
ABSTRACT
Glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR) is part of a system of signals involved in controlling T-cell activation. Targeting and agonizing GITR in mice promotes antitumor immunity by enhancing the function of effector T cells and inhibiting regulatory T cells. Here, we describe MEDI1873, a novel hexameric human GITR agonist comprising an IgG1 Fc domain, a coronin 1A trimerization domain and the human GITRL extracellular domain (ECD). MEDI1873 was optimized through systematic testing of different trimerization domains, aglycosylation of the GITRL ECD and comparison of different Fc isotypes. MEDI1873 exhibits oligomeric heterogeneity and superiority to an anti-GITR antibody with respect to evoking robust GITR agonism, T-cell activation and clustering of Fc gamma receptors. Further, it recapitulates, in vitro, several aspects of GITR targeting described in mice, including modulation of regulatory T-cell suppression and the ability to increase the CD8+:CD4+ T-cell ratio via antibody-dependent T-cell cytotoxicity. To support translation into a therapeutic setting, we demonstrate that MEDI1873 is a potent T-cell agonist in vivo in non-human primates, inducing marked enhancement of humoral and T-cell proliferative responses against protein antigen, and demonstrate the presence of GITR- and FoxP3-expressing infiltrating lymphocytes in a range of human tumors. Overall our data provide compelling evidence that MEDI1873 is a novel, potent GITR agonist with the ability to modulate T-cell responses, and suggest that previously described GITR biology in mice may translate to the human setting, reinforcing the potential of targeting the GITR pathway as a therapeutic approach to cancer.
Introduction
Promoting antitumor immunity through immune checkpoint blockade is now a recognized approach in the treatment of cancer, exemplified by antibodies directed against cytotoxic T-lymphocyte antigen (CTLA)-4, programmed death ligand 1 (PD-L1) and programmed cell death 1 (PD-1), which block inhibitory signals to T cells and are now approved for the treatment of multiple forms of cancer.Citation1-9
An alternative approach to promoting antitumor immunity is through the use of agonistic molecules that deliver signals through co-stimulatory receptors expressed on T cells. Glucocorticoid-induced tumor necrosis factor receptor (TNFR)-related protein (GITR), also known as tumor necrosis factor superfamily receptor 18 (TNFSFR18), activation-inducible TNFR family receptor (AITR), or CD357, is expressed constitutively on regulatory T cells (Tregs) and upregulated on effector T cells following activation. GITR ligand (GITRL) is expressed predominantly on macrophages, dendritic cells and B cells, and delivers signals through GITR that enhance effector T-cell proliferation and activation,Citation10 and has been shown to reprogram and inhibit Tregs through effects on FoxP3 expression.Citation11 Extensive studies in mice have illustrated the potential of targeting and agonism of GITR to promote antitumor immunity through their ability to drive these signals,Citation12 and through localized depletion of suppressive Tregs within the tumor.Citation13
The minimally active form of human (h)GITRL is trimeric in structure, and promotes multimerization of the GITR receptor, which, in turn, allows the recruitment of downstream signaling adaptors called TNFR-associated factors (TRAFs) that mediate the activation of signaling pathways such as nuclear factor kappa B (NFκB) and mitogen-activated protein kinase (MAPK).Citation10 Based on this model of GITR signaling, we reasoned that dimeric antibodies or Fc-fusion proteins, typically used as therapeutics, would represent sub-optimal GITR agonists, and that a minimally trimeric structure would confer optimal GITR agonism.
Here, we describe the generation and characterization of MEDI1873, a hexameric GITRL fusion protein (GITRL FP) designed on the principles of trimer stabilization, optimal clustering of GITR and Fc receptor engagement. Each MEDI1873 monomer comprises three distinct domains ():Citation1 a human immunoglobulin G1 (IgG1) fragment crystallizable (Fc) domain,Citation2 an isoleucine zipper trimerization domain from human coronin 1A,Citation3 and the hGITRL extracellular domain (ECD), with a point mutation (N161D) that eliminates the sole occupied glycosylation site. We demonstrate that MEDI1873 can be purified to homogeneity and is a significantly more potent agonist of hGITR than a monoclonal antibody. We show in human primary lymphocyte assays, in vitro, that this agonism results in enhanced T-cell activation and a reduction in regulatory T-cell suppression and that MEDI1873 is capable of mediating ADCC against activated T cells, resulting in an increased CD8+:CD4+ T-cell ratio. Further, we demonstrate that in non-human primates MEDI1873 increases T-cell activation and response to immunization in vivo. Finally, we detail flow cytometric and histological analyses of patient tumor samples that indicate the presence of GITR positive and FoxP3 positive tumor-infiltrating lymphocytes (TILs). Overall these data confirm the translation of previous mouse GITR biology to a human setting and support the targeting of GITR, via high-valency agonists, as a potential therapeutic approach for the treatment of cancer.
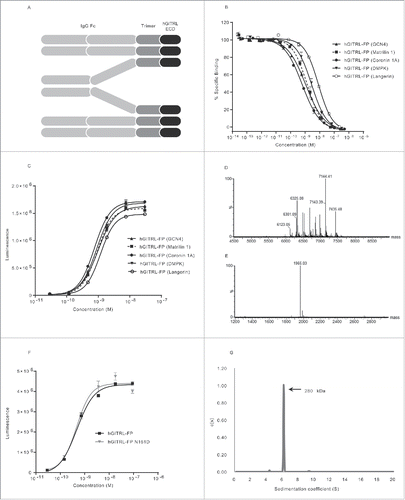
Figure 1. MEDI1873 is a homogenous hexameric GITRL fusion protein with potent agonistic activity. (A) Schematic of the domain structure of the GITRL FP, consisting of IgG Fc domain, trimerization domain and extracellular domain (ECD) of human GITRL. (B) Percentage-specific binding of GITRL to GITR in an HTRF-based assay in the presence and absence of GITRL FP variants. Error bars represent the standard error of the mean (SEM) from duplicates. (C) Luminescence signal downstream of an NFκB-luciferase reporter gene in hGITR-transfected Jurkat cells following addition of GITRL FP variants. Error bars represent the SEM from triplicates. (D and E) Combined, deconvoluted mass spectra for tryptic peptide (T42–43) which contains the N-glycosylation consensus sequence at position 161 of hGITRL for hGITRL(wt) FP (coronin 1A) (D) and hGITRL(N161D) FP (coronin 1A) (E). (F) Luminescence signal downstream of an NFκB-luciferase reporter gene in hGITR-transfected Jurkat cells following addition of hGITRL(wt) FP (coronin 1A) and hGITRL(N161D) FP (coronin 1A). Error bars represent SEM from triplicates. (G) Distribution of sedimentation coefficients (measured in Svedberg) for MEDI1873 following analytical ultracentrifugation analysis.
Materials and methods
Reagents and cell culture
Anti-hGITR antibody (A18) is a fully human IgG1 antibody identified through immunization of VelocImmune® mice followed by hybridoma generation and subsequent screening. Full details of its isolation are provided in supplementary methods. All cell culture was conducted at 37°C and 5% CO2 unless otherwise stated.
Recombinant GITRL FP protein cloning, expression, purification and quantification
DNA fragments encoding a haemagglutinin signal peptide sequence, an IgG1 or IgG4P Fc domain, a trimerization domain and human GITRL ECD (Geneart) were cloned into pDest12.2oriP (Thermo Fisher). CHO cells, grown in a proprietary chemically defined media similar to CD-CHO™ (Life Technologies Ltd, Paisley, UK), were transfected with the resulting plasmids and cultured for 8 d in shake flasks. Proteins were purified using MabSelect SuRe affinity chromatography (GE Healthcare) followed by size-exclusion chromatography (SEC). Alternatively cells were cultured for 10–12 d by fed-batch in wave bags and protein purified using MabSelectSure™ resin (GE Healthcare) followed by Hydroxyapatite Type 1 resin and elution with a salt gradient.
Receptor/ligand inhibition assay
Trimeric GITRL (with an N-terminal HA tag; RnD systems) and anti-HA-XL665 (Cisbio) diluted in assay buffer (100 mM HEPES pH7.4, 0.8 M potassium fluoride and 0.1% BSA) was added to all wells of a 384-well plate (Corning). GITRL FP molecules were diluted 5-fold in PBS/0.1% BSA and added to the assay plate in duplicate. Total binding and non-specific binding (NSB) were determined by addition of assay buffer or anti-GITR antibody MAB689 (R&D Systems), respectively, to control wells. Europium-conjugated GITR-Fc was added and plates incubated overnight. Fluorescence was measured using an EnVision plate reader (PerkinElmer). Emission ratios were expressed as a percentage of the control wells and data analyzed using a four-parameter logistic equation with GraphPad Prism (GraphPad Software, Inc.) to give apparent IC50 values.
GITR agonism NFκB reporter assay
Jurkat cells (ECACC) were transduced with lentivirus encoding an NFκB luciferase reporter cassette (Promega) and subsequently transfected or transduced with plasmid or lentivirus encoding hGITR or cynomolgus monkey (cyno) GITR. The resulting Jurkat reporter cells were mixed with test reagents in RPMI-1640 containing 10% fetal bovine serum (Gibco) and incubated for 3 h after which luciferase expression was detected by addition of Steady-Glo (Promega). In select assays, TILs were added to Jurkat reporter cells before the addition of test reagents. TILs were isolated from primary human tumors, following collagenase digestion, via positive selection with CD45 microbeads. Luminescence was measured using an EnVision plate reader. EC50 values were determined using non-linear regression analysis (log [agonist] vs. response, three-parameter fit curve) in GraphPad Prism.
Tryptic peptide mapping
Samples were incubated in denaturing buffer (8 M Guanidine, 130 mM Tris, 1 mM EDTA, 17 mM of Dithiothreitol, pH 7.6) for 30 min at 37°C before addition of 38.5 mM 2-iodoacetamide and incubation for 30 min at room temperature in darkness. Samples were buffer exchanged into 2 M Urea, 100 mM Tris Aminomethane, pH 7.6 and digested with Trypsin at an E:S ratio of 1:20 at 37°C for 4 h. The reaction was stopped by addition of 4% TFA, and samples analyzed by RP LC-MS using an Acquity UPLC coupled to a Synapt G2 QToF mass spectrometer (Waters). Digested samples were injected onto a BEH300 C18 column held at 55°C (Waters). Peptides were eluted at a constant flow rate of 0.2 mL/min using a 75 min binary gradient; solvent B was increased from 0% to 35%. Solvents A (water) and B (acetonitrile) were supplemented with 0.02% (v/v) trifluoroacetic acid. Spectra were acquired between 50 and 2500 m/z using a data-independent mode of acquisition. Low- and high-energy spectra were processed using MassLynx (Waters).
Analytical ultracentrifugation
Ultracentrifugation was performed in a ProteomeLab XL-A/XL-I analytical ultracentrifuge (rotor speed 42,000 rpm, running temperature 20°C) and monitored with absorbance at 280 nm. Data were analyzed and a c(s) profile generated using SEDFIT software.Citation14
Solution-based affinity determination
GITRL FP was titrated with recombinant hGITR and equilibrated overnight at 25°C. Samples were transferred to the KinExA 3200 (Sapidyne Instruments), temperature controlled at 25°C. Equilibrated mixtures were sampled using biotinylated human GITR bound to azlactone beads via streptavidin. DyLight 650 labeled Protein G’ (Sigma) was used for detection and data processed using KinExAPro software (version 3.6.8.).
Cell-based affinity determination
MEDI1873 was diluted in FACS buffer (eBiosciences) over a 10-fold dilution series added to 1×106 hGITR or cynoGITR-overexpressing Jurkat cells in duplicate and incubated at 4°C overnight with shaking. Cells were washed 3× with ice cold FACS buffer and resuspended in FACS buffer containing phycoerythrin-labeled goat anti-human Fcγ-specific secondary antibody (Jackson Immunoresearch). Plates were incubated for 40 min at 4°C, cells were washed as before and resuspended in FACS buffer before acquisition on a BD FACSCanto II (Becton-Dickinson). Flow Jo software (TreeStar) was used to gate live cells and to quantify the mean fluorescence intensity (MFI) of each sample. Prism software (GraphPad) was used to plot the measured MFI as a function of MEDI1873 concentration (pM). The resulting binding isotherm was analyzed with a non-linear regression model describing one-site (specific) binding.
FcγRIIIa affinity determination
Anti-histidine tag antibody (AbD Serotec) was immobilized using amine coupling chemistry onto a CM5 sensor chip (GE Healthcare) followed by the capture of histidine-tagged recombinant FcγRIIIa (158V). A dilution series of MEDI1873 or an IgG1 isotype control antibody were prepared in HBS-EP+ buffer and injected across the sensor surface, using a Biacore T100 (GE Healthcare), at a flow rate of 40 μL/minute for a contact time of 60 sec before allowing the samples to dissociate. Resultant sensorgrams were fitted onto a simple 1:1 bimolecular binding model to derive the kinetic parameters. The equilibrium dissociation constant (KD) was calculated as a ratio of rate of dissociation and the rate of association. The experiment was performed three times to obtain the standard deviation of KD.
T-cell co-stimulation assay
Human peripheral blood mononuclear cells (PBMCs) were isolated from leukocyte cones (supplied by NHS Blood and Transplant Service (NHSBT, UK) as anonymized samples from consenting donors) by layering over Ficoll-Paque Plus per manufacturer's instructions (GE Healthcare). CD3+ T cells were isolated from PBMCs by negative selection using a Robosep and Easysep CD3+ enrichment kit per manufacturer's instructions (Stem Cell). Isolated CD3+ T cells were resuspended in assay media (RPMI1640 Glutamax I culture media with 5% human AB serum and 1% penicillin/streptomycin) and added to six-well plates coated with 0.2 µg/mL anti-CD3 (OKT3, eBioscience). Cells were cultured for 4 d, harvested, resuspended in fresh media and cultured for two further days. Cells were harvested, resuspended in media and added to 96-well plates coated with 2 µg/mL anti-CD3 and MEDI1873, or control. Plates were incubated for 4 d, supplemented with 0.5 μCi of tritiated thymidine and incubated for a further 18 h, after which cells were harvested onto 96-well glass fiber plates using a cell harvester (Brandel), and thymidine incorporation quantitated by addition of Microscint 20 into each well followed by reading on a Topcount (PerkinElmer).
EC50 values were determined using non-linear regression analysis (log [agonist] vs. response, curve fit) in GraphPad Prism, version 5.01 (GraphPad Software). The bottom of the curve was fixed at the value observed for the isotype control. The top of the curve was fixed at the maximum observed value in the dose response.
Regulatory T-cell suppression assay
PBMCs were isolated as described above. Human CD4+ effector and Treg cells were isolated from PBMCs using a human Treg isolation kit per manufacturer's instructions (Life Technologies). Effector T cells were labeled using CellTrace™ CFSE cell proliferation kit per manufacturer's instructions (Life Technologies) and were co-cultured with Tregs at a 1:1 ratio in 96-well plates coated with 0.5 µg/mL anti-CD3 and MEDI1873 or control. Culture media was as per co-stimulation assays, but supplemented with 1 µg/mL anti-CD28 (CD28.2, eBiosciences). Plates were incubated for 4 d after which cells were washed once in PBS, stained with Fixable Viability Dye eFluor® 780 per manufacturer's instructions (eBiosciences), fixed by resuspension in CellFix™ (Becton Dickinson) and analyzed using a FACSCanto II flow cytometer. The percentage of effector T cells that had undergone cell division (CFSE low) was assessed using FlowJo software. Non-viable cells and Tregs (CFSE negative) were identified and excluded from the analysis.
Antibody-dependent cell cytotoxicity (ADCC) assay
Culture media for all stages was RPMI1640 supplemented with 10% FBS. CD3+, CD4+ and CD8+ human T cells or NK cells were isolated from heparin anti-coagulated whole blood (obtained under informed consent from the MedImmune Blood Donor Program) using RosetteSep per manufacturer's instructions (Stem Cell). T cells were activated by culture for 48 h in 96-well plates coated with 5 µg/mL anti-CD3 and anti-CD28. Activated T cells, and control T cells, were labeled with VyBrant DiO per manufacturer's instructions (Life Technologies). Labeled cells were mixed with NK cells at the effector to target ratio indicated and MEDI1873, IgG4 GITRL FP or control at the concentrations indicated. Plates were incubated for 14–24 h after which cells were removed, stained with fluorophore conjugated anti-CD4+ and anti-CD8+ antibodies, resuspended in FACs buffer containing propidium iodide and analyzed on an LSRII flow cytometer (BD Biosciences). Non-viable (propidium iodide-positive) cells among DiO-positive target cells were identified using FlowJo software. MEDI1873-specific cytotoxicity was calculated by subtracting the mean percentage of non-viable cells in the non-antibody-containing control wells from all other wells.
Flow cytometric assessment of GITR expression on TIL
Surgical resection samples from lung cancer patients were analyzed by Caprion Biosciences Inc. (Québec, Canada). This work was approved by the Comité éthique en recherche du Center Hospitalier de l'Université de Montréal and informed consent was obtained using informed consent forms. The tumor samples were fragmented using a scalpel and a single-cell suspension was obtained by tissue disaggregation in a Medimachine system (BD Biosciences), per manufacturer's instructions. Cell suspensions were filtered using a 100-µm pore size, washed in PBS and filtered again using a 40-µm pore size. Cells were then stained with fixable live/dead stain (Life Technologies) and fluorophore conjugated antibodies against CD4+, CD8+, CD3 (BD Biosciences), FoxP3 and GITR (Biolegend). Stained cells were acquired using a BD LSR Fortessa cytometer and resulting data analyzed using FlowJo.
Immunohistochemical assessment of GITR and FoxP3
Tissue samples from 76 cancer patients, with NSCLC, colorectal cancer (CRC), bladder cancer, or squamous cell cancer of the head and neck (SCCHN), were obtained from commercial sources (Asterand, ConversantBio, ProteoGenex, National Disease Research Interchange (NDRI) or ILSBio) and used in accordance with the ethical principles originating in the Declaration of Helsinki and in compliance with all national and local regulatory guidelines. Four micron tissue sections were cut from formalin-fixed paraffin-embedded (FFPE) samples and placed on StarFrost® microscope slides.
To assess GITR and FoxP3 expression, two separate (i.e., monoplex) immunohistochemical assays were developed using a rabbit anti-human TNFRSF18 (GITR) antibody (Sigma Aldrich, HPA-008025; 0.3 ug/mL, 20 min), and a rabbit anti-human FoxP3 monoclonal antibody (Spring Bioscience, SP97/M3974; 1:25, 32 min). Slides were stained using a Ventana Discovery Ultra IHC/ISH research slide staining system with a heated antigen retrieval pretreatment step (Cell Conditioner 1). Signal was detected using OmniMap anti-rabbit HRP and DAB chromogen substrate; and the slides were counterstained with hematoxylin. Human tonsil sections, incubated with anti-GITR, anti-FoxP3 or non-immune isotype antibody (Roche, 790–4795; rabbit monoclonal IgG), were included as controls, and performed as expected with GITR+ membrane staining seen on a subset of lymphocytes, FoxP3+ nuclear staining seen in a subset of lymphocytes, and no staining seen with the isotype antibody.
Stained sections were digitally scanned at 20× magnification using an Aperio ScanScope AT Turbo brightfield scanner (Leica Biosystems Inc., Buffalo Grove, IL) and each digital slide was viewed using Aperio ImageScope v12.1.0.5029. For each slide, 10 fields of view (FOVs) were chosen across areas of low, medium and high expression from the tumor center and invasive margin, as permitted, to represent the section-wide expression pattern. Areas of artifact, necrosis, peri-tumoral connective tissue and other non-tumoral tissues were avoided. The area of each FOV was measured at 20× magnification, a grid mask overlay was applied, and the number of marker-positive TILs enumerated. The 10 FOV areas and marker-positive TIL counts were summed and the number of marker-positive TILs/mm2 was calculated.
Pharmacodynamic study in cynomolgus monkeys
The study was conducted at Covance Preclinical Services GmbH (Münster, Germany), which is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. All procedures were in compliance with the German Animal Welfare Act and approved by the local Institutional Animal Care and Use Committee. The study was performed in line with relevant ECC and ICH guidelines.
Purpose bred, experimentally naive male cynomolgus monkeys (Macaca fascicularis) of Asian origin were used (five animals per group). Weight and age at study start ranged from 3.2 to 5.2 kg and 3.2 to 4.2 y, respectively. All animals were immunized on Day 1 of the study by intramuscular (IM) injection of 0.5 mL tetanus vaccine (Tetanol®, Novartis). MEDI1873 was administered at 1 or 10 mg/kg by intravenous (IV) bolus injection on Days 1, 3 and 5. Control group animals received vehicle (20 mM sodium phosphate, 230 mM sucrose, 0.02% polysorbate 80, pH 7.5) via the same dose route and schedule. MEDI1873 concentrations in serum samples were determined using an indirect enzyme-linked immunosorbent assay (ELISA) with a lower limit of quantitation (LLOQ) of 100 ng/mL. For determination of anti-tetanus toxoid IgM and IgG antibodies, 0.5 mL blood samples were collected from the cephalic vein on Day 1 (pre-dose), 2, 15, 22 and 31. These samples were allowed to coagulate at room temperature for a minimum of 30 min, and then centrifuged at 1500 × g for 10 min. Resultant serum was analyzed using total monkey IgM and IgG, or anti-tetanus toxoid IgM or lgG ELISA kits (α Diagnostic International). For flow cytometry analysis, 0.5 mL blood samples were collected from the cephalic vein on Day −21, −13 and −7, 1 (pre-dose), 3, 5, 9, 11, 15, 18, 22 and 31; details of the antibodies used are detailed in Table S1.
Statistical analysis
The extra sum of squares F test was used to compare EC50 values between different dose response curves for in vitro assays. A two-tailed unpaired T test was used to compare maximum signals between dose response curves in the reporter gene assay. Results and associated p values are reported in the text and associated figure legends.
One-way ANOVA, with Tukey's correction for multiple comparisons, was used to compare values, at the highest concentration used, between A18, MEDI1873 and isotype control in the primary T-cell proliferation assay. One-way ANOVA, with Dunnett's correction for multiple comparisons was used to compare treatment values to control values in assays of regulatory T-cell inhibition. Two-way ANOVA, with Dunnett's correction for multiple comparisons was used to compare treatment values to control values in ADCC assays. One-way ANOVA, with Dunnett's correction for multiple comparisons was used to compare treatment values to control values for the ex vivo Ki67 flow cytometric assay using cynomolgus monkey whole blood. In all cases, multiplicity adjusted p-values are reported in the associated text and/or figure legends.
Results
MEDI1873, an optimized hexameric GITRL FP, contains the human coronin 1A trimerization domain
Due to the limited number of hydrophobic residues in its trimer interface, hGITRL has a lower propensity to form stable trimers than other TNFR family members,Citation15,16 and consequently exists in a monomer-dimer-trimer equilibrium in solution. We sought to stabilize the hGITRL trimer, through the inclusion of different human trimeric coiled-coil domains, which were expected to stabilize the trimeric form based on previous studies.Citation15,17 Four coiled-coil sequences were selected following a search of the Protein Data Bank. The first two were the coiled-coil domains from human langerinCitation18 and myotonic dystrophy protein kinase (DMPK).Citation19 The second two were the human orthologues of coiled-coil domains from mouse coronin 1ACitation20 and chicken matrilin 1.Citation21 The sequences encoding each of the trimerization motifs (Table S2), in addition to the yeast GCN4 pII sequence, which has previously been used to stabilize GITRL,Citation15 were joined at the C terminus to the ECD of hGITRL and at the N terminus to a hIgG Fc domain using linker domains ( and Fig. S1). The combination of these three domains was predicted to result in the formation of hexameric oligomers, as has been demonstrated previously for a chicken tenascin C trimerization domain-containing GITRL FP,Citation17 and for the GCN4 pII variant (Fig. S2). The five GITRL FPs were expressed in CHO cells, purified to homogeneity and tested in a panel of in vitro assays (). The variant containing the coronin 1A trimerization domain gave the highest expression yield as well as significantly lower IC50 and EC50 values relative to the next most potent construct, containing GCN4, in the biochemical receptor/ligand competition assay (, 0.6 nM vs. 1.1 nM, p < 0.0001) and NFκB reporter assay (, 0.75 nM vs. 0.89 nM, p = 0.04), respectively. Consequently, this variant was taken forward for further characterization. Interestingly, the GITRL FP langerin variant had a significantly higher IC50 value (p < 0.0001) in the competition assay and a lower melting temperature compared with the other tested variants (Fig. S3); specifically, a 13-fold IC50 difference and a melting temperature of 47.5 vs. 64 °C compared with coronin 1A.
Table 1. Properties of hGITRL FP molecules with different trimerization domains.
GITRL activity in vitro is independent of glycosylation and Fc isotype
Peptide mapping of the GITRL FP demonstrated that the IgG Fc N-glycosylation site (N78) was glycosylated and that the carbohydrate structures were typical for IgG Fc regions expressed in CHO cells (data not shown). The GITRL ECD sequence contained two canonical N-glycosylation sites (N129 and N161) (Fig. S1), the glycosylation status of which has not been previously determined. Peptide mapping revealed that the N-glycosylation consensus site at N161 (), but not N129 (Fig. S4A), was occupied with primarily neutral and charged, biantennary complex-type oligosaccharides.
The N161 residue of hGITRL ECD was replaced with an aspartate to eliminate the consensus N-glycosylation site. Peptide mapping confirmed the N161D amino acid substitution and no N-linked glycans were detected at D161 (). An assessment of the potency of the wild-type (N161) and mutant (D161) proteins with respect to binding (Fig. S4B) and GITR agonism (), revealed no significant differences (p = 0.85), and no impact on expression yields was observed (data not shown). Based on these data the D161 mutant form of the GITRL ECD was incorporated into an IgG1 GITRL FP designated MEDI1873.
The IgG1 Fc domain was selected following mouse studies conducted by ourselvesCitation35 and othersCitation13 that demonstrated that the enhanced activity of GITR targeting agents was dependent upon maximizing Fc gamma receptor engagement. The Jurkat-based NFκB reporter gene assay was used to confirm that no significant differences in potency were observed between MEDI1873 and an IgG4 Fc-containing GITRL FP (EC50 0.2 vs. 0.3 nM, respectively) (Fig. S5).
The anticipated quaternary structure of MEDI1873, a hexamer with a predicted mass of ∼283 kDa, was confirmed by the orthogonal techniques of analytical ultracentrifugation (AUC) and size-exclusion chromatography-multi-angle light scattering (SEC-MALS) analysis. Both confirmed a high degree of homogeneity (95%) and a molecular weight of ∼280 kDa, consistent with a hexameric structure ( and Fig. S6).
MEDI1873 binds avidly to GITR and FcɣRIIIA to effectively enhance GITR agonism
A kinetic exclusion assay (KinExA) estimated a solution KD of 82 nM for the binding of MEDI1873 to recombinant hGITR (), while a cell-based measure of affinity, encompassing any avidity affects due to receptor clustering, estimated an apparent equilibrium binding constant (KDapp) of 12.8 pM (). MEDI1873 demonstrated concentration-dependent agonism of hGITR in the context of a Jurkat NFκB reporter gene assay, with an average EC50 of 0.4 nM across four independent assays (Table S3). In contrast, a monoclonal antibody directed against GITR (A18) demonstrated a significantly (p < 0.0001) reduced EC50 of 2.2 nM, and a significant (p < 0.0001), 50-fold, reduced maximal signal ().
Figure 2. In vitro activity of MEDI1873. (A) Binding of MEDI1873 to recombinant hGITR in a solution-based affinity measurement assay using KinExA. (B) Binding of MEDI1873 to hGITR-transfected Jurkats, as measured by mean fluorescence intensity (MFI) in a FACS-based assay. The binding isotherm was analyzed and dissociation constant estimated using a non-linear regression model describing one-site (specific) binding. (C) Luminescence signal downstream of an NFκB-luciferase reporter gene in hGITR-transfected Jurkat cells following addition of MEDI1873, anti-GITR (A18) or appropriate isotype matched controls. Error bars represent the SEM from triplicates. p value represents results of an unpaired T test between values at the highest concentrations of MEDI1873 and A18. (D) Sensorgrams showing MEDI1873 binding to hFcgRIIIA-158V in an SPR-based assay. Data were fitted using a simple 1:1 bimolecular binding model to derive the kinetic parameters. (E) Luminescence signal (RLU) downstream of an NFκB-luciferase reporter gene in hGITR-transfected Jurkat cells following addition of MEDI1873, or appropriate isotype matched control, in the presence or absence of CD45+ TIL from primary human tumors. Error bars represent the SEM from quadruplicates. p value represents results of an unpaired T test between values at the highest concentration of MEDI1873 in the presence and absence of CD45+ TIL.
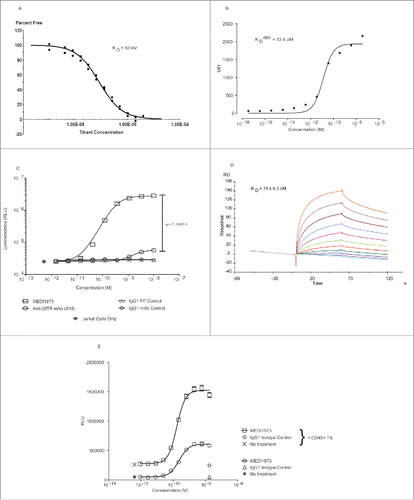
Surface plasmon resonance confirmed that the IgG1 Fc of MEDI1873 could bind to FcγRs, specifically FcγRIIIA, and that, as might be predicted from the increased valency, this binding occurred with a higher affinity than that of a standard IgG1 monoclonal antibody; KD of 75 nM for MEDI1873 compared with 175 nM for an IgG1 isotype control antibody ( and Fig. S7).
The level of cross-linking via FcγRs to further increase the potency of MEDI1873 was tested by the addition of CD45+ cells, extracted from primary human tumor tissue, to the Jurkat NFκB reporter gene assay system. The addition of these cells resulted in a significant, 1.4-fold, increase in MEDI1873 potency (EC50 of 231 pM vs. 165 pM, p = 0.0004) and a significant, 2.5-fold, increase in the maximum observed signal (1.5 × 107 vs. 0.6 × 107, p < 0.0001) ().
MEDI1873 enhances T-cell proliferation and inhibits suppression by regulatory T cells
The ability of MEDI1873 to enhance the proliferation of human T cells was assessed in a restimulation assay in which CD3+ T cells were activated with anti-CD3, subsequently rested and then restimulated. In this context, both MEDI1873 and A18, were able to minimally enhance the proliferation of T cells. The maximal proliferative signal reached by the anti-GITR antibody A18 (7290) was not significantly (p = 0.09) greater than that seen with isotype control (4272). In contrast, MEDI1873 generated a significantly (p = 0.03) greater proliferative signal (8474) as compared with isotype control, though this was not significantly different to A18 (p = 0.6). Similarly the EC50s for MEDI1873 and A18 (4.7 and 7.9 nM, respectively) were not significantly different (p = 0.5). When cross-linked via plate binding the EC50 of MEDI1873 was similar to the soluble form (2.6 nM), and not significantly different to cross linked A18 (p = 0.8). However, the maximal proliferative signal generated increased significantly (p = 0.0009) by 2.5-fold to 21,048. In contrast, the activity of the anti-GITR antibody A18 was not notably affected ().
Figure 3. Functional outcome of MEDI1873 in primary T cells. (A) Thymidine incorporation in CD3+ human T cells restimulated with anti-CD3 and anti-CD28 antibodies in the presence of MEDI1873, anti-GITR (A18) or isotype control in solution of plate bound. Error bars represent the SEM from triplicates. (B) Expression of GITR (solid histograms) on CD4+ FoxP3+ Tregs (purple), CD4+ FoxP3− helper cells (red) and CD8+ cytotoxic T cells (blue) within the TIL of a NSCLC tumor. Semi-transparent, dotted line histograms represent isotype control staining. Data is representative of three independent samples. (C) Percentage lysis of anti-CD3 and anti-CD28 activated CD3+ T cells by allogeneic NK cells in the presence of MEDI1873 and IgG4 GITRL FP. Error bars represent the SEM from triplicates. Two-way ANOVA, with Dunnett's correction for multiple comparisons was used to compare treatment values to control values. ****p < 0.0001, **p = 0.0015, +p = 0.0138, ns = not significant. (D) Percentage of CD4+ and CD8+ T cells present within the total CD3+ population following treatment with no drug or with MEDI1873 at 3.6 nM at the end of the assay illustrated in (C). (E) Percentage of divided CD4+ FoxP3− effector T cells, measured via CFSE dilution, following activation with anti-CD3 and anti-CD28 in the presence or absence of CD4+ FoxP3+ Tregs with or without MEDI1873 at the concentrations indicated. Error bars, where present, represent the SEM from duplicates. One-way ANOVA, with Dunnett's correction for multiple comparisons was used to compare treatment values to control values. p values, where significant are indicated as follows ***p = 0.0005, **p < 0.006.
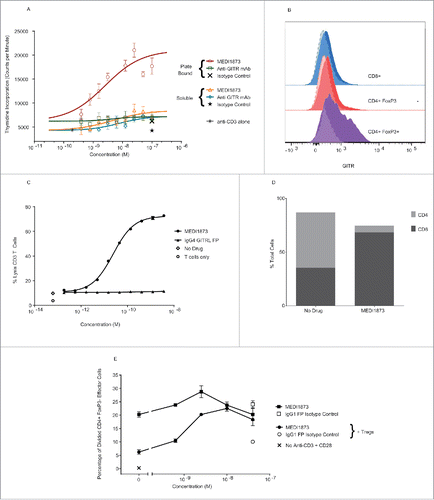
While effector T-cell activation is a critical component of the mechanism of action for GITR targeting agents, a degree of the activity seen for these agents in mice appears to be due to their ability to deplete intratumoral Tregs in an Fc-dependent manner due to the increased expression of GITR on Tregs vs. other T cells. A similar expression hierarchy to that seen in mice was also observed on ex-vivo T cells isolated from NSCLC tissue (), and on in vitro activated human T cells (Fig. S8), with human CD4+ FoxP3+ Tregs demonstrating notably higher levels of expression than CD8+ or CD4+ FoxP3− cells. In a flow cytometric assay of NK cell-mediated ADCC, MEDI1873 was able to mediate a significant concentration-dependent increase in the lysis of activated CD3+ T cells at every concentration tested; in contrast, an IgG4-Fc GITRL FP showed only very minimal, but significant, activity at the highest concentration tested (). A higher ratio of CD8+:CD4+ cells was observed at the end of the assay (10.6) as compared with the beginning of the assay (0.68) (), suggesting preferential depletion of GITR high cells.
A potential role for GITR in overcoming the suppressive function of Tregs, either through direct effects on Tregs or induced resistance in effector T cells, has been widely reported in mice.Citation22-25 In suppression assays conducted using primary human regulatory and helper CD4+ T cells, the addition of Tregs significantly (p = 0.0003) inhibited helper T-cell proliferation. MEDI1873 did not significantly increase the proliferation of helper T cells alone at any concentration tested, but at concentrations at, or above, 2.5 nM was able to significantly increase the proliferation of helper T cells in the presence of Tregs (p = 0.06 to 0.0005 depending on concentration), such that proliferation was comparable to that observed in the absence of Tregs ().
MEDI1873 induces proliferation of circulating T-cell subsets and enhances humoral immune responses in cynomolgus monkeys
To establish in vivo proof of mechanism and identify candidate pharmacodynamic (PD) biomarkers, species cross-reactivity of MEDI1873 was investigated. As anticipated MEDI1873 did not bind mouse or rat GITR in vitro (data not shown), but did bind to cynomolgus monkey (cyno) GITR, with a KD of 107 nM, and demonstrated an EC50 of 422 pM in a Jurkat NFκB reporter gene assay measuring agonism of cyno GITR (). These values are comparable to the affinity (82 nM) and potency (400 pM) observed for human GITR, supporting the validity of the cynomolgus monkey as a relevant species for assessing the in vivo PD of MEDI1873.
Figure 4. Functional activity of MEDI1873 in vivo. (A) Luminescence signal (RLU) downstream of an NFκB-luciferase reporter gene in cynomolgus monkey GITR-transfected Jurkat cells following addition of MEDI1873, or appropriate isotype matched control. Error bars represent the SEM from triplicates. (B) The pharmacokinetic profile of MEDI1873 in cynomolgus monkeys upon IV bolus administration at 1 or 10 mg/kg on Day 1, 3 and 5. (C) Serum tetanus toxoid-specific IgM in cynomolgus monkeys treated as described in (B). (D) Serum tetanus toxoid-specific IgG in cynomolgus monkeys treated as described in (B). (E) percentage of circulating CD3+CD4+CD95highCD28+/dim/− cells expressing Ki67 in cynomolgus monkeys treated as described in (B). Arrows indicate days of administration. Stars indicate study days on which statistical significance (p < 0.05; ANOVA) compared with control was achieved.
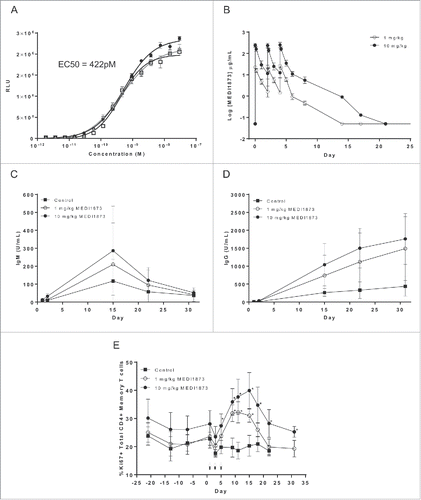
In a subsequent in vivo study, cynomolgus monkeys (5 males/group) were immunized intramuscularly with tetanus vaccine on Day 1, followed by IV bolus administration of 0 (vehicle control), 1 or 10 mg/kg of MEDI1873 on Day 1, 3 and 5. The observed PK exposure of MEDI1873 () was approximately dose proportional across the 1–10 mg/kg dose groups, with a terminal half-life ranging between 0.66 and 0.92 d.
With respect to humoral immunity, treatment with MEDI1873 was associated with a trend toward an increase in the levels of circulating anti-tetanus IgM and IgG antibodies (). Although differences between the two MEDI1873-treated groups were not statistically significant, there was evidence of an apparent dose-relationship, with average increases in anti-tetanus toxoid IgM and IgG antibodies generally being higher in animals dosed at 10 compared with 1 mg/kg MEDI1873.
As a measure of PD, upregulation of the proliferation marker Ki67 in defined T-cell subpopulations was monitored by flow cytometry until Day 31. A substantial, statistically significant (p < 0.05) increase in Ki67 expression was observed in circulating CD3+CD4+CD95highCD28+/dim/− memory T cells, with the response peaking between Day 10 and 15 and then declining (). An apparent dose dependency in response was observed with the magnitude of the Ki67 increase being greater in animals treated with 10 compared with 1 mg/kg of MEDI1873. Similar, though less robust, changes in Ki67 expression were also observed in circulating CD8+ memory T cells (data not shown). There was evidence of a PK/PD disconnect, with MEDI1873 being largely cleared from the systemic periphery following both the 1 and 10 mg/kg dose at the time of the peak Ki67 response. Using PK/PD modeling, the PK of MEDI1873 could be adequately described using a two-compartment model with first-order elimination from the central compartment, with the PD response being described by a T-cell lifespan-based indirect response model (data not shown). Overall, the data generated in cynomolgus monkeys are indicative of MEDI1873 being pharmacodynamically active in this species, and are supportive of its ability to enhance cellular and humoral immunity.
GITR and FoxP3 expression is frequent and highly correlated in human tumors
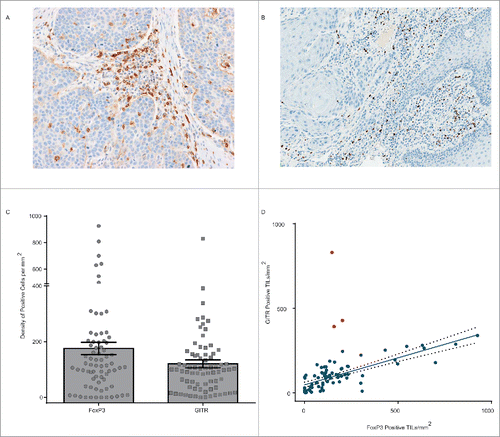
Our initial flow cytometry assessments () indicated that the expression of GITR on human NSCLC TIL demonstrates a similar pattern to that observed in mouse tumor models, with expression being highest on FoxP3+, CD4+ Tregs. To assess the expression of GITR and FoxP3, and their relationship to one another, in a broader range of human tumors, and across a larger sample set, we developed optimized IHC methods and used them to assess the expression of GITR and FoxP3 () in 76 sections of tumor tissue from squamous NSCLC, CRC, SCCHN and bladder cancer patients. Nuclear FoxP3 expression was identified on TIL in 71 of 76 (93%) samples assessed and membranous GITR was detected on TIL in 75 of 76 (99%) samples. The density of FoxP3 positive TIL varied considerably from 1.1 to 925.7 positive cells per mm2, with a mean of 175.5; the density of GITR-positive TIL showed similar variation, from 0.6 to 830.1 positive cells per mm2, with a mean of 120.4 (). The level of GITR expression did not vary significantly across tumor types (Fig. S9). In contrast, the level of FoxP3 expression in SCCHN samples was significantly higher than that in NSCLC and bladder cancer samples (Fig. S9). Assessment of the correlation between FoxP3 and GITR expression in sections from the same sample () indicated that, in general, samples with higher FoxP3 also had higher GITR (Spearman correlation coefficient 0.7). However, this was not always the case, and three samples in particular demonstrated high levels of GITR in the presence of limited levels of FoxP3. In the presence of these samples, the linear correlation between GITR and FoxP3 was modest (Pearson correlation coefficient 0.5), however, when these outlying samples were excluded, the linear correlation became significantly stronger (Pearson correlation coefficient 0.8).
Figure 5. Relationship between GITR and FoxP3 expression in primary human tumors. (A) GITR positive tumor-infiltrating lymphocytes, at 20× magnification, in squamous cell cancer of the head and neck identified by immunohistochemical staining. (B) FoxP3 positive tumor-infiltrating lymphocytes, at 20× magnification, in squamous cell cancer of the head and neck identified by immunohistochemical staining. (C) Density of FoxP3 (left) and GITR (right) expressing cells per mm3, assessed by manual counting, of sections from 76 human FFPE tumor samples. Error bars represent the SEM. (D) Correlation between the density of GITR positive and FoxP3 positive cells within the samples illustrated in ©. Red dots indicate outlying samples with respect to the overall pattern of correlation.
Discussion
Targeting of GITR in mice has been shown to promote potent antitumor immunity in a range of models through the activation of T cells,Citation12 and the localized modulation and/or depletion of intratumoral Tregs.Citation13,26 Based on this encouraging data, we set out to design an effective agonist of hGITR for use as a potential cancer immunotherapy. Mouse GITR has been shown to signal effectively as a dimer,Citation16,27,28 while, in contrast, human GITR requires trimerization. Given this difference, we reasoned that while a bivalent antibody, such as the anti-mouse GITR antibody DTA-1, might be a highly effective agonist in mice, a molecule with a trimer-based structure would be required for optimal agonism of hGITR.
To achieve such a structure, we designed a series of fusion proteins, composed of the ECD of hGITRL linked, at its N-terminus, to a trimerization domain and a human IgG Fc domain. By combining these domains, a hexameric quaternary structure is predicted, due to Fc-mediated dimerization of stabilized GITRL trimers. We conducted a systematic comparison of four human-derived trimerization domains, and selected the coronin 1A-containing molecule for further characterization based on superior expression yield, and potency. Interestingly, the langerin variant showed a higher IC50 compared with the coronin 1a variant, in addition to a significantly lower melting temperature. It could be hypothesized that the reduced thermal stability is a result of partial protein unfolding, which affects engagement of the langerin GITRL FP with its receptor. The differences in activity of the variants cannot be readily explained by solely taking into account the strength of the coiled-coil interaction as determined by the PROCOIL algorithm.Citation29 This suggests that the context in which a coiled-coil domain is placed influences its strength and stability, and that empirical testing of multiple variants remains a critical process to arrive at an optimal multivalent molecule. We report here, for the first time, that of the two putative N-linked glycosylation sites within the hGITRL (N129KS and N161NT), only N161 is occupied; being heterogeneously glycosylated with neutral and charged, biantennary complex-type oligosaccharides. Previous studiesCitation15 have indicated that N161 comprises part of the GITRL-GITR interface and when mutated to an alanine, together with L159, the binding of GITRL to GITR is ablated. However, when we mutated N161 to the charged variant, aspartate, we did not observe any impact on GITR binding or agonism, suggesting either a critical role for L159 or the conservation of size at residue 161 in preserving receptor binding. Given the potential for variation in glycan composition during cell culture that may impact manufacturability, and for terminal glycans such as mannose to impact pharmacokinetics through binding to mannose receptors,Citation30 this glycosylation site was removed, with no impact on activity.
Assessment of the agonistic potency and manufacturability revealed no significant differences between IgG1 and IgG4 containing GITRL FPs. However, our own preclinical studies in mice using surrogate mouse GITRL FPsCitation35 together with other published studies,Citation13 indicate a role for FcγR engagement and Fc-mediated depletion of Tregs in maximizing the activity of GITR targeting agents. Given this potentially important biology, an IgG1 Fc domain containing GITRL FP was selected for further development, and designated MEDI1873.
In keeping with our initial hypothesis with respect to the optimal format for GITR agonists, MEDI1873 demonstrated significantly greater agonistic potential than A18, an anti-GITR antibody, in reporter gene assays of GITR signaling. Previous studies in mice have indicated the critical nature of higher order clustering for effective GITR agonism in primary cells. In keeping with this finding, when present in solution both MEDI1873 and A18 had limited effects on the proliferation of primary human T cells, which express far less GITR than the transfected Jurkat lines used in the reporter assay. However, when cross-linked MEDI1873 dramatically enhanced T-cell proliferation, while A18 did not, suggesting that the increased valency, and/or the natural ligand structure, of MEDI1873 provides for more effective GITR agonism.
The T-cell co-stimulatory properties of MEDI1873 were also demonstrated in vivo in a PD study in cynomolgus monkeys. In this study, MEDI1873 demonstrated the ability to enhance proliferation of select T-cell subpopulations as well as antibody production in response to a protein antigen. Importantly, an apparent dose-relationship in these responses was observed suggesting that these endpoints may have utility as PD markers to confirm proof of mechanism and establish pharmacokinetic/PD relationships in humans; as has been seen for agonists of similar pathways previously.Citation31
As well as a role in activating T cells, GITR has been shown to both modulate the function of Tregs and to mediate their selective depletion. The latter property is dependent, in mice, upon the differential expression of GITR observed in Tregs vs. effector.Citation13 We observed a similar hierarchy of GITR expression in human T-cell subsets within NSCLC tumors and showed that MEDI1873 is capable of depleting GITR expressing T cells in vitro leading to an increased CD8+:CD4+ ratio within cultures, suggesting that the biology previously only observed in mice may translate to humans. Further, we showed that MEDI1873 has increased affinity for FcγRIIIA compared with an IgG1 mAb, and as such may be a more effective Treg-depleting agent. The ability of MEDI1873 to modulate Treg function was assessed in a suppression assay, in which MEDI1873 was able to overcome the suppressive effects of Tregs on effector T-cell proliferation. MEDI1873 demonstrates minimal ability to stimulate effector T-cell proliferation when given in the absence of Tregs in this particular assay, given the lack of prior stimulation provided to the cells. This suggests that the reduction in Treg suppression observed is at least in part due to direct effects on Treg function, however, further assays are required to firmly conclude this.
While the expression of GITR on intratumoral T cells, and particularly Tregs, is well established in mice, a greater understanding of these same expression patterns in cancer patients with different tumor types could provide useful insights with respect to where GITR targeting agents might be most effective. Since no such assessment has been undertaken previously we assessed 76 primary human tumor samples by IHC for expression of both GITR and FoxP3. We found that the majority of tumors demonstrated GITR and FoxP3 staining and that the two were highly correlated in most cases (Pearson correlation coefficient 0.8). No significant difference in GITR expression was observed across the four tumor types tested; however, SCCHN demonstrated a significantly higher expression of FoxP3 than either NSCLC or bladder cancer samples. This result suggests that Tregs may play a more dominant role in the setting of SCCHN. The use of individual IHC stains on separate sections precludes us from saying for certain whether GITR and FoxP3 are co-expressed on Tregs, although previous studiesCitation32-34 would support this conclusion. In general, we observed a higher frequency of FoxP3 positive as compared with GITR positive TIL in most tumors, suggesting that if GITR is expressed on Tregs then it is only a sub-set. In addition, the presence of three outlying samples that demonstrate a higher density of GITR positive as compared with FoxP3 positive TIL, suggests that GITR is not restricted to only Tregs, but is expressed on tumor-infiltrating effector cells as well. This conclusion is supported by our flow cytometric assessment of expression on T cells within NSCLC TIL, which showed predominant expression of GITR on Tregs, but did indicate some level of expression on CD4+ FoxP3− and CD8+ T cells. Ultimately, assessment of further tumor samples by flow cytometry or multiplex immunohistochemical/immunofluorescent approaches will be required to fully elucidate the expression pattern of GITR within TIL from different cancer types. However, once established, this data might inform as to the dominant mechanism, effector cell activation vs. Treg modulation, for a GITR targeting agent in different settings, and potentially inform optimal combination partners in each setting.
Overall our data support the conclusion that GITR agonists with higher valency, such as MEDI1873, show improved potency as compared with antibodies in human systems. Further, the totality of in vitro and in vivo pharmacology, together with the flow cytometry and IHC data on patient samples, support the conclusion that the majority of biology observed in mice appears to translate to human and non-human primate systems, and that targeting of GITR in human tumors has the potential to both co-stimulate effector T cells and modulate Tregs. Based on these data, this novel hexameric GITR agonist is currently being assessed in a phase 1 clinical study in patients with advanced solid tumors (NCT02583165).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Confidentiality statement
All authors are current or former salaried employees of MedImmune Ltd or MedImmune LLC (a member of the AstraZeneca Group) and have/had stock or stock options in AstraZeneca, the parent company of MedImmune.
KONI_A_1280645_supplementary_data.zip
Download Zip (960.4 KB)Acknowledgments
The authors would like to thank the following: Janette Dillon and Catherine Huntington for cell line generation, the Biologics Expression team for small scale protein expression and purification, the Early Expression Supply and Purification Process Sciences teams for large-scale expression and purification, respectively, and the DNA Chemistry and Core Tissue Culture teams. The authors would also like to acknowledge Caprion Biosciences Inc. (Québec, Canada) for support in the generation of TIL-based flow cytometry data.
References
- Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med 2015; 373(17):1627-39; PMID:26412456; http://dx.doi.org/10.1056/NEJMoa1507643
- Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med 2015; 373(2):123-35; PMID:26028407; http://dx.doi.org/10.1056/NEJMoa1504627
- Cameron F, Whiteside G, Perry C. Ipilimumab: first global approval. Drugs 2011; 71(8):1093-104; PMID:21668044; http://dx.doi.org/10.2165/11594010-000000000-00000
- Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, Molina J, Kim JH, Arvis CD, Ahn MJ et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 2015; 387(10027):1540-50; PMID:26712084; http://dx.doi.org/10.1016/S0140-6736(15)01281-7
- Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 2015; 373(19):1803-13; PMID:26406148; http://dx.doi.org/10.1056/NEJMoa1510665
- Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, Hodi FS, Schachter J, Pavlick AC, Lewis KD et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol 2015; 16(8):908-18; PMID:26115796; http://dx.doi.org/10.1016/S1470-2045(15)00083-2
- Rizvi NA, Mazieres J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, Horn L, Lena H, Minenza E, Mennecier B et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol 2015; 16(3):257-65; PMID:25704439; http://dx.doi.org/10.1016/S1470-2045(15)70054-9
- Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O'Donnell PH, Balmanoukian A, Loriot Y et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016; 387(10031):1909-20; PMID:26952546; http://dx.doi.org/10.1016/S0140-6736(16)00561-4
- Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr, Lao CD et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015; 16(4):375-84; PMID:25795410; http://dx.doi.org/10.1016/S1470-2045(15)70076-8
- Clouthier DL, Watts TH. Cell-specific and context-dependent effects of GITR in cancer, autoimmunity, and infection. Cytokine Growth Factor Rev 2014; 25(2):91-106; PMID:24484736; http://dx.doi.org/10.1016/j.cytogfr.2013.12.003
- Schaer DA, Budhu S, Liu C, Bryson C, Malandro N, Cohen A, Zhong H, Yang X, Houghton AN, Merghoub T et al. GITR pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability. Cancer Immunol Res 2013; 1(5):320-31; PMID:24416730; http://dx.doi.org/10.1158/2326-6066.CIR-13-0086
- Schaer DA, Murphy JT, Wolchok JD. Modulation of GITR for cancer immunotherapy. Curr Opin Immunol 2012; 24(2):217-24; PMID:22245556; http://dx.doi.org/10.1016/j.coi.2011.12.011
- Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, Wilson NS, Dranoff G, Brogdon JL. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med 2013; 210(9):1685-93; PMID:23897982; http://dx.doi.org/10.1084/jem.20130573
- Dam J, Schuck P. Calculating sedimentation coefficient distributions by direct modeling of sedimentation velocity concentration profiles. Methods Enzymol 2004; 384:185-212; PMID:15081688; http://dx.doi.org/10.1016/S0076-6879(04)84012-6
- Chattopadhyay K, Ramagopal UA, Mukhopadhaya A, Malashkevich VN, Dilorenzo TP, Brenowitz M, Nathenson SG, Almo SC. Assembly and structural properties of glucocorticoid-induced TNF receptor ligand: Implications for function. Proc Natl Acad Sci U S A 2007; 104(49):19452-7; PMID:18040044; http://dx.doi.org/10.1073/pnas.0709264104
- Zhou Z, Song X, Berezov A, Zhang G, Li Y, Zhang H, Murali R, Li B, Greene MI. Human glucocorticoid-induced TNF receptor ligand regulates its signaling activity through multiple oligomerization states. Proc Natl Acad Sci U S A 2008; 105(14):5465-70; PMID:18378892; http://dx.doi.org/10.1073/pnas.0711350105
- Wyzgol A, Muller N, Fick A, Munkel S, Grigoleit GU, Pfizenmaier K, Wajant H. Trimer stabilization, oligomerization, and antibody-mediated cell surface immobilization improve the activity of soluble trimers of CD27L, CD40L, 41BBL, and glucocorticoid-induced TNF receptor ligand. J Immunol 2009; 183(3):1851-61; PMID:19596991; http://dx.doi.org/10.4049/jimmunol.0802597
- Feinberg H, Powlesland AS, Taylor ME, Weis WI. Trimeric structure of langerin. J Biol Chem 2010; 285(17):13285-93; PMID:20181944; http://dx.doi.org/10.1074/jbc.M109.086058
- Garcia P, Ucurum Z, Bucher R, Svergun DI, Huber T, Lustig A, Konarev PV, Marino M, Mayans O. Molecular insights into the self-assembly mechanism of dystrophia myotonica kinase. FASEB J 2006; 20(8):1142-51; PMID:16770013; http://dx.doi.org/10.1096/fj.05-5262com
- Kammerer RA, Kostrewa D, Progias P, Honnappa S, Avila D, Lustig A, Winkler FK, Pieters J, Steinmetz MO. A conserved trimerization motif controls the topology of short coiled coils. Proc Natl Acad Sci U S A 2005; 102(39):13891-6; PMID:16172398; http://dx.doi.org/10.1073/pnas.0502390102
- Dames SA, Kammerer RA, Wiltscheck R, Engel J, Alexandrescu AT. NMR structure of a parallel homotrimeric coiled coil. Nat Struct Biol 1998; 5(8):687-91; PMID:9699631; http://dx.doi.org/10.1038/90444
- Stephens GL, McHugh RS, Whitters MJ, Young DA, Luxenberg D, Carreno BM, Collins M, Shevach EM. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol 2004; 173(8):5008-20; PMID:15470044; http://dx.doi.org/10.4049/jimmunol.173.8.5008
- Ephrem A, Epstein AL, Stephens GL, Thornton AM, Glass D, Shevach EM. Modulation of Treg cells/T effector function by GITR signaling is context-dependent. Eur J Immunol 2013; 43(9):2421-9; PMID:23722868; http://dx.doi.org/10.1002/eji.201343451
- Ji HB, Liao G, Faubion WA, Abadia-Molina AC, Cozzo C, Laroux FS, Caton A, Terhorst C. Cutting edge: the natural ligand for glucocorticoid-induced TNF receptor-related protein abrogates regulatory T cell suppression. J Immunol 2004; 172(10):5823-7; PMID:15128759; http://dx.doi.org/10.4049/jimmunol.172.10.5823
- Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol 2002; 3(2):135-42; PMID:11812990; http://dx.doi.org/10.1038/ni759
- Schaer DA, Budhu S, Liu C, Bryson C, Malandro N, Cohen A, Zhong H, Yang X, Houghton AN, Merghoub T et al. GITR pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability. Cancer Immunol Res 2013; 1(5):320-31; PMID:24416730; http://dx.doi.org/10.1158/2326-6066.CIR-13-0086
- Chattopadhyay K, Ramagopal UA, Nathenson SG, Almo SC. 1.8 A structure of murine GITR ligand dimer expressed in Drosophila melanogaster S2 cells. Acta Crystallogr D Biol Crystallogr 2009; 65(Pt 5):434-9; PMID:19390148; http://dx.doi.org/10.1107/S0907444909005721
- Chattopadhyay K, Ramagopal UA, Brenowitz M, Nathenson SG, Almo SC. Evolution of GITRL immune function: murine GITRL exhibits unique structural and biochemical properties within the TNF superfamily. Proc Natl Acad Sci U S A 2008; 105(2):635-40; PMID:18182486; http://dx.doi.org/10.1073/pnas.0710529105
- Mahrenholz CC, Abfalter IG, Bodenhofer U, Volkmer R, Hochreiter S. Complex networks govern coiled-coil oligomerization–predicting and profiling by means of a machine learning approach. Mol Cell Proteomics 2011; 10(5):M110.004994; PMID:21311038; http://dx.doi.org/10.1074/mcp.M110.004994
- Sola RJ, Griebenow K. Glycosylation of therapeutic proteins: an effective strategy to optimize efficacy. BioDrugs 2010; 24(1):9-21; PMID:20055529; http://dx.doi.org/10.2165/11530550-000000000-00000
- Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, Walker J, Gonzalez I, Meeuwsen T, Fox BA et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res 2013; 73(24):7189-98; PMID:24177180; http://dx.doi.org/10.1158/0008-5472.CAN-12-4174
- Benevides L, Cardoso CR, Tiezzi DG, Marana HR, Andrade JM, Silva JS. Enrichment of regulatory T cells in invasive breast tumor correlates with the upregulation of IL-17A expression and invasiveness of the tumor. Eur J Immunol 2013; 43(6):1518-28; PMID:23529839; http://dx.doi.org/10.1002/eji.201242951
- Pedroza-Gonzalez A, Verhoef C, Ijzermans JN, Peppelenbosch MP, Kwekkeboom J, Verheij J, Janssen HL, Sprengers D. Activated tumor-infiltrating CD4+ regulatory T cells restrain antitumor immunity in patients with primary or metastatic liver cancer. Hepatology 2013; 57(1):183-94; PMID:22911397; http://dx.doi.org/10.1002/hep.26013
- Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, Whiteside TL. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin Cancer Res 2007; 13(15 Pt 1):4345-54; PMID:17671115; http://dx.doi.org/10.1158/1078-0432.CCR-07-0472
- Leyland R, Watkins A, Mulgrew K, Holoweckyj N, Bamber L, Tigue NJ, Offer E, Andrews J, Yan L, Mullins S et al. A novel murine GITR ligand fusion protein induces antitumor activity as a monotherapy, which is further enhanced in combination with an OX40 agonist. Clin Cancer Res. 2017 Jan 9; PMID:28069723; http://dx.doi.org/10.1158/1078-0432.CCR-16-2000