
ABSTRACT
We developed cergutuzumab amunaleukin (CEA-IL2v, RG7813), a novel monomeric CEA-targeted immunocytokine, that comprises a single IL-2 variant (IL2v) moiety with abolished CD25 binding, fused to the C-terminus of a high affinity, bivalent carcinoembryonic antigen (CEA)-specific antibody devoid of Fc-mediated effector functions. Its molecular design aims to (i) avoid preferential activation of regulatory T-cells vs. immune effector cells by removing CD25 binding; (ii) increase the therapeutic index of IL-2 therapy by (a) preferential retention at the tumor by having a lower dissociation rate from CEA-expressing cancer cells vs. IL-2R-expressing cells, (b) avoiding any FcγR-binding and Fc effector functions and (c) reduced binding to endothelial cells expressing CD25; and (iii) improve the pharmacokinetics, and thus convenience of administration, of IL-2. The crystal structure of the IL2v-IL-2Rβγ complex was determined and CEA-IL2v activity was assessed using human immune effector cells. Tumor targeting was investigated in tumor-bearing mice using 89Zr-labeled CEA-IL2v. Efficacy studies were performed in (a) syngeneic mouse models as monotherapy and combined with anti-PD-L1, and in (b) xenograft mouse models in combination with ADCC-mediating antibodies. CEA-IL2v binds to CEA with pM avidity but not to CD25, and consequently did not preferentially activate Tregs. In vivo, CEA-IL2v demonstrated superior pharmacokinetics and tumor targeting compared with a wild-type IL-2-based CEA immunocytokine (CEA-IL2wt). CEA-IL2v strongly expanded NK and CD8+ T cells, skewing the CD8+:CD4+ ratio toward CD8+ T cells both in the periphery and in the tumor, and mediated single agent efficacy in syngeneic MC38-CEA and PancO2-CEA models. Combination with trastuzumab, cetuximab and imgatuzumab, all of human IgG1 isotype, resulted in superior efficacy compared with the monotherapies alone. Combined with anti-PD-L1, CEA-IL2v mediated superior efficacy over the respective monotherapies, and over the combination with an untargeted control immunocytokine. These preclinical data support the ongoing clinical investigation of the cergutuzumab amunaleukin immunocytokine with abolished CD25 binding for the treatment of CEA-positive solid tumors in combination with PD-L1 checkpoint blockade and ADCC competent antibodies.
Introduction
IL-2 plays a central role in the generation, differentiation, survival and homeostasis of immune effector cells. IL-2 is synthesized by activated CD4+ helper T cells, and has multiple immunological effects.Citation1-5 IL-2 acts by binding to IL-2 receptors (IL-2R). Association of the α (CD25), β (CD122) and γ (γc, CD132) subunits results in the trimeric high-affinity IL-2R. The dimeric intermediate affinity IL-2Rβγ consists of the β- and γ-subunits and binds IL-2 with 50-fold lower affinity. CD25 is not required for IL-2 signaling, but confers high affinity binding, whereas the β and the γ subunits mediate signal transduction.Citation3,4,6 IL-2Rβγ is expressed on NK cells, monocytes, macrophages and resting CD4+ and CD8+ T cells, while IL-2Rαβγ is transiently induced on activated T and NK cells, and is constitutively expressed on CD4+FoxP3+ Tregs.Citation7 (Fig. S1). The ability of IL-2 to expand and activate innate and adaptive effector cells is the basis of its antitumor activity, however, as a regulatory mechanism that can prevent excessive immune responses and autoimmunity, IL-2 leads to activation-induced cell death (AICD) rendering activated T cells susceptible to Fas-mediated apoptosis. Moreover, IL-2 is essential for the maintenance and expansion of immunosuppressive CD4+ CD25+ Treg cells.Citation7-9
Based on its antitumor efficacy, high-dose IL-2 (aldesleukin) has been approved as first immunotherapy for patients with metastatic renal cell carcinoma and malignant melanoma. However, its antitumor immunity can be compromised by the induction of Tregs and AICD. In addition, systemic IL-2 treatment has been associated with severe cardiovascular, pulmonary, hepatic, gastrointestinal, neurologic and hematological side effects, such that it is only given to patients at specialized centers.Citation10 Preclinical experiments showed that IL-2-induced pulmonary edema is caused by interaction of IL-2 with CD25 on lung endothelial cells and that it can be abrogated by a CD25 blocking antibody, genetic disruption, or the use of IL-2-antibody complexes.Citation11 Thus, we reasoned that an IL-2 variant (IL2v) with abolished CD25 binding may show reduced toxicity and vascular leak syndrome while still being able to activate effector cells with antitumor potential through retained IL-2Rβγ signaling (Fig. S1).
Various tumor-targeted IL-2 immunocytokines have been developed during the last decade(s) based on an immunoglobulin backbone or using non-Fc containing antibody fragments.Citation12-16 First-generation immunocytokines have shown inadequate pharmacokinetic (PK) properties and a toxicity profile similar to aldesleukin. Moreover, fusing one or two wild-type IL-2 moieties to an antibody results in low pM affinity for IL-2Rαβγ and thus preferential binding to immune cells in the circulation rather than to the tumor. So far, no immunocytokine has successfully passed pivotal trials. Wittrup and colleagues recently demonstrated the limitations of a wild-type IL-2 based gp75 (TRP1)-targeted immunocytokine in C56BL/6 mice, which resulted in peripheral activation and lack of tumor targeting,Citation17 but showed the potential of half-life extended monomeric wild-type IL-2 Fc protein combined with ADCC competent antibodies and adoptive T-cell therapy.Citation18 When combining a tumor-antigen-targeting antibody with such a half-life extended interleukin-2, PD-1 checkpoint blockade and a powerful T cell vaccine large tumors could be eliminated in syngeneic and genetically engineered mouse models.Citation19
As therapeutic approaches using IL-2 can only be useful if the limitations associated with its application can be overcome, we designed a IL2v characterized by three mutations that abolish binding to CD25. To enable half-life extension and preferential targeting to the tumor, the IL2v moiety was fused to CH1A1A-2F1, a carcinoembryonic antigen (CEA)-specific antibody, resulting in the immunocytokine cergutuzumab amunaleukin (CEA-IL2v, RG7813). CH1A1A-2F1 was affinity matured and stability enhanced by phage display based on the murine PR1A3 antibody, and recognizes a membrane-proximal epitope in the B3 domain of CEA (CEACAM5, Cd66e), whereas it does not recognize shed CEA as found in the circulation.Citation20-22 This antibody is also basis of CEA TCB (RG7802), a CEA-CD3 T cell bispecific antibody currently in Phase 1/1b clinical trials.Citation23-25 CEA is a glycophosphatidylinositol-anchored glycoprotein that serves as a cancer biomarker and has been used as a target for tumor imaging, radioimmunotherapy, antibody drug conjugates or T cell bispecific antibodies.Citation26-30 Prevalence of CEA expression in major tumor types was confirmed by immunohistochemistry to be high with ca. 95% of colorectal, 90% of pancreatic, 80% of gastric, 60% of non-small cell lung and 40% of breast cancer specimens staining positive for CEA with moderate or high intensity (data not shown), whereas in normal tissues only low-level CEA expression is found on the apical surface of glandular epithelia in the gastrointestinal tract (GI) where it may not be accessible for antibody binding.Citation31,32 As tumor specific CEA-expression is broad, CEA-IL2v was designed to overcome local tumor immunosuppression in combination therapy e.g., with ADCC-mediating or T cell bispecific antibodies, adoptive T cell therapy and PD-1/PD-L1 checkpoint blockade in a variety of tumors.
CEA-IL2v was characterized on immune effector cells by assessing the activation of P-STAT5, cell proliferation, effects on Fas-induced apoptosis, expression of activation markers and cytokine release. Tumor targeting was determined with 89Zr-labeled CEA-IL2v in tumor-bearing mice since this radiolabeled compound can easily be used in clinical Positron Emission Tomography (PET) studies to confirm tumor targeting visually and quantitatively in first in human studies.Citation33 PK, pharmacodynamics and antitumor efficacy were analyzed in fully immunocompetent mice as monotherapy and combined with PD-L1 checkpoint blockade, and combined with ADCC competent antibodies in human CD16-transgenic SCID mice.
Results
IL2v design and IL2v/IL-2Rβγ co-crystal structure
We aimed to design an immunocytokine incapable of binding to CD25 (IL-2Rα) by structure-based mutation of key residues, namely F42A, Y45A, L72G building the CD25 binding interface, which is conserved between species, e.g., human, mouse and non-human primates (NHP).Citation34,35 To avoid heterogeneity, the O-glycosylation site was removed by a T3A mutation. In addition, like in aldesleukin, the cysteine residue was mutated to avoid aggregation by a C125A mutation (numbering based on UniProt ID P60568 excluding the signal peptide).
To confirm that the mutations in IL2v indeed only interfere with CD25 binding, and do not affect the interaction with IL-2Rβγ, we determined the co-crystal structure of the IL2v-IL-2Rβγ ternary complex at a resolution of 2.3 Å (, Table S1). The structure contains one ternary complex molecule in the asymmetric unit and consists of the β-chain residues 6–208, γ-chain residues 33–225 and IL2v residues 4–30, 37–133. Although the protein was treated with PNGase F before crystallization, electron density for 5 N-glycosylated Asn residues on the receptor chains was observed (data not shown). In addition, we identified two covalently attached mannose moieties at W194 and W197 within the class I cytokine receptor WSXWS motif of the IL-2Rβ chain.Citation36 The overall structure of the IL2v-IL-2Rβγ complex is essentially identical to the wild-type quaternary complex (pdb code 2b5i) with only minor changes observed within the βγ interface and within IL-2 (superposition of all atoms with a root mean square deviation (rmsd) of 1.02 Å) (). Binding of IL-2 to CD25 is mediated by two hydrophobic patches around F42, Y45 and L72 on the IL-2 surface.Citation34,35 F42 and Y45 are located in the loop AB, whereas L72 is part of helix B. The mutations F42A, Y45A and L72G remove a large part of the Van’ der Waal interaction surface and thereby prevent binding of IL2v to CD25 without affecting the IL-2 structure () or the interaction with IL-2Rβγ.
Figure 1. (A) IL2v/IL-2Rβγ complex structure. IL2v is shown yellow, the mutations in blue. The IL-2Rβ chain is colored in cyan and the γc chain in magenta. (B) Close-up view onto the loop region between helix A and helix B and the binding site of IL-2 with IL-2Rα. The picture shows an overlay of the IL2v-IL-2Rβγ (yellow) structure with 2B5I (gray representation). Mutations in the two hydrophobic patches with F42A, Y45A and L72G are highlighted in blue and circled. (C) Superposition of all atoms of IL2v-IL-2Rβγ with the quaternary complex IL-2, IL-2Rα, IL-2Rβ, γc (2erj). IL2v-IL-2Rβγ is colored as in . 2B5I is shown in gray. (D) Schematic representation of CEA-IL2v and its key design features.
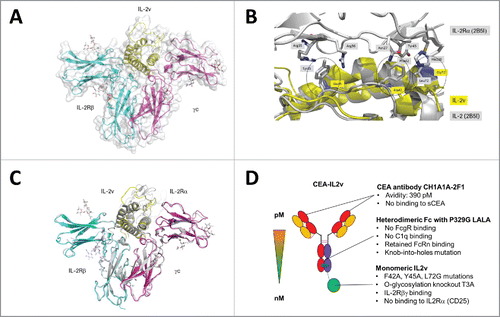
CEA-IL2v design
IL2v was used for the construction of a monomeric CEA-IL2v immunocytokine. depicts the key features.Citation1 CEA-IL2v is based on the humanized CH1A1A-2F1 IgG1 antibody, and like the CH1A1A-2F1 parental antibody binds the CEA B3 domain with monovalent affinity of 40 nM and in a bivalent setting with 390 pM avidity (Table S2) while it does not recognize shed CEA in a sandwich binding assay (data not shown). The parental CEA antibody does neither recognize CEACAM1 or CEACAM6, nor internalize on CEA+ tumor cell lines, and has no functional effects, e.g., on proliferation (data not shown).Citation2 CEA-IL2v was designed to avoid unspecific and systemic FcγR mediated co-activation of innate immune effector cells, e.g., NK cells, macrophages/monocytes through simultaneous binding to Fcγ- and IL2-receptors by introducing P329G, L234A, L235A mutations into the Fc portion. These mutations completely abolish binding to FcγRs and C1q, but do not affect FcRn binding or PK.Citation37 3) IL2v is fused via a flexible and presumably non-immunogenic (G4S)2 connector to the C-terminus of the knob-containing heterodimeric heavy chain engineered using the knob-into-holes technology.Citation38 Consequently, CEA-IL2v binds monovalently to IL-2Rβγ and when soluble does not exhibit avidity effects for IL-2R-expressing cells. Thus, monomeric CEA-IL2v exhibits a higher relative, functional affinity for CEA on tumor cells than for the IL-2Rβγ on immune cells as compared with wild-type IL-2-based dimeric immunocytokines. Due to the removal of the O-glycosylation site, CEA-IL2v carries only the typical Fc carbohydrates. Whereas CEA-IL2v was designed to no longer bind to CD25 on activated T effector and Treg cells, it should retain affinity and functional activity for the dimeric IL-2Rβγ receptor. Table S2 shows the retained affinity of CEA-IL2v for a pre-formed human and mouse IL-2Rβγ-Fc heterodimer, whereas no binding to CD25 is detectable by surface plasmon resonance. Notably, the mutations affect affinity of human IL2v human and murine IL-2R in a similar manner enabling use in rodent models. Tables S3 and S4 give an overview about the immunocytokines used in this manuscript and their design features.
Cellular activity in vitro
CEA-IL2v was subsequently characterized in several cellular and functional assays. CEA-IL2v bound efficiently to CEA-expressing A549 tumor cells with EC50 value in the low nM range comparable to the parental CEA antibody CH1A1A-2F1 indicating that antibody-IL2v fusion does not affect CEA binding (Fig. S2A, Table S5). Cellular binding of the CH1A1A-2F1 antibody was only affected by shed CEA starting from 5 μg/mL range (data not shown), which is significantly above the levels typically observed in plasma of healthy donors (median 1.49 ng/mL) and cancer patients (median 3.86 ng/mL).Citation39 Similarly, CEA-IL2v bound efficiently to IL-2R expressing (and CEA negative) immune cells including NK92 cells, a human IL-2Rαβγ-positive NK cell line, and to NK and T cells within peripheral blood mononuclear cells (PBMC) (Fig. S2B–D, Table S5). The ability for bi-specific binding was assessed in a “sandwich binding” assay in which simultaneous binding to CEA+ target cells and IL-2Rβγ was confirmed using the IL-2Rβγ-Fc heterodimer for detection (Fig. S2E, Table S5). Internalization studies with IL-2Rβγ+ NK92 cells showed that CEA-IL2v was rapidly internalized upon binding to IL-2R (Fig. S2F, Table S5).
The ability of CEA-IL2v to induce activation of IL-2R signaling in human NK, CD4+ and CD8+ T cells was measured by a dose-dependent increase of STAT5 phosphorylation compared with wild-type IL-2-based CEA-IL2wt and aldesleukin (, Table S5). CEA-IL2v, CEA-IL2wt and aldesleukin were equally effective in inducing STAT5 phosphorylation in NK and CD8+ T cells and similarly in CD4+ T cells. However, CEA-IL2v was significantly less potent than CEA-IL2wt and aldesleukin in activating suppressive Tregs that express high levels of CD25. Thus, CEA-IL2v no longer preferentially activated Tregs, but only activated Tregs at concentrations when CD8+ T and NK cells were also activated, a desired property when attempting to restore the immune cell activation and attenuate local tumor immunosuppression. In addition, T cells were more resistant to CEA-IL2v regarding Fas-mediated apoptosis induction than to CEA-IL2wt and aldesleukin due to abolished CD25 binding ().
Figure 2. (A–D) FACS analysis of STAT5 phosphorylation on (A) Treg cells (CD4+CD25+FOXP3+), (B) CD4+ T cells (CD4+CD25−), C) CD8+ T cells (CD3+CD8+) and (D) NK cells (CD3−CD56+) within freshly isolated PBMCs 20 min after treatment with CEA-IL2v, CEA-IL2wt and aldesleukin; (E–H) Fas-mediated apoptosis induction (E, F) and proliferation (G, H) of CD4+ and CD8+ T cells following treatment with CEA-IL2v and CEA-IL2wt after overnight pre-activation with PHA. Exemplary data from ≥ 3 independent experiments. Shown are means from triplicates and standard deviation.
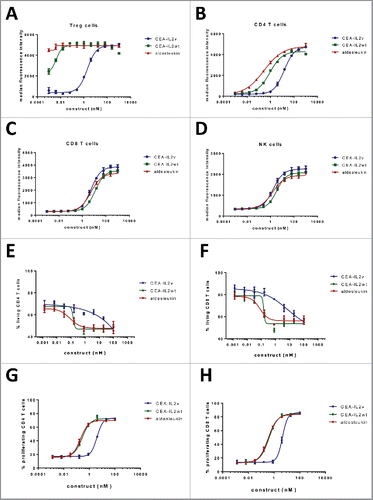
Treatment of immune effector cells (PBMC derived NK and T cells) with CEA-IL2v induced proliferation and upregulation of activation markers (Fig. S3). CEA-IL2v, CEA-IL2wt and aldesleukin showed similar activity in the induction of proliferation of resting NK cells and CD8+ T cells within PBMC, whereas the CEA-IL2wt and aledleukin were more active on resting CD4+ T cells (including Tregs) than CEA-IL2v. Pre-activated T cells were more responsive to CEA-IL2v treatment than resting T cells, resulting in EC50 values that were approximately 10-fold lower (). In general, NK cells responded promptly to CEA-IL2v treatment and most NK cells proliferated after 5 d of treatment. Similarly to NK cells, CD8+ T cells underwent proliferation after 5 d of treatment whereas CD4+ T cells responded more slowly with only a proportion of cells proliferating after the same period of time (data not shown). Compared to NK cells, higher concentrations of CEA-IL2v were required to stimulate resting T cells (Fig. S3).
Immune cell activation was assessed by measuring the dose dependent induction of expression of early (CD69) and late (CD25) activation markers within 3 d post treatment. Treatment with CEA-IL2v triggered similar immune cell activation in NK and CD8+ T cells (Fig. S3 D–G). CD69 (early activation marker) upregulation was detected as early as 6 h after antibody treatment and was maintained for up to 72 h post treatment on NK cells and 48 h post treatment on CD8+ T cells. CD25 upregulation was strongest at later time points than CD69, in agreement with the fact that CD25 is considered to be a late activation marker. Whereas CD69 was induced to a similar extent by CEA-IL2v and CEA-IL2wt, CD25 was more strongly upregulated on NK cells and CD8+ T by CEA-IL2v than by CEA-IL2wt. This can be explained by the lack of binding of CEA-IL2v to CD25 and a subsequent lack of internalization and/or shedding of CD25. Taken together, NK cells responded promptly to CEA-IL2v stimulation and rapidly upregulated activation markers, followed at later time points by CD8+ and CD4+ T cells (Fig. S3, data not shown). The activation of NK cells by CEA-IL2v translated into increased killing of the CEA-positive colon cancer cell line LS180 accompanied by increased release of Granzyme B, IFNγ and RANTES and upregulation of CD25 and CD69 on NK cells. Like before no difference between CEA-IL2v and CEA-IL2wt was observed in inducing activation of resting NK cells as analyzed by measuring tumor cell killing and cytokine release (Fig. S4).
Importantly, CEA-IL2v-mediated activity was not affected by artificial crosslinking to other cells via a secondary anti-Fc antibody, and presence of CEA-positive cells did not influence IL-2R-related activity (data not shown). In line with known biology, IL-2Rβγ signaling only requires the formation of the IL-2Rβγ heterodimer upon binding but not crosslinking and formation of higher oligomers, thus it cannot be expected that binding of CEA-IL2v to CEA on tumor cells and crosslinking results in an enhanced induction of IL-2R signaling.
Pharmacokinetics and tumor targeting in vivo
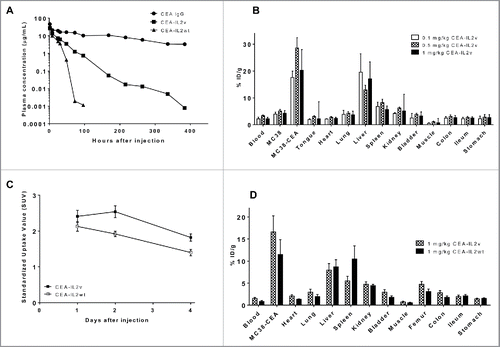
CEA-IL2v was designed to target CEA positive tumors more efficiently compared with the corresponding CEA-IL2wt due to the lack of CD25 binding and by being monovalent for the intermediate affinity IL-2Rβγ compared with classical immunocytokine. This design with a higher relative affinity for CEA compared with IL-2R should avoid sequestration of the construct in a peripheral high-affinity IL-2Rαβγ sink and lead to reduced clearance, and subsequently a relatively higher uptake in CEA-positive tumors than CEA-IL2wt. As a non-rodent homologue of human CEA does not exist, CEA-transgenic C57BL/6 mice were used in PK, biodistribution, and efficacy experiments to mimic human organ specific CEA expression and to allow for engraftment of syngeneic tumors stably expressing human CEA. PK studies demonstrated that indeed CEA-IL2v showed approximately 2-fold (1.7–2.3-fold) reduced clearance as compared with CEA-IL2wt by reducing IL-2Rαβγ-mediated clearance (). These PK properties should ultimately enable efficient tumor targeting of CEA-IL2v, acknowledging that CEA-IL2v was still cleared faster than the parental CEA antibody ().
Figure 3. (A) Mean plasma concentration-time profiles of CEA antibody CH1A1A1–2F1, CEA-IL2v and CEA- IL2wt in CEA-transgenic C57BL/6 mice as determined by ELISA. (B) Organ biodistribution of 89Zr-CEA-IL2v 3 d after injection at different doses in immunocompetent CEA-transgenic C57BL/6 mice bearing syngeneic CEA-positive MC38-CEA tumors and CEA-negative MC38 tumors on contra-lateral site. All values are expressed as %ID/g. Data represent mean value ± SD from at least four determinations. (C) Standardized Uptake Values (SUVs) of tumors derived from PET imaging of immunocompetent CEA-transgenic C57BL/6 mice bearing syngeneic CEA-positive MC38-CEA tumors after injection of 89Zr-CEA-IL2v or 89Zr-CEA-IL2wt. Data represent mean value ± SD from at least four determinations. (D) Biodistribution of 89Zr-CEA-IL2v compared with 89Zr-CEA-IL2wt 4 d after injection in immunocompetent CEA-transgenic C57BL/6 mice bearing syngeneic MC38-CE tumors. Data represent mean value ± SD from > 4 determinations.
Immunohistochemistry of CEA expression in CEA-transgenic C57BL/6 mice showed that CEA is heterogeneously expressed in the GI tract, and at low levels in the brain and testis, but not in other tissues, so that these mice do not completely reflect human normal tissue CEA expression, but can still provide valuable insights about potential accumulation in organs other than tumor (data not shown). CEA-IL2v PK in wild-type and CEA-transgenic C57BL/6 mice was comparable and dose proportional indicating that either CEA is not accessible from the blood stream and/or that the low normal tissue CEA expression in transgenic mice does not affect antibody biodistribution (data not shown).
Radioactivity-based organ biodistribution studies were performed in CEA expressing models with 89Zr-labeled immunocytokines. To demonstrate CEA-mediated tumor uptake, immunocompetent CEA-transgenic C57BL/6 mice bearing one syngeneic MC38-CEA tumor and one CEA-negative MC38 tumor on contralateral sites were injected with 89Zr-CEA-IL2v mixed with unlabeled CEA-IL2v to a total concentration of 0.1, 0.5 and 1 mg/kg. Biodistribution analysis performed at 3 d after injection demonstrated significantly higher tumor uptake in MC38-CEA tumors than MC38 tumors (p > 0.001) for all doses. In addition to MC38-CEA tumors, 89Zr-CEA-IL2v also accumulated in the liver and the spleen ().
To demonstrate differences in biodistribution between CEA-IL2v and CEA-IL2wt, a PET study was performed in the MC38-CEA model upon injection with 89Zr-CEA-IL2v or 89Zr-CEA-IL2wt mixed with the corresponding unlabeled immunocytokine to a total concentration of 1 mg/kg. Scans were performed at 1, 2 and 4 d after injection; ex vivo biodistribution was assessed immediately after the last scan at 4 d. This PET study demonstrated higher tumor accumulation of 89Zr-CEA-IL2v than for 89Zr-CEA-IL2wt ( and ). At days 2 and 4 post injection, Standardized Uptake Values (SUVs) in the tumor were significantly higher for CEA-IL2v than for CEA-IL2wt (p = 0.007 at 2 d and p = 0.011 at 4 d). Ex vivo assessment of the biodistribution at 4 d after injection revealed that the spleen uptake of 89Zr-CEA-IL2wt was significantly higher than of 89Zr-CEA-IL2v (p = 0.007), while the tumor uptake of 89Zr-CEA-IL2v was significantly higher than of 89Zr-CEA-IL2wt as also observed by PET imaging (p = 0.020) (Fig. S5). These data confirmed that adding the IL2v moiety to the parental CEA antibody CH1A1A-2F1 antibody did not abolish tumor targeting characteristics. Finally, greater tumor and lower spleen uptake of CEA-IL2v compared with CEA-IL2wt demonstrated the advantages of IL2v over IL-2 for better tumor targeting. However, it should be noted that CEA-IL2v in addition to being targeted to the tumor still exhibits peripheral binding to immune cells in lymphoid tissues/peripheral blood as can be seen from immune-pharmacodynamic studies (see below).
Pharmacodynamics in vivo
The mechanism of action and immuno-pharmacodynamics of CEA-IL2v and/or its murinized surrogate muCEA-IL2v was studied in fully immunocompetent tumor-free C57BL/6 mice or tumor-bearing C57BL/6 mice transgenic for CEA.Citation40
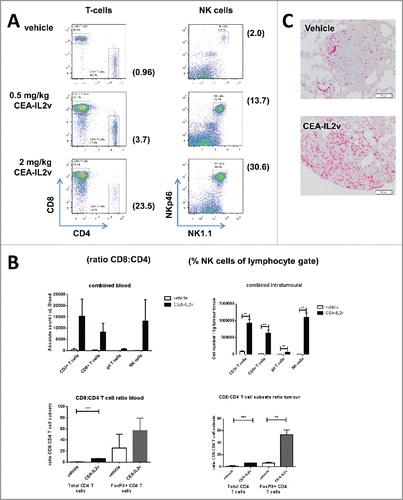
In tumor-free mice, a strong expansion of peripheral CD8+ T and NK cells after treatment with 0.5 and 2 mg/kg CEA-IL2v was observed (). A more detailed analysis using different doses of CEA-IL2v showed that after an initial and rapid drop in cell numbers, putatively a re-distribution phenomenon, CD8+, γδ T cells and NKp46+ NK cells underwent a strong expansion in the blood that peaked around days 4 to 7, and returned to baseline levels ca. 2 weeks post treatment (Fig. S6A). The increase in cell numbers was accompanied by a corresponding increase in the expression of the proliferation marker Ki67 (Fig. S6B). As total CD4+ T cell numbers did not significantly change, the preferential expansion of the CD8+ T cells skewed the T cell compartment in favor of this subset (Fig. S6B). These data are in line with experiments using IL-2-antibody complexes that no longer interact with CD25 and caused a strong preferential expansion of CD8+ T-memory over CD4+ T cells.Citation41
Figure 4. Immuno-pharmacodynamics in tumor-free and tumor-bearing C57BL/6 mice: (A) Peripheral T and NK cell expansion by CEA-IL2v. Shown are lymphocytes in blood 7 d after a single i.v. dose of CEA-IL2v. (B) Increase in the numbers of circulating (per μL blood) and intratumoral (per g tissue) CD8+ T cells, γδ T cells and NK cells in LLC1-CEA syngeneic tumor model 5 d after injection of 0.5 or 2 mg/kg muCEA-IL2v as determined by flow cytometry (upper panels). A skewing of the T cell compartment in favor of CD8+ T cells as shown by the ratio of CD8+ T to total CD4+ T and Treg in the blood and tumors of LLC1-CEA tumor-bearing mice (lower panels). (C) Increase of CD3 T cells in LLC1-CEA syngeneic tumor model 5 d after injection of 1.0 mg/kg CEA-IL2v as determined by immunohistochemistry.
In a separate study, the kinetics of CD8+ and NK cell proliferation following treatment with muCEA-IL2v was compared with muCEA-IL2wt. An initial drop in circulating CD8+ T and NK cell numbers followed by the rapid dose-dependent expansion of the CD8+ T and NK cells by day 5 post treatment was observed for both immunocytokines. However, the magnitude and duration of the expansion differed. muCEA-IL2v induced a 10-fold expansion of NK and CD8+ T cells that was sustained for more than 14 d for CD8+ T cells and 7 d for NK cells, whereas muCEA-IL2wt induced a shorter lived and lower expansion in these cell populations (Fig. S7A,B). Interestingly, whether the muCEA-IL2v was administered intravenously or subcutaneously had no effect on the final T cell or NK cell responses (data not shown). As previously, muCEA-IL2v had a more pronounced effect on CD8+ T cells than on CD4+ T cells and a skewing of the T cell compartment in favor of the CD8+ T cells in the CEA-IL2v treated group was observed resulting in a ratio of CD8+:CD4+ T cells of ∼8:1 by day 5 post treatment. In contrast, there was no T cell skewing in the muCEA-IL2wt treated groups as this construct induced a similar level of expansion in both T cell subsets (Fig. S7C).
To determine whether the preferential expansion of CD8+ T and NK cells observed in the tumor-free animals also occurred in tumor-bearing mice, CEA-transgenic C57/BL6 mice were injected intravenously with LLC1-CEA and once the tumors were established in the lungs, treated with muCEA-IL2v. Five days after treatment, mice were bled and sacrificed, tumors and lymphoid tissues were removed and immune cell subsets were quantified by flow cytometry. As observed previously in tumor-free animals, there was a significant increase in the numbers of CD8+ T and NK cells in blood after treatment with muCEA-IL2v, an expansion that was also observed in the tumor (), whereas there was only a marginal increase in the number of the CD4+ or CD4+Foxp3+ Tregs in blood and tumor (data not shown). Accordingly, the preferential expansion of CD8+ T over CD4+ T cells, including Tregs, skewed the T cell compartment in favor of CD8+ T cells (, lower panels). The increase in CD8+ T and NK cell counts correlated with an increase in Ki67 expression on cells isolated from blood and tumors. Notably, Ki67+ NK and CD8+ T cells expanded not only in peripheral blood, but also in lymphoid tissues of mice bearing subcutaneous MC38-CEA or LLC1-CEA lung tumors (Fig. S8A, B). A transient splenomegaly was observed in muCEA-IL2v-treated animals compared with the vehicle-treated group, but spleens returned to their normal size once immune cell numbers in blood returned to baseline (data not shown). In addition to CD8+ T and NK cells, an expansion of innate γδ-TCR+ T cells was observed in blood and in tumors of treated animals. Thus, in both tumor models, a strong intratumoral expansion of CD3 T cells (driven mostly by CD8+ T cells), NK cells and γδ T cells was observed after treatment with muCEA-IL2v. The expansion of CD3 T cells following treatment with CEA-IL2v was in addition confirmed by immunohistochemistry ().
In summary, CEA-IL2v, in contrast to CEA-IL2wt, was able to induce a preferential (vs. CD4+ T cells including Tregs) expansion of both CD8+ T cells and NK cells in the blood, lymphoid tissues and tumors, thereby significantly altering the overall composition of the peripheral and tumor-associated lymphocytes. This increase in tumor infiltrating immune cells forms the basis for the rationale to combine CEA-IL2v with ADCC-competent and T cell bispecific antibodies and/or PD-L1 checkpoint blockade. Due to the nature of IL-2R expression and biology, CEA-IL2v also mediates systemic effects on peripheral and immune cells residing in lymphoid tissues that are independent of targeting to CEA.
Monotherapy efficacy
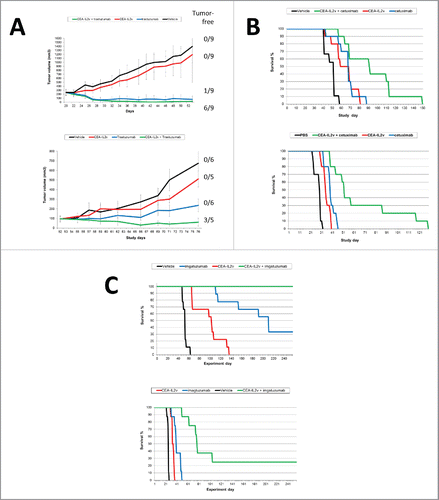
The monotherapy efficacy of CEA-IL2v was subsequently tested using the syngeneic MC38-CEA cell line stably transfected with CEA injected intrasplenically into CEA-transgenic C57BL/6 mice. Animals treated with CEA-IL2v (2 mg/kg, q1w3) showed a statistically significant increase in median survival of 63 d vs. 42 d for vehicle treated animals (p < 0.039), and increased overall long-term survival indicating absence of tumor in 3/8 animals (). Similarly, muCEA-IL2v was tested in the syngeneic pancreatic PancO2-CEA model injected intrapancreatically into C57BL/6 mice. Animals treated with muCEA-IL2v (0.5 mg/kg, q1w3) showed a statistically significant increase in median survival of 42 d vs. 30 d for vehicle treated animals (p = 0.0385) (). In this model, Alexa-647 labeled CEA-IL2v accumulated specifically in the CEA positive pancreatic cancer regions, but not in normal pancreas; followed by enhanced T cell proliferation in regions where CEA-IL2v was present (Fig. S9A).
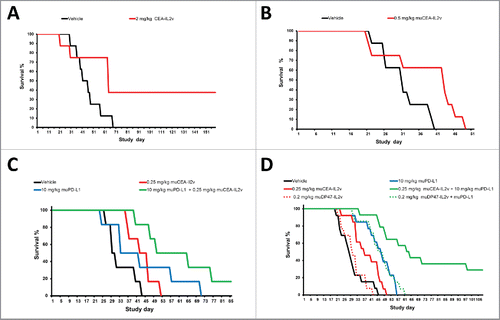
Figure 5. (A, B) Monotherapy efficacy: (A) Efficacy of 2 mg/kg CEA-IL2v (q1w3 starting on day 7 after tumor inocculation) as single agent in the syngeneic intrasplenic MC38-CEA model in CEA-transgenic C57/BL6 mice. (B) Efficacy of 0.5 mg/kg muCEA-IL2v (q1w3 starting on day 7 after tumor inocculation) as single agent in the syngeneic orthotopic PancO2-CEA model in CEA-transgenic C57/BL6 mice. (C, D) Combination of CEA-IL2v with PD-L1 checkpoint blockade in syngeneic models in CEA-transgenic C57BL/6 mice. (C) Efficacy of 0.25 mg/kg muCEA-IL2v (q1w5) combined with 10 mg/kg <muPD-L1> antibody 6E11 (q1w5) concomitantly starting on day 7 vs. vehicle and the respective monotherapies in the orthotopic PancO2-CEA pancreatic syngeneic model. (D) Efficacy of 0.25 mg/kg muCEA-IL2v or a matched dose of 0.2 mg/kg untargeted muDP47-IL2v control immunocytokine (q1w4) combined with 10 mg/kg <muPD-L1> antibody 6E11 (q1w4) concomitantly starting on day 7 vs. vehicle and the respective monotherapies. Pooled data from two independent studies are shown.
In contrast to tumor and lymphoid tissues, no T cell infiltration or inflammation was observed by histology in CEA-positive regions of the GI tract of non-tumor-bearing CEA-transgenic C57BL/6 mice upon treatment with 1 or 2 mg/kg CEA-IL2v (3q7d) (Fig. S9B). Likewise, no obvious GI toxicity was observed in tumor-bearing animals treated with CEA-IL2v or muCEA-IL2v, respectively. In line with this, immunohistochemical analysis after perfusion with Alexa-647 labeled CEA antibody CH1A1A-2F1 and CEA-IL2v showed that they did not accumulate in the GI (Fig. S9C). This supports the view that CEA expressed on the apical surface of normal tissues in the GI tract may not be easily accessible to CEA-IL2v delivered systemically by injection.
Combination with PD-L1 checkpoint blockade
PD-L1 is found in various tumors where it is induced by IFNγ. It interferes with the immune episode of cancer cells by interacting with the inhibitory PD-1 receptor on T cells.Citation42 In vitro studies showed that CEA-IL2v induces PD-L1 upregulation on tumor cells (Fig. S10). CEA-IL2v alone did not induce PD-L1 on A549 tumor cells; but in the presence of PBMC, PD-L1 was upregulated on A549 cells presumably mediated by the release of IFNγ. Expression levels of PD-L1 increased with increasing concentration of CEA-IL2v and a higher effector (E): target (T) ratio.
We then investigated in syngeneic mouse models whether CEA-IL2v enhanced the efficacy of PD-L1 checkpoint blockade. In the intra-pancreatic syngeneic PancO2-CEA model concomitant dosing of muCEA-IL2v (0.25 mg/kg, q1w5) and anti-muPD-L1 (6E11) (10 mg/kg, q1w5) resulted in superior median survival of 57 d (p = 0.0043) vs. 29 d for vehicle treated animals and 1/6 tumor-free mice, vs. day 43 (p = 0.030, 0/6) for muCEA-IL2v and day 36 (p = 0.057, 0/6) (, Table S6A).
In the same model, a combination study compared the benefit of muCEA-IL2v (0.25 mg/kg, q1w4) combined with concomitant PD-L1 blockade to the combination with the untargeted muDP47-IL2v control immunocytokine (, Table S6B) at a dose (0.20 mg/kg, q1w4) matched for equivalent exposure and peripheral pharmacodynamics in a pre-experiment (data not shown). Notably, muCEA-IL2v plus anti-muPD-L1 (6E11) (10 mg/kg, q1w4) showed a combination effect resulting in superior median survival of 61 d (p < 0.0001) with long-term survival in 5/14 animals vs. 27 d for vehicle treated animals (0/13), whereas the combination with untargeted muDP47-IL2v at the matched dose showed a median survival of 44 d (p < 0.0001) and 0/14 tumor-free animals, and failed to show a significant difference from the muPD-L1 single agent group with 46 d (0/13) (p = 0.0001 vs. vehicle; p = 0.7260 vs. muDP47-IL2v + muPD-L1). These data confirm that CEA-targeting of CEA-IL2v results in superior efficacy in combination with PD-L1 checkpoint blockade and support the benefit of targeting the immunocytokine to CEA.
Combination with ADCC competent antibodies
A possible application of CEA-IL2v includes its combination with ADCC-competent or -enhanced IgG1 antibodies. PBMC stimulated by CEA-IL2v in co-cultures with tumor cells showed dose-dependent killing of tumor cells and dose dependent upregulation of CD69 and CD25 activation markers on NK cells. The addition of trastuzumab or cetuximab enhanced the cytotoxic activity of NK cells, expression of CD69 and CD25 and release of cytokines (Fig. S11A–B).
The combination of CEA-IL2v with cetuximab, trastuzumab and glycoengineered imgatuzumabCitation43 was subsequently investigated in human CD16-transgenic SCID mice (, Table S7). As a single agent, CEA-IL2v (1 mg/kg, q1w3) showed only minor tumor growth inhibition (TGI) in the s.c. N87 gastric and the orthotopic KPL4 breast cancer xenograft models. Combination with trastuzumab (25 mg/kg, q1w3 in N87 and 10 mg/kg, q1w3 in KPL-4) resulted in superior TGI compared with the respective monotherapies including complete tumor remission in several animals (, Table S7A, B). In the intravenously injected A549 lung cancer and intrasplenically injected LS174T colorectal xenograft models, CEA-IL2v (1 mg/kg, q1w3) mediated only a moderate effect, whereas combination with cetuximab (25 mg/kg, q1w3) resulted in increased median and overall survival (OS) including long-term surviving mice (, Table S7C, D). Similarly, in these xenograft models, combination with imgatuzumab (25 mg/kg, q1w3) resulted in superior median and OS compared with the respective monotherapies, and in long-term survival of all treated animals in the A549 model (, Table S7E, F). When comparing studies, the combination with glycoengineered imgatuzumab appeared superior in outcome compared with the cetuximab combination. Thus, combining CEA-IL2v with ADCC competent antibodies resulted in at least additive enhancement of TGI including remission and increased median/OS (, Table S7D).
Figure 6. (A–C) Combination of CEA-IL2v with ADCC competent/ADCC enhanced antibodies in human CD16-transgenic SCID mice: (A) Combination of CEA-IL2v (1 mg/kg, q1w3) with <HER2> antibody trastuzumab in N87 (25 mg/kg, q1w3) (top) and KPL4 (10 mg/kg, q1w3) (bottom) xenograft models; (B) Combination of CEA-IL2v (1 mg/kg, q1w3) with <EGFR> antibody cetuxiumab (25 mg/kg, q1w3) in A549 (top) and Ls174t (bottom) xenograft models; (C) Combination of CEA-IL2v (1 mg/kg, q1w3) with glycoengineered <EGFR> antibody imgatuzumab (25 mg/kg, q1w3) in A549 (top) and LS174t (bottom) xenograft models.
Discussion
Various approaches have been described to modify the affinity of IL-2 to its receptor. Boyman and Sprent have shown that IL-2 complexed with the S4B6 antibody that sterically blocks the IL-2:CD25 interaction, mediates a preferential expansion and activation of IL-2Rβ(hi) effector CD8+ T and NK cells, but not CD4+ and Tregs cells as observed with wild-type IL-2 (41), without exhibiting the toxicity of the latter.Citation11,44 Garcia and colleagues have recently described a natural conformational switch to engineer an IL-2 superkine with increased binding affinity for IL-2Rβ. Crystal structures show that IL-2 superkine exhibits a conformation resembling that of CD25-bound IL-2. The IL-2 superkine induced superior expansion of cytotoxic T cells, leading to improved antitumor responses, and elicited less expansion of Tregs and reduced pulmonary edema.Citation45
Alternatively, NKTR-214, a wild-type IL-2 prodrug conjugated to releasable polyethylene glycol (PEG) chains was described. In tumor models, NKTR-214 shifted the ratio of T cells toward CD8+ T cells, consistent with preferential binding to IL-2Rβγ over CD25, and mediated antitumor efficacy. Furthermore, NKTR-214 showed higher exposure in tumors as compared with aldesleukin.Citation46 EMD 521873 (Selectikine) is an immunocytokine that targets the necrotic core of tumors fused with two D20T mutated IL-2 moieties with increased affinity for CD25 that binds preferentially the high affinity IL-2R.Citation47,48 Carmenate and colleagues described an untargeted IL-2 mutein with reduced CD25 affinity using different mutations than the ones used in IL2v.Citation49 This mutein differs from IL-2 by four mutations in the CD25 interface, and its CD25 binding is not completely abolished. The IL-2 mutein induced in vitro proliferation of CD8+ T and NK cells, but showed reduced capacity to induce proliferation of Tregs. In tumor models, it showed higher efficacy and tolerability than wild-type IL-2.Citation49 Recently, RDB 1450 was described, an engineered fusion protein of permuted IL-2 and CD25 that is selective for IL-2Rβγ and was found to have improved PK properties and to mediate a marked expansion of NK and CD8+ T cells, but not Tregs, and exhibited reduced toxicity in mice.Citation50 Most recently, Sim and colleagues showed that IL2 F42K circumvented Treg expansion and promoted NK activation,Citation51 and Ghasemi and colleagues described a fusion protein of an IL-2 R38A/F42K double mutant with reduced CD25 binding fused to cowpox virus encoded NKG2D binding protein (OMCP) to target IL-2 to NKG2D positive immune cells such as NK cells.Citation52
Different from these approaches and based on the rationale above, we designed an IL2v with completely abolished CD25 binding and fused it to a CEA-specific antibody to enhance its half-life and enable preferential targeting to the tumor. X-ray co-crystallography of the ternary complex confirmed the binding mode to IL-2Rβγ with no major impact on both the IL-2Rβγ and IL2v structures as compared with the wild-type IL-2/IL-2Rβγ complex and the lack of an interface allowing binding to CD25.Citation34,35 Indeed, surface plasmon resonance and P-STAT5 data using immune effector cells and Tregs from PBMC are supportive of a complete absence of CD25 affinity. CEA-IL2v exhibits a similar mechanism as described for the IL-2-S4B6 complexCitation11,53 and the human CD25-mimobody described recently,Citation54 including the lack of preferential activation of Tregs, reduced potency in triggering Fas-mediated apoptosis (AICD), superior tolerability compared with a wild-type IL-2-based immunocytokine and reduced clearance resulting in superior PK due to the lack of binding to the peripheral and endothelial CD25 sink.Citation11
Previously described immunocytokines were largely based on wild-type IL-2, resulting in low tumor targeting due to high affinity of IL-2 for the high affinity IL-2R as opposed to the tumor antigen. Indeed they show poor tumor targeting with antigen specificity being irrelevant for efficacy and biodistribution.Citation17 A consequence of engineering IL2v with reduced relative IL-2R affinity, CEA-IL2v shows superior tumor targeting in comparison with CEA-IL2wt in the syngeneic MC38-CEA model. The radiolabeled CEA-IL2v immunocytokine was excreted via the hepatobiliary pathway and therefore in addition to the uptake in spleen (IL2v-mediated) and CEA + tumor (CEA-mediated) a moderate to high liver uptake was observed in line with uptake typically seen with other antibodies due to excretion.Citation55 In line with this, no signs of liver toxicity were observed in mice and using a cynomolgus specific cyCEA-IL2v surrogate immunocytokine in cynomolgus monkeys (data not shown).
No preclinical safety studies in the NHP were done with the clinical CEA-IL2v construct due to lack of cross-reactivity with cynomolgus monkey CEA (cyCEA). Single dose and repeat dose safety studies were performed with a bivalent anti-cyCEA homolog fused to the same IL2v as in CEA-IL2v. In the cynomolgus monkey, there was no evidence for hepatotoxicity or any adverse effects mediated by cyCEA-targeting in any of the tissues known to express CEA (e.g., GI). Effects observed in the above studies were consistent with the known safety profile of wild-type IL-2 with the exception that no vascular leak syndrome was observed with cyCEA-IL2v under the given study conditions (data no shown). Immuno-pharmacodynamic effects were similar to those observed with muCEA-IL2v in the mouse. After a transient reduction in circulating lymphocytes, cyCEA-IL2v induced T and NK cell activation, increased numbers with skewing toward the CD8+ T cell population (data no shown).
Further mechanistic support for the benefit of targeting comes from in vivo efficacy studies, where we demonstrated that CEA-IL2v mediates superior efficacy including long-term survival for some of the mice treated, whereas an untargeted control immunocytokine did not exhibit such efficacy when combined with PD-L1 checkpoint blockade. Different from FcγRIII-mediated or CD3-induced ADCC with antibodies or T cell bispecific antibodies, IL-2R signaling does not require crosslinking of IL-2R upon antigen binding on the target cell so that no strictly target-dependent IL-2R signaling activity can be expected. Thus, CEA-IL2v on top of being preferentially retained at the tumor also exhibits a systemic effect on peripheral immune cells as can be seen from the strong peripheral CD8+ T and NK cell expansion. Similarly to aldesleukin, untargeted IL-2 superkine, IL-2 mutein or NKTR-214, this additional, untargeted systemic effect contributes to the antitumoral effect of CEA-IL2v. Nevertheless, while systemic immune activation can be achieved with untargeted IL-2 based therapies, CEA-IL2v has the added potential of targeting and retention in the tumor, thus enhancing the local, tumoral concentration for a given, tolerated systemic exposure.
CEA-IL2v can be considered and IL-15-like cytokine in the sense that both cytokines activate IL-2Rβγ signaling and that preferential activation of Treg vs. effector cells is avoided.Citation56,57 However, IL-15 as it exhibits considerable toxicity in human trialsCitation58 so that for IL-15 based cytokines a similar approach either mutating the interface or the use of the IL-15-IL-15Rα (Sushi) domain is required to avoid high affinity binding to the IL-15Rα chain expressed, for example, on dendritic cells and fibroblasts. ALT-803, a clinical stage untargeted complex of an IL-15 superagonist mutant, fusions of ALT-803 to single-chain variable fragment (scFv) recognizing CD20 and a dimeric IL-15/IL-15 receptor α Sushi-Fc fusion protein, was found to exhibit significant biologic activity on NK and T cells including activity in syngeneic models, expansion of CD8+ T cells and boosting of ADCC competent antibodies in line with findings for CEA-IL2v.Citation59-64 Overall, the immuno-pharmacodynamics of these IL-15 based cytokines as well as the IL-2 based cytokine above are quite comparable to those of CEA-IL2v as they all activate signaling through the IL-2Rβγ complex.
The combination of CEA-IL2v with checkpoint blockade was tested in syngeneic mouse models in fully immunocompetent CEA-transgenic C57BL/6 mice tolerant to human CEA using PancO2 cells expressing CEA. The used models are known to be immunogenic,Citation65,66 having antigen specific T cells present in the tumor at the start of treatment and showing moderate single agent efficacy mediated by PD-L1 checkpoint blockade. We observed that the combination of CEA-IL2v with an anti-PD-L1 antibody can further improve outcome and induce complete tumor remission/long-term survival in a significant proportion of treated animals as compared with the respective monotherapies.
The combination effect of a diabody-based wild-type IL-2 immunocytokine with rituximab has been shown.Citation67 Given its enhanced features in terms of PK, targeting and tolerability, CEA-IL2v was developed as a combination partner for ADCC-competent, ADCC-enhanced and T cell bispecific antibodies. The preclinical data shown in this paper support this combination rationale: CEA-IL2v is able to strongly enhance the antitumoral effect of cetuximab, trastuzumab and particularily the ADCC enhanced glycoengineered antibody imgatuzumab in xenograft models relying on innate immunity mediated by NK cells, macrophages and monocytes. As CEA is absent in rodents and no syngeneic cell lines co-expressing human CEA and EGFR/HER2 and the respective transgenic mice tolerant for CEA and EGFR/HER2 are available, the ADCC combination of CEA-IL2v could only be studied in B and T cell deficient models. Combination studies using an analogous FAP-targeted FAP-IL2v immunocytokine and antibodies specific for antigens on syngeneic cell lines confirmed these findings in fully immunocompetent mice (manuscript in preparation). These data are in line with the findings on the benefit of the combination of a half-life enhanced wild-type IL-2 Fc fusion with the gp75 specific antibody TRP1 in the B16 model or adoptive T cell therapy.Citation18
Taken together, cergutuzumab amunaleukin (CEA-IL2v, RG7813) is a novel IL2v-based, half-life enhanced immunocytokine with the added benefit of tumor targeting, designed for abolished CD25 binding for improved tolerability, PK and tumor targeting as well as the preferential activation of CD8+ and NK immune effector cells over Tregs as compared with wild-type IL-2-based immunocytokines. As a result of the IL2v design with preferential binding to CEA over IL-2R resulting in better accumulation and retention at the tumor site, cergutuzumab amunaleukin is able to work through tumor targeting-dependent mechanisms, together with targeting-independent mechanisms including the preferential peripheral expansion of CD8+ and NK immune effector cells through IL-2Rβγ while avoiding a preferential expansion of Tregs. Preclinical data support the use of cergutuzumab amunaleukin for combination immunotherapy with ADCC-competent or -enhanced antibodies of the IgG1 isotype, T cell bispecific antibodies that rely on CD8+ T effector cells and also with PD-L1 checkpoint blockade in immunogenic tumors. CEA-IL2v (RG7813) is currently in phase I clinical trial in CEA positive solid tumors as a single agent (NCT02004106) and in combination with atezolizumab (NCT02350673).
Materials and methods
Crystal structure analysis
Crystallization, data collection and structure determination were performed as described in the supplement. Data collection and refinement statistics are summarized in Table S2. Coordinates are deposited in RCBS under the PDB ID 5M5E.
Cell lines
An overview about cell lines used and their origin is given in the supplement.
Immunocytokines and antibodies
The supplement gives an overview about the immunocytokines, mouse surrogate immunocytokines and antibodies used in this study and their design in Tables S3 and S4. All immunocytokines were produced by transient production using HEK293 or stable CHO clones followed by purification using Protein A affinity chromatography and size exclusion/ion exchange chromatography. 6E11, a muPD-L1 specific muIgG1 surrogate antibody, was kindly provided by Dr. Jeong Kim, Genentech, San Francisco.
STAT5 phosphorylation
Freshly isolated PBMCs were incubated for 20 min at 37°C with IL-2-containing molecules. After incubation, cells were immediately fixed with Cytofix buffer (BD Bioscience) to preserve the phosphorylation status for 10 min at 37°C and permeabilized with Phosflow Perm buffer III (BD Bioscience) for 30 min at 4°C. The cells were stained with indicated antibodies and analyzed by flow cytometry.
Proliferation
PBMCs were labeled with 100 nM CFSE (Sigma-Aldrich) and stimulated with IL-2 containing molecules. After the indicated time, PBMCs were stained for immune cell subsets and proliferation was measured by determining CFSE dilution.
Activation induced cell death
Freshly isolated PBMCs were stimulated with 1 μg/mL PHA overnight. Pre-activated PBMCs were stimulated with IL-2 containing molecules for 4 d before the anti-Fas antibody (clone CH11, Millipore) was added for additional 16 h. The percentage of living cells was measured by using Annexin V (Roche Applied Science) and the LIVE/DEAD Fixable Viability Stain eFluor 660 (eBioscience).
Mouse xenograft and syngeneic models
Subcutaneous and orthotopic xenograft and syngeneic models were used to assess the effect of combinations on the in vivo efficacy of CEA-IL2v. Briefly, for the NSCLC xenograft lung model, female human CD16-transgenic SCID mice (Charles River Laboratories, Lyon, France) were inoculated with 3 × 106 A549 cells injected intravenously. For the colorectal liver metastases models, the CRC cell line LS174T was injected into the spleen (3 × 106 cells). For the gastric sc model, the N87 cells were injected subcutaneously (1 × 106 cells). For the breast orthotopic model, the cell line KPL-4 was injected into the mammary fat pad (5 × 106 cells). For the pancreatic syngeneic model, female CEA-transgenic C57BL/6-CEA mice (Charles River Laboratories, Lyon, France) were inoculated with 1 × 105 Panc02-CEA cells injected intrapancreatically. Mice were maintained under specific-pathogen-free conditions with daily cycles of 12 h light/darkness according to guidelines (GV-SOLAS; FELASA) and food and water were provided ad libitum. Continuous health monitoring was performed and the experimental study protocol was reviewed and approved by the Veterinary Department of Kanton Zurich.
Mice were randomized into different treatment groups and therapy started when evidence of tumor growth was visible in the target organ of killed scout animals (days indicated in figure legends). All treatments were administered IV. The termination criterion for sacrificing animals was sickness with locomotion impairment, and median OS was defined as the experimental day by which 50% of animals had been killed. Kaplan–Meier survival curves and the Pairwise Log-Rank test were used to compare survival between animals.
Ex vivo immune cell analysis
The syngeneic MC38-CEA and LLC1-CEA mouse models were used as previously stated to assess the effects of CEA-IL2v and muCEA-IL2v on the major immune effector cell subsets. Once the tumors were established (> 250 mm2 for MC38-CEA and day 21 post injection for LLC-CEA), treatment was given, and the mice killed 5 d post treatment. The mice were bled, the tumors and lymphoid tissue removed, and the immune effector cells isolated (see Supplemental Materials and Methods). The different immune cell subsets were identified, quantified and the activation/proliferation status determined by flow cytometry on the BD LSR Fortessa flow cytometer. Cytometry analysis was performed using Flowjo and statistical analysis with Graphpad Prism.
Disclosure of potential conflicts of interest
All authors are (or were) Roche employees (at the time when the study was conducted), except the authors of VU University Medical Center.
Supplemental_materials.zip
Download Zip (1.6 MB)Acknowledgments
We thank Martina Carola Birk for supporting the crystallization efforts, Dr Jeong Kim, Genentech for providing the anti-muPD-L1 surrogate antibody 6E11, and all project team members contributing to the preclinical, technical and clinical development of CEA-IL2v.
References
- Malek TR, Yu A, Zhu L, Matsutani T, Adeegbe D, Bayer A. L. IL-2 family of cytokines in T regulatory cell development and homeostasis. J Clin Immunol 2008; 28:635-9; PMID:18726679; http://dx.doi.org/10.1007/s10875-008-9235-y
- Kasprzak A, Olejniczak K, Przybyszewska W, Zabel M. Cellular expression of interleukin 2 (IL-2) and its receptor (IL-2R, CD25) in lung tumours. Folia Morphol (Warsz) 2007; 66:159-66; PMID:17985312
- Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol 2006; 6:595-601; PMID:16868550; http://dx.doi.org/10.1038/nri1901
- Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol 2012; 12:180-90; PMID:22343569; http://dx.doi.org/10.1038/nri3156
- Arenas-Ramirez N, Woytschak J, Boyman O. Interleukin-2: Biology, Design and Application. Trends Immunol 2015; 36:763-77; PMID:26572555; http://dx.doi.org/10.1016/j.it.2015.10.003
- Boyman O, Krieg C, Homann D, Sprent J. Homeostatic maintenance of T cells and natural killer cells. Cell Mol Life Sci 2012; 69:1597-608; PMID:22460580; http://dx.doi.org/10.1007/s00018-012-0968-7
- Fontenot JD, Rasmussen JP, Gavin MA, Rudensky A. Y. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol 2005; 6:1142-51; PMID:16227984; http://dx.doi.org/10.1038/ni1263
- D'Cruz LM, Klein L. Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat Immunol 2005; 6:1152-59; PMID:16227983; http://dx.doi.org/10.1038/ni1264
- Maloy KJ, Powrie F. Fueling regulation: IL-2 keeps CD4+ Treg cells fit. Nat Immunol 2005; 6:1071-72; PMID:16239920; http://dx.doi.org/10.1038/ni1105-1071
- Rosenberg SA IL-2: the first effective immunotherapy for human cancer. J Immunol 2014; 192:5451-58; PMID:24907378; http://dx.doi.org/10.4049/jimmunol.1490019
- Krieg C, Letourneau S, Pantaleo G, Boyman O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc Natl Acad Sci U S A 2010; 107:11906-11; PMID:20547866; http://dx.doi.org/10.1073/pnas.1002569107
- Kontermann RE. Antibody-cytokine fusion proteins. Arch Biochem Biophys 2012; 526:194-205; PMID:22445675; http://dx.doi.org/10.1016/j.abb.2012.03.001
- Pasche N, Neri D. Immunocytokines: a novel class of potent armed antibodies. Drug Discov Today 2012; 17:583-90; PMID:22289353; http://dx.doi.org/10.1016/j.drudis.2012.01.007
- Ortiz-Sanchez E, Helguera G, Daniels TR, Penichet ML. Antibody-cytokine fusion proteins: applications in cancer therapy. Expert Opin Biol Ther 2008; 8:609632; PMID:18407765; http://dx.doi.org/10.1517/14712598.8.5.609
- Lode HN, Xiang R, Perri P, Pertl U, Lode A, Gillies SD, Reisfeld RA. What to do with targeted IL-2. Drugs Today (Barc) 2000; 36:321-36; PMID:12861355; http://dx.doi.org/10.1358/dot.2000.36.5.575044
- Lode HN, Xiang R, Becker JC, Gillies SD, Reisfeld R A. Immunocytokines: a promising approach to cancer immunotherapy. Pharmacol Ther 1998; 80:277-92; PMID:9888698; http://dx.doi.org/10.1016/S0163-7258(98)00033-3
- Tzeng A, Kwan BH, Opel CF, Navaratna T, Wittrup KD. Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proc Natl Acad Sci U S A 2015; 112:3320-3325; PMID:25733854; http://dx.doi.org/10.1073/pnas.1416159112
- Zhu EF, Gai SA, Opel CF, Kwan BH, Surana R, Mihm MC, Kauke MJ, Moynihan KD, Angelini A, Williams RT et al. Synergistic innate and adaptive immune response to combination immunotherapy with anti-tumor antigen antibodies and extended serum half-life IL-2. Cancer Cell 2015; 27:489-501; PMID:25873172; http://dx.doi.org/10.1016/j.ccell.2015.03.004
- Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu E F, Engreitz JM, Williams RT, Rakhra K, Zhang MH, Rothschilds AM et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med 2016; 22(12):1402-10; PMID:27775706; http://dx.doi.org/10.1038/nm.4200
- Ashraf SQ, Umana P, Mossner E, Ntouroupi T, Brunker P, Schmidt C, Wilding JL, Mortensen NJ, Bodmer WF. Humanised IgG1 antibody variants targeting membrane-bound carcinoembryonic antigen by antibody-dependent cellular cytotoxicity and phagocytosis. Br J Cancer 2009; 101:1758-68; PMID:19904275; http://dx.doi.org/10.1038/sj.bjc.6605355
- Durbin H, Young S, Stewart LM, Wrba F, Rowan A J, Snary D, Bodmer WF. An epitope on carcinoembryonic antigen defined by the clinically relevant antibody PR1A3. Proc Natl Acad Sci U S A 1994; 91:4313-17; PMID:7514303; http://dx.doi.org/10.1073/pnas.91.10.4313
- Wilkinson RW, Ross EL, Poulsom R, Ilyas M, Straub J, Snary D, Bodmer WF, Mather SJ. Antibody targeting studies in a transgenic murine model of spontaneous colorectal tumors. Proc Natl Acad Sci U S A 2001; 98:10256-260; PMID:11517330; http://dx.doi.org/10.1073/pnas.181353498
- Bacac M, Klein C, Umana P. CEA TCB: A novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. Oncoimmunology 2016; 5:e1203498; PMID:27622073; http://dx.doi.org/10.1080/2162402X.2016.1203498
- Lehmann S, Perera R, Grimm HP, Sam J, Colombetti S, Fauti T, Fahrni L, Schaller T, Freimoser-Grundschober A, Zielonka J et al. In Vivo Fluorescence Imaging of the Activity of CEA TCB, a Novel T-Cell Bispecific Antibody, Reveals Highly Specific Tumor Targeting and Fast Induction of T-Cell-Mediated Tumor Killing. Clin Cancer Res 2016; 22:4417-27; PMID:27117182; http://dx.doi.org/10.1158/1078-0432.CCR-15-2622
- Bacac M, Fauti T, Sam J, Colombetti S, Weinzierl T, Ouaret D, Bodmer W, Lehmann S, Hofer T, Hosse RJ et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin Cancer Res 2016; 22:3286-97; PMID:26861458; http://dx.doi.org/10.1158/1078-0432.CCR-15-1696
- Goldstein MJ, Mitchell EP. Carcinoembryonic antigen in the staging and follow-up of patients with colorectal cancer. Cancer Invest 2005; 23:338-51; PMID:16100946; http://dx.doi.org/10.1081/CNV-58878
- Oberst MD, Fuhrmann S, Mulgrew K, Amann M, Cheng L, Lutterbuese P, Richman L, Coats S, Baeuerle PA, Hammond SA. CEA/CD3 bispecific antibody MEDI-565/AMG 211 activation of T cells and subsequent killing of human tumors is independent of mutations commonly found in colorectal adenocarcinomas. MAbs 2014; 6:1571-84; PMID:25484061; http://dx.doi.org/10.4161/19420862.2014.975660
- Peng L, Oberst MD, Huang J, Brohawn P, Morehouse C, Lekstrom K, Baeuerle PA, Wu H, Yao Y, Coats SR et al. The CEA/CD3-bispecific antibody MEDI-565 (MT111) binds a nonlinear epitope in the full-length but not a short splice variant of CEA. PLoS One 2012; 7:e36412; PMID:22574157; http://dx.doi.org/10.1371/journal.pone.0036412
- Bacac M, Fauti T, Sam J, Colombetti S, Weinzierl T, Ouaret D, Bodmer WF, Lehmann S, Hofer T, Hosse RJ et al. A Novel Carcinoembryonic Antigen T Cell Bispecific Antibody (Cea Tcb) for the Treatment of Solid Tumors. Clin Cancer Res 2016; 22(13):3286-97; PMID:26861458; http://dx.doi.org/10.1158/1078-0432.CCR-15-1696
- Govindan SV, Cardillo TM, Rossi EA, Trisal P, McBride WJ, Sharkey RM, Goldenberg DM. Improving the therapeutic index in cancer therapy by using antibody-drug conjugates designed with a moderately cytotoxic drug. Mol Pharm 2015; 12:1836-47; PMID:25402018; http://dx.doi.org/10.1021/mp5006195
- Dabbs DJ, Sturtz K, Zaino RJ. The immunohistochemical discrimination of endometrioid adenocarcinomas. Hum Pathol 1996; 27:172-7; PMID:8617459; http://dx.doi.org/10.1016/S0046-8177(96)90371-8
- Thompson JA, Grunert F, Zimmermann W. Carcinoembryonic antigen gene family: molecular biology and clinical perspectives. J Clin Lab Anal 1991; 5:344-66; PMID:1941355; http://dx.doi.org/10.1002/jcla.1860050510
- Van Dongen GA, Huisman MC, Boellaard R, Harry Hendrikse N, Windhorst AD, Visser GW, Molthoff CF, Vugts DJ. 89Zr-immuno-PET for imaging of long circulating drugs and disease targets: why, how and when to be applied? Q J Nucl Med Mol Imaging 2015; 59:18-38; PMID:25517081
- Rickert M, Wang X, Boulanger MJ, Goriatcheva N, Garcia KC. The structure of interleukin-2 complexed with its alpha receptor. Science 2005; 308:1477-80; PMID:15933202; http://dx.doi.org/10.1126/science.1109745
- Wang X, Rickert M, Garcia KC. Structure of the quaternary complex of interleukin-2 with its alpha, beta, gammac receptors. Science 2005; 310:1159-63; PMID:16293754; http://dx.doi.org/10.1126/science.1117893
- Hilton DJ, Watowich SS, Katz L, Lodish HF. Saturation mutagenesis of the WSXWS motif of the erythropoietin receptor. J Biol Chem 1996; 271:4699-708; PMID:8617735; http://dx.doi.org/10.1074/jbc.271.23.13754
- Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umana P, Mossner E. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel 2016; 29(10):457-66; PMID:27578889; http://dx.doi.org/10.1093/protein/gzw040
- Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, Carter P. An efficient route to human bispecific IgG. Nat Biotechnol 1998; 16:677-81 PMID: 9661204; PMID:9661204; http://dx.doi.org/10.1038/nbt0798-677
- Wild N, Andres H, Rollinger W, Krause F, Dilba P, Tacke M, Karl J. A combination of serum markers for the early detection of colorectal cancer. Clin Cancer Res 2010; 16:6111-6121; PMID:20798228; http://dx.doi.org/10.1158/1078-0432.CCR-10-0119
- Clarke P, Mann J, Simpson JF, Rickard-Dickson K, Primus FJ. Mice transgenic for human carcinoembryonic antigen as a model for immunotherapy. Cancer Res 1998; 58:1469-1477; PMID:9537250
- Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 2006; 311:1924-27; PMID:16484453; http://dx.doi.org/10.1126/science.1122927
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, combinations. Sci Transl Med 2016; 8:328rv324; PMID:26936508; http://dx.doi.org/10.1126/scitranslmed.aad7118
- Gerdes CA, Umana P. GA201: a novel humanized and glycoengineered anti-EGFR antibody–response. Clin Cancer Res 2014; 20; 1055; PMID:24536075; http://dx.doi.org/10.1158/1078-0432.CCR-13-2699
- Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, Votavova P, Pepper M, Kovar M, Garcia KC. Antibodies to Interleukin-2 Elicit Selective T Cell Subset Potentiation through Distinct Conformational Mechanisms. Immunity 2015; 42:815-25; PMID:25992858; http://dx.doi.org/10.1016/j.immuni.2015.04.015
- Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, Moraga I, Raeber ME, Bowman GR, Novick P et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature 2012; 484:529-33; PMID:22446627; http://dx.doi.org/10.1038/nature10975
- Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, Sheng DW, Liu XF, Sims PW, VanderVeen LA et al. NKTR-214, an Engineered Cytokine with Biased IL2 Receptor Binding, Increased Tumor Exposure, Marked Efficacy in Mouse Tumor Models. Clinical Cancer Research 2016; 22:680-90; PMID:26832745; http://dx.doi.org/10.1158/1078-0432.CCR-15-1631
- Gillies SD, Lan Y, Hettmann T, Brunkhorst B, Sun Y, Mueller SO, Lo KM. A low-toxicity IL-2-based immunocytokine retains antitumor activity despite its high degree of IL-2 receptor selectivity. Clin Cancer Res 2011; 17:3673-85; PMID:21531812; http://dx.doi.org/10.1158/1078-0432.CCR-10-2921
- Gillessen S, Gnad-Vogt US, Gallerani E, Beck J, Sessa C, Omlin A, Mattiacci MR, Liedert B, Kramer D, Laurent J et al. A phase I dose-escalation study of the immunocytokine EMD 521873 (Selectikine) in patients with advanced solid tumours. Eur J Cancer 2013; 49:35-44; PMID:22918078; http://dx.doi.org/10.1016/j.ejca.2012.07.015
- Carmenate T, Pacios A, Enamorado M, Moreno E, Garcia-Martinez K, Fuente D, Leon K. Human IL-2 mutein with higher antitumor efficacy than wild type IL-2. J Immunol 2013; 190:6230-38; PMID:23677467; http://dx.doi.org/10.4049/jimmunol.1201895
- Losey HC, Lopes JE, Dean RL, Flick H, Gomes M, Huff MR, Moroso RA, Sun L, Wang C, Waters JF et al. Utilizing a selective agonist of the intermediate-affinity IL-2 receptor with an improved pharmacokinetic profile leads to an enhanced immunostimulatory response with reduced toxicity in mice, Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA). Cancer Res 2015 2001; 75(15 Suppl), Abstract nr 3158.
- Sim GC, Liu C, Wang E, Liu H, Creasy C, Dai Z, Overwijk WW, Roszik J, Marincola FM, Hwu P et al. IL-2 variant circumvents ICOS+ regulatory T cell expansion and promotes NK cell activation. Cancer Immunol Res 2016; PMID:27697858; http://dx.doi.org/10.1158/2326-6066.CIR-15-0195
- Ghasemi R, Lazear E, Wang X, Arefanian S, Zheleznyak A, Carreno BM, Higashikubo R, Gelman AE, Kreisel D, Fremont DH et al. Selective targeting of IL-2 to NKG2D bearing cells for improved immunotherapy. Nat Commun 2016; 7:12878; PMID:27650575; http://dx.doi.org/10.1038/ncomms12878
- Boyman O, Surh CD, Sprent J. Potential use of IL-2/anti-IL-2 antibody immune complexes for the treatment of cancer and autoimmune disease. Expert Opin Biol Ther 2006: 6:1323-1331; PMID:17223740; http://dx.doi.org/10.1517/14712598.6.12.1323
- Arenas-Ramirez N, Zou C, Popp S, Zingg D, Brannetti B, Wirth E, Calzascia T, Kovarik J, Sommer L, Zenke G et al. Improved cancer immunotherapy by a CD25-mimobody conferring selectivity to human interleukin-2. Sci Transl Med 2016; 8:367ra166; PMID:27903862; http://dx.doi.org/10.1126/scitranslmed.aag3187
- Perk LR, Visser GW, Vosjan MJ, Stigter-van Walsum M, Tijink BM, Leemans CR, van Dongen GA. (89)Zr as a PET surrogate radioisotope for scouting biodistribution of the therapeutic radiometals (90)Y and (177)Lu in tumor-bearing nude mice after coupling to the internalizing antibody cetuximab. J Nucl Med 2005; 46:1898-1906; PMID:16269605
- Ring AM, Lin JX, Feng D, Mitra S, Rickert M, Bowman GR, Pande VS, Li P, Moraga I, Spolski R et al. Mechanistic and structural insight into the functional dichotomy between IL-2 and IL-15. Nat Immunol 2012; 13:1187-95; PMID:23104097; http://dx.doi.org/10.1038/ni.2449
- Waldmann TA. The shared and contrasting roles of IL2 and IL15 in the life and death of normal and neoplastic lymphocytes: implications for cancer therapy. Cancer Immunol Res 2015; 3:219-227; PMID:25736261; http://dx.doi.org/10.1158/2326-6066.CIR-15-0009
- Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, Fleisher TA, Dubois SP, Perera LP, Stewart DM et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2015; 33:74-82; PMID:25403209; http://dx.doi.org/10.1200/JCO.2014.57.3329
- Otegbeye F, Mackowski N, Ojo E, De Lima M, Wald DN. The IL-15 Super-Agonist ALT-803 Promotes Superior Activation and Cytotoxicity of Ex Vivo Expanded NK Cells Against AML. Blood 2015; 126:3090.
- Rosario M, Liu B, Kong L, Collins LI, Schneider SE, Chen X, Han K, Jeng EK, Rhode PR, Leong JW et al. The IL-15-Based ALT-803 Complex Enhances FcγRIIIa-Triggered NK Cell Responses and In Vivo Clearance of B Cell Lymphomas. Clin Cancer Res. 2016; 22:596-608; PMID:26423796; http://dx.doi.org/10.1158/1078-0432.CCR-15-1419
- Wong HC, Jeng EK, Rhode PR. The IL-15-based superagonist ALT-803 promotes the antigen-independent conversion of memory CD8(+) T cells into innate-like effector cells with antitumor activity. Oncoimmunology 2013; 2:e26442; PMID:24404427; http://dx.doi.org/10.4161/onci.26442
- Xu WX, Jones M, Liu B, Zhu XY, Johnson CB, Edwards AC, Kong L, Jeng EK, Han KP, Marcus WD et al. Efficacy and Mechanism-of-Action of a Novel Superagonist Interleukin-15: Interleukin-15 Receptor alpha Su/Fc Fusion Complex in Syngeneic Murine Models of Multiple Myeloma. Cancer Res 2013; 73:3075-86; PMID:23644531; http://dx.doi.org/10.1158/0008-5472.CAN-12-2357
- Miller JS, Cooley S, Holtan S, Arora M, Ustun C, Jeng E, Wong HC, Verneris MR, Wagner JE, Weisdorf DJ et al. 'First-in-human' phase I dose escalation trial of IL-15N72D/IL-15R alpha-Fc superagonist complex (ALT-803) demonstrates immune activation with anti-tumor activity in patients with relapsed hematological malignancy. Blood 2015; 126:1957; http://dx.doi.org/10.1182/blood-2015-02-625574
- Liu B, Kong L, Han K, Hong H, Marcus WD, Chen X, Jeng EK, Alter S, Zhu X, Rubinstein MP et al. A novel fusion of ALT-803 (IL-15 Superagonist) with an antibody demonstrates antigen-specific antitumor responses. J Biol Chem 2016; 291(46):23869-881; PMID:27650494; http://dx.doi.org/10.1074/jbc.M116.733600
- Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014; 515:572-6; PMID:25428506; http://dx.doi.org/10.1038/nature14001
- Lechner MG, Karimi SS, Barry-Holson K, Angell TE, Murphy KA, Church CH, Ohlfest JR, Hu P, Epstein AL. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J Immunother 2013; 36:477-89; PMID:24145359; http://dx.doi.org/10.1097/01.cji.0000436722.46675.4a
- Schliemann C, Palumbo A, Zuberbuhler K, Villa A, Kaspar M, Trachsel E, Klapper W, Menssen HD, Neri D. Complete eradication of human B-cell lymphoma xenografts using rituximab in combination with the immunocytokine L19-IL2. Blood 2009; 113:2275-83; PMID:19005180; http://dx.doi.org/10.1182/blood-2008-05-160747