24 For this reason, we also investigated a potential role of this caspase in HHP-induced ecto-CALR.
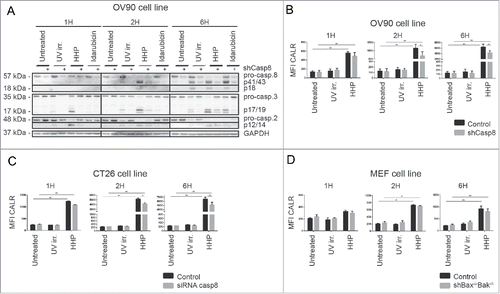
To confirm a role of caspase-2, a stable OV-90 clone expressing a caspase-2 small hairpin (shRNA) in which caspase-2 was significantly depleted (clone 06 in Fig. S3A) was prepared. We observed cleavage of pro-caspase-2 in OV-90 cells 1 h after HHP treatment (). Our data suggest that caspase-2 processing could be an upstream event that precedes caspase-8 processing.
Figure 5. Caspase-2 is involved in the CALR cell surface exposure induced by HHP. (A) The kinetics of pro-caspase-8, pro-caspase-3, and pro-caspase-2 cleavage upon UV-B, HHP, or idarubicin treatment in OV-90 control and OV-90 shcaspase-2 cells at the indicated time points (1, 2, and 6 h). Caspase-2 knockdown was verified by western blotting and equal protein loading was demonstrated by using GAPDH as a loading control. Experiments were performed in triplicate. (B) CALR surface exposure was measured in OV-90 control and OV-90 shcaspase-2 cells 1, 2, and 6 h after UV-B or HHP treatment by flow cytometry. Data are presented as the mean ± SD for three independent experiments. Significant differences are shown (*p < 0.05, **p < 0.01).
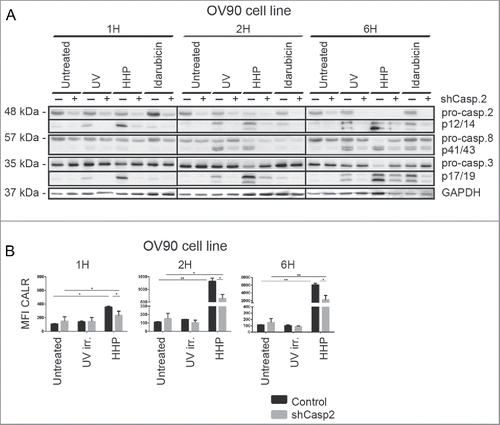
Caspase-2 was also activated in OV-90 cells upon idarubicin and UV-B treatment. Furthermore, OV-90 shcaspase-2 expressing cells had reduced capacity to cleave caspase-3 (6 h) upon idarubicin treatment and these cells exhibited decreased cleavage of pro-caspase-8 (at 2 and 6 h) after UV-B irradiation ().
Next, we analyzed the role of caspase-2 in ecto-CALR exposure induced by HHP. The involvement of caspase-2 in this process was demonstrated by relatively lower capacity of OV-90 shcaspase-2 cells to expose CALR in response to HHP treatment (). On the contrary, caspase-2 knockdown had no effect on CALR induced by idarubicin (Fig. S3A). The kinetics of cell death of OV-90 shcaspase-2 cells was similar to control cells upon HHP, idarubicin and also UV-B treatment (Fig. S3A).
Taken together, our results indicate that caspase-2 plays a role in HHP-mediated ecto-CALR exposure.
CALR on HHP treated cells is important for phagocytosis by dendritic cells
Considering the established role of ecto-CALR as an “eat me” signal,Citation25 we further investigated the possible effect of ecto-CALR on phagocytosis of HHP treated tumor cells by dendritic cells, an important antigen-presenting cells of the innate immune system.Citation26 For this purpose, we used specific antibody to block CALRCitation16 and we also investigated the interaction of dendritic cells with OV-90 shPERK, shcaspase-8 and shcaspase-2 expressing cells which had lower capacity to expose CALR (depending upon the signaling molecule, i.e., caspases-2 shRNA > PERK shRNA > caspases-8 shRNA) after HHP treatment ( and ). In general, HHP treated cancer cells were phagocytosed faster and to a greater extent than tumor cells killed by UV-B or by idarubicin. After 3 h, the rate of phagocytosis of HHP treated cancer cells was 2.5-fold higher compared with UV-B irradiated cells and 1.8-fold higher in comparison to cells killed by idarubicin (Figs. 6A and B and S4A). The analysis of the interaction of dendritic cells with HHP treated OV-90 shPERK, shcaspase-8 and shcaspase-2 cells revealed strong linear correlation between phagocytosis of HHP treated tumor cells and their CALR levels 6 h after HHP treatment (). Among the modified OV-90 cells chosen for this experiment, HHP treated OV-90 shcaspase-2 expressing cells were phagocytosed to a significantly lower extent compared with OV-90 control cells ( and ). These results correlated with the finding that, OV-90 shcaspase-2 expressing cells had the most prominent decrease in CALR expression compared with OV-90 control cells (2.7-fold lower MFI values of CALR) relative to shPERK or shcaspase-8 cells (). Thus, although the depletion of caspase-2 affects ecto-CALR exposure in both a statistically significant and biologically relevant manner (as it limits uptake by DCs), the reduction of CALR caused by PERK or caspase-8 knockdown is not biologically relevant.
Figure 6. CALR surface expression is important for phagocytosis of HHP treated tumor cells by population of dendritic cells. (A) Flow cytometry analysis of phagocytosis. DiO-labeled immature dendritic cells were co-cultured with DiD-labeled OV-90 control, shPERK, shcaspase-2, or shcaspase-8 cells treated with UV-B and idarubicin for 24 h and with HHP for 6 h at 37 °C for 3 h. A fraction of DiO-labeled dendritic cells was loaded with DiD-labeled OV-90 control cells which were pre-incubated with blocking anti-CALR antibody or relevant isotype control antibody. As a control, dendritic cells and tumor cells were co-cultured on ice for 3 h (Fig. S4B). Dendritic cells that phagocytosed killed tumor cells were identified as DiO+DiD+ double-positive cells. Data are presented as the mean ± SD for three independent experiments. Significant differences are shown (*p < 0.05, **p < 0.01). (B) Representative dot plots of flow cytometry and fluorescence microscopy analysis of the phagocytosis. DiO-labeled immature dendritic cells were co-cultured for 3 h with DiI-labeled HHP and idarubicin treated tumor cells and the engulfment of tumor cells was verified by fluorescence microscopy. (C) Correlation between phagocytosis of HHP treated tumor cells and their CALR levels. The percentage of dendritic cells taking up HHP killed tumor cells was determined and correlated with CALR levels on tumor cells 6 h after HHP treatment.
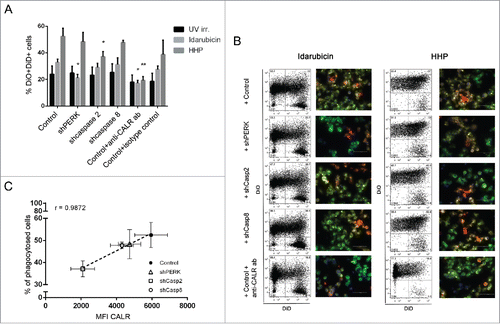
When the engulfment of idarubicin treated cells was investigated, we detected significantly lower rate of phagocytosis in the case of OV-90 shPERK expressing cells ( and ). This result is in the line with the fact that these cells treated by idarubicin for 24 h had the greatest reduction of ecto-CALR compared with OV-90 control cells (2.7-fold lower MFI values of CALR) (data not shown). Moreover, blockade of the CALR on HHP and also idarubicin treated tumor cells by a specific antibody inhibited their phagocytosis by dendritic cells ( and ).
Therefore, CALR is required not only for phagocytosis of idarubicin killed tumor cells but also for phagocytosis of HHP treated cells by dendritic cells and for HHP, this process is strongly compromised in absence of caspase-2.
Discussion
HHP was identified as an inducer of anticancer immunity in a wide range of human tumor cell lines and primary tumor cells in our previous study. This physical modality was, due to its immunogenic properties, standardized and validated for incorporation into manufacturing protocols for cancer immunotherapy products.Citation20 We have initiated multiple clinical trials for prostate, ovarian and lung cancer evaluating the potential of DCs loaded with HHP treated cancer cells to induce tumor cell-specific immune responses.Citation20,27 Nevertheless, HHP-treated cancer cells were utilized so far solely for pulsing of ex vivo cultured DCs but their direct use to induce antitumor immunity has not been validated. Here, we show in two different mouse tumor models that HHP-treated tumor cells evoke protective immune memory response when s.c. injected per se, i.e., without any adjuvant. Since we proved that immunization with HHP-treated tumor cells induce CD4+ and CD8+ T cell-dependent immune memory, it is most likely that HHP-treated tumor cells are immunogenic in vivo because they are effectively taken up and processed by DCs. Furthermore, we have shown that HHP-treated cancer cells also induce maturation of DCs due to the release of “danger” signaling molecules like HMGB1 or ATP as shown in in vitro studies.Citation20
Considering the importance of ecto-CALR for tumor cells phagocytosis,Citation25 induction of tumor specific immune response in vaccinated patientsCitation28 and patient prognosis,Citation13 we aimed to unravel a sequence of key events that are required (but not necessarily sufficient) to transform cellular stress into immunogenic signaling (similar to previously described anthracyclines and Hyp-PDTCitation7). Therefore it was not the main aim of the study to characterize in details the cell death mechanisms triggered by HHP but mostly to identify the key components responsible for ecto-CALR exposure. The ecto-CALR inducing capacity of ICD inducers has been shown to depend on the properties of ER stress and ROS production.Citation16,29,30 We observed that HHP treatment results in ROS production, a proximal event triggering other downstream signaling pathways, such as ISR and cleavage of caspase-2, caspase-8, and caspase-3. The application of various ROS scavengers suggested that for HHP-induced CALR, ROS production is one of the pre-requisites, an observation that is in line with previous studies concerning Hyp-PDT, mitoxantrone, oxaliplatin, UV-C or oncolytic coxsackievirus B3.Citation7,15,31 However, the suppression of ROS production was not sufficient to completely abolish HHP-induced CALR exposure suggesting that ROS-independent mechanisms may also participate in this process.
The danger signaling pathways causing the translocation of CALR to the cell surface in response to various chemotherapeutic treatments or Hyp-PDT overlap, but are not identical.Citation7,15 We showed that HHP-induced pathway leading to ecto-CALR exposure also overlaps with those induced by chemotherapy or Hyp-PDT but is not entirely identical to either. More specifically, HHP-mediated ROS production, possibly impinging on ER homeostasis, further triggers early PERK activation, eIF2α phosphorylation, and caspase-2 cleavage (but not Bax/Bak), which are important for HHP-induced danger signaling involving CALR induction.
Recent studies have shown that ROS production as well as ER stress mayCitation22–24,32 or may notCitation33 induce caspase-2 cleavage, depending on the context. In our context, we found that caspase-2 cleavage following HHP is downstream of ROS-based ISR. Our study also shows that compromising PERK expression mitigates processing of caspase-2—an interesting observation requiring further mechanistic studies. Moreover, in the current study, we report for the first time, a role of caspase-2 in CALR trafficking during ICD. While it is not yet clear how caspase-2 might be regulating CALR exposure, the localization of this caspase in the ER/Golgi system suggests possibility of participation in regulating trafficking mechanisms—a hypothesis that needs verification in near future.Citation34
It was shown that caspase-8 activation is not required for the CALR induction by Hyp-PDT, although it is necessary for mitoxantrone/oxaliplatin-induced CALR.Citation7,15 In our study HHP-induced CALR was found to be at least partially mediated by caspase-8. Unfortunately, we did not observe any reduction of CALR in either of these cells upon idarubicin treatment, which might be due to differences in mode of action between idarubicin and mitoxantrone which was used in previous studies,Citation16 showing the context-dependent importance of caspase-8 for proper CALR exposure even for different anthracyclines.Citation7,15 Moreover, Bax/Bak were previously classified as “core molecular entities” participating in ICD-associated danger signaling because of their role in both Hyp-PDT and chemotherapy-induced CALR. However, in contrast to these results, we observed that Bax/Bak deficiency does not affect CALR trafficking induced by HHP. Thus, our results taken together with previous publicationsCitation7,15 clearly show that across HHP, Hyp-PDT and immunogenic chemotherapy, PERK is apparently an important molecular entity involved in danger signaling pathways in an ICD-inducer independent fashion while all the other molecules tend to show an ICD-inducer dependent involvement in CALR induction.
CALR and another “eat me” signals like translocation of phosphatidylserine from inside the cell to the surface, together with the downregulation of “don't eat me” signals such as CD47, elicit the recognition and removal of dying cells by phagocytes, most importantly by professional phagocytes such as dendritic cells.Citation25,35 We have previously shown that the interaction of immature dendritic cells with HHP killed tumor cells led to the increased uptake of tumor cells by dendritic cells and induced the expression of maturation-associated markers on dendritic cells and the production of IL-12p70 and proinflammatory cytokines, demonstrating that HHP-treated tumor cells provide a potent activation stimulus to dendritic cells.Citation20 Faster phagocytosis was similarly observed in this study and correlated with the rapid induction of CALR after HHP treatment. Moreover, the blockade of CALR by specific antibody or depletion of caspase-2, an important player in CALR surface exposure, significantly inhibited their engulfment by dendritic cells.
In conclusion, we characterized the molecular mechanism of ecto-CALR exposure pathway induced by HHP which is more analogous to that induced by anthracycline treatment rather than that induced by Hyp-PDT. Moreover, we characterized a novel role of PERK-caspase-2 axis in mediating CALR induction. Altogether, our findings reveal that the PERK–eIF2α phosphorylation–caspase-2 axis plays an important role in the HHP-mediated CALR exposure. This kind of information might help to prepare more efficient cancer vaccine in which the relative expression levels of proteins required for full inactivation of tumor cells and stress-elicited immunogenic signaling can be used with respect to their prognostic and predictive impact in cancer patient's treatment.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_materials.zip
Download Zip (6.9 MB)Funding
This project was supported by the research grant IGA NT12402-5, student research grant GAUK 682214, the Ministry of Education, Youth and Sports of CR within the LQ1604 National Sustainability Program II (Project BIOCEV-FAR) and by the project “BIOCEV” (CZ.1.05/1.1.00/02.0109).
References
- Galluzzi L, Vacchelli E, Bravo-San Pedro JM, Buque A, Senovilla L, Baracco EE, Bloy N, Castoldi F, Abastado JP, Agostinis P et al. Classification of current anticancer immunotherapies. Oncotarget 2014; 5(24):12472-508; PMID:25537519; http://dx.doi.org/10.18632/oncotarget.2998
- Fucikova J, Kralikova P, Fialova A, Brtnicky T, Rob L, Bartunkova J, Spísek R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res 2011; 71(14):4821-33; PMID:21602432; http://dx.doi.org/10.1158/0008-5472.CAN-11-0950
- Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010; 29(4):482-91; PMID:19881547; http://dx.doi.org/10.1038/onc.2009.356
- Obeid M, Panaretakis T, Joza N, Tufi R, Tesniere A, van Endert P, Zitvogel L, Kroemer G. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ 2007; 14(10):1848-50; PMID:17657249; http://dx.doi.org/10.1038/sj.cdd.4402201
- Koks C, Garg AD, Ehrhardt M, Riva M, Vandenberk L, Boon L, De Vleeschouwer S, Agostinis P, Graf N, Van Gool SW. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int J Cancer 2014; 136(5):E313-25; PMID:25208916; http://dx.doi.org/10.1002/ijc.29202
- Vacchelli E, Eggermont A, Sautes-Fridman C, Galon J, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: oncolytic viruses for cancer therapy. Oncoimmunology 2013; 2(6):e24612; PMID:23894720; http://dx.doi.org/10.4161/onci.24612
- Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJ et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J 2012; 31(5):1062-79; PMID:22252128; http://dx.doi.org/10.1038/emboj.2011.497
- Garg AD, Vandenberk L, Koks C, Verschuere T, Boon L, Van Gool SW, Agostinis P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med 2016; 8(328):328ra327; PMID:26936504; http://dx.doi.org/10.1126/scitranslmed.aae0105
- Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014; 3(9):e955691; PMID:25941621; http://dx.doi.org/10.4161/21624011.2014.955691
- Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM, Breckpot K, Brough D, Chaurio R, Cirone M et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol 2015; 6: 588; PMID:26635802; http://dx.doi.org/10.3389/fimmu.2015.00588
- Cao C, Han Y, Ren Y, Wang Y. Mitoxantrone-mediated apoptotic B16-F1 cells induce specific anti-tumor immune response. Cell Mol Immunol 2009; 6(6):469-75; PMID:20003823; http://dx.doi.org/10.1038/cmi.2009.59
- Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood 2007; 109(11):4839-45; PMID:17299090; http://dx.doi.org/10.1182/blood-2006-10-054221
- Garg AD, Elsen S, Krysko DV, Vandenabeele P, de Witte P, Agostinis P. Resistance to anticancer vaccination effect is controlled by a cancer cell-autonomous phenotype that disrupts immunogenic phagocytic removal. Oncotarget 2015; 6(29):26841-60; PMID:26314964; http://dx.doi.org/10.18632/oncotarget.4754
- Martins I, Kepp O, Galluzzi L, Senovilla L, Schlemmer F, Adjemian S, Menger L, Michaud M, Zitvogel L, Kroemer G. Surface-exposed calreticulin in the interaction between dying cells and phagocytes. Ann NY Acad Sci 2010; 1209: 77-82; PMID:20958319; http://dx.doi.org/10.1111/j.1749-6632.2010.05740.x
- Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, Durchschlag M, Joza N, Pierron G, van Endert P et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J 2009; 28(5):578-90; PMID:19165151; http://dx.doi.org/10.1038/emboj.2009.1
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007; 13(1):54-61; PMID:17187072; http://dx.doi.org/10.1038/nm1523
- van Vliet AR, Martin S, Garg AD, Agostinis P. The PERKs of damage-associated molecular patterns mediating cancer immunogenicity: from sensor to the plasma membrane and beyond. Semin Cancer Biol 2015; 33: 74-85; PMID:25882379; http://dx.doi.org/10.1016/j.semcancer.2015.03.010
- Garg AD, Dudek AM, Ferreira GB, Verfaillie T, Vandenabeele P, Krysko DV, Mathieu C, Agostinis P. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy 2013; 9(9):1292-307; PMID:23800749; http://dx.doi.org/10.4161/auto.25399
- Galluzzi L, Kepp O, Kroemer G. Enlightening the impact of immunogenic cell death in photodynamic cancer therapy. EMBO J 2012; 31(5):1055-7; PMID:22252132; http://dx.doi.org/10.1038/emboj.2012.2
- Fucikova J, Moserova I, Truxova I, Hermanova I, Vancurova I, Partlova S, Fialova A, Sojka L, Cartron PF, Houska M et al. High hydrostatic pressure induces immunogenic cell death in human tumor cells. Int J Cancer 2014; 135(5):1165-77; PMID:24500981; http://dx.doi.org/10.1002/ijc.28766
- Adkins I, Fucikova J, Garg AD, Agostinis P, Spisek R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology 2014; 3(12):e968434; PMID:25964865; http://dx.doi.org/10.4161/21624011.2014.968434
- Mishra R, Karande AA. Endoplasmic reticulum stress-mediated activation of p38 MAPK, caspase-2 and caspase-8 leads to abrin-induced apoptosis. PLoS One 2014; 9(3):e92586; PMID:24664279; http://dx.doi.org/10.1371/journal.pone.0092586
- Uchibayashi R, Tsuruma K, Inokuchi Y, Shimazawa M, Hara H. Involvement of Bid and caspase-2 in endoplasmic reticulum stress- and oxidative stress-induced retinal ganglion cell death. J Neurosci Res 2011; 89(11):1783-94; PMID:21805492; http://dx.doi.org/10.1002/jnr.22691
- Upton JP, Austgen K, Nishino M, Coakley KM, Hagen A, Han D, Papa FR, Oakes SA. Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol Cellular Biol 2008; 28(12):3943-51; PMID:18426910; http://dx.doi.org/10.1128/MCB.00013-08
- Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005; 123(2):321-34; PMID:16239148; http://dx.doi.org/10.1016/j.cell.2005.08.032
- Blachere NE, Darnell RB, Albert ML. Apoptotic cells deliver processed antigen to dendritic cells for cross-presentation. PLoS Biol 2005; 3(6):e185; PMID:15839733; http://dx.doi.org/10.1371/journal.pbio.0030185
- Podrazil M, Horvath R, Becht E, Rozkova D, Bilkova P, Sochorova K, Hromadkova H, Kayserova J, Vavrova K, Lastovicka J et al. Phase I/II clinical trial of dendritic-cell based immunotherapy (DCVAC/PCa) combined with chemotherapy in patients with metastatic, castration-resistant prostate cancer. Oncotarget 2015; 6(20):18192-205; PMID:26078335; http://dx.doi.org/10.18632/oncotarget.4145
- Zappasodi R, de Braud F, Di Nicola M. Lymphoma immunotherapy: current status. Front Immunol 2015; 6: 448; PMID:26388871; http://dx.doi.org/10.3389/fimmu.2015.00448
- Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S, Yamazaki T, Sukkurwala AQ, Michaud M, Mignot G et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med 2012; 4(143):143ra199; PMID:22814852; http://dx.doi.org/10.1126/scitranslmed.3003807
- Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, Michaud M, Menger L, Gdoura A, Tajeddine N et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene 2011; 30(10):1147-58; PMID:21151176; http://dx.doi.org/10.1038/onc.2010.500
- Miyamoto S, Inoue H, Nakamura T, Yamada M, Sakamoto C, Urata Y, Okazaki T, Marumoto T, Takahashi A, Takayama K et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res 2012; 72(10):2609-21; PMID:22461509; http://dx.doi.org/10.1158/0008-5472.CAN-11-3185
- Prasad V, Chandele A, Jagtap JC, Sudheer Kumar P, Shastry P. ROS-triggered caspase 2 activation and feedback amplification loop in beta-carotene-induced apoptosis. Free Radic Biol Med 2006; 41(3):431-42; PMID:16843824; http://dx.doi.org/10.1016/j.freeradbiomed.2006.03.009
- Sandow JJ, Dorstyn L, O'Reilly LA, Tailler M, Kumar S, Strasser A, Ekert PG. ER stress does not cause upregulation and activation of caspase-2 to initiate apoptosis. Cell Death Differ 2014; 21(3):475-80; PMID:24292555; http://dx.doi.org/10.1038/cdd.2013.168
- Krumschnabel G, Sohm B, Bock F, Manzl C, Villunger A. The enigma of caspase-2: the laymen's view. Cell Death Differ 2009; 16(2):195-207; PMID:19023332; http://dx.doi.org/10.1038/cdd.2008.170
- Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Clearance of apoptotic cells: getting rid of the corpses. Mol Cell 2004; 14(3):277-87; PMID:15125832; http://dx.doi.org/10.1016/S1097-2765(04)00237-0