
ABSTRACT
Recent clinical studies indicate that adoptive T-cell therapy and especially chimeric antigen receptor (CAR) T-cell therapy is a very potent and potentially curative treatment for B-lineage hematologic malignancies. Currently, autologous peripheral blood T cells are used for adoptive T-cell therapy. Adoptive T cells derived from healthy allogeneic donors may have several advantages; however, the expected occurrence of graft versus host disease (GvHD) as a consequence of the diverse allogeneic T-cell receptor (TCR) repertoire expressed by these cells compromises this approach. Here, we generated T cells from cord blood hematopoietic progenitor cells (HPCs) that were transduced to express an antigen receptor (AR): either a CAR or a TCR with or without built-in CD28 co-stimulatory domains. These AR-transgenic HPCs were culture-expanded on an OP9-DL1 feeder layer and subsequently differentiated to CD5+CD7+ T-lineage precursors, to CD4+ CD8+ double positive cells and finally to mature AR+ T cells. The AR+ T cells were largely naive CD45RA+CD62L+ T cells. These T cells had mostly germline TCRα and TCRβ loci and therefore lacked surface-expressed CD3/TCRαβ complexes. The CD3− AR-transgenic cells were mono-specific, functional T cells as they displayed specific cytotoxic activity. Cytokine production, including IL-2, was prominent in those cells bearing ARs with built-in CD28 domains. Data sustain the concept that cord blood HPC derived, in vitro generated allogeneic CD3− AR+ T cells can be used to more effectively eliminate malignant cells, while at the same time limiting the occurrence of GvHD.
Introduction
Adoptive transfer of gene-modified T cells expressing either chimeric antigen receptors (CARs) or tumor-specific T-cell receptors (TCRs) constitutes a novel and, under certain conditions, a highly effective strategy to treat malignancies. Clinical trials involving CAR T-cell therapy have demonstrated impressive clinical responses in CD19+ acute lymphoblastic leukemia with reported complete remission rate of about 90%.Citation1-3
CARs are artificial modular proteins consisting of a single chain variable fragment for antigen recognition, a spacer, a transmembrane region and one or more intracellular functional sequences derived from CD3ζ and co-stimulatory molecules such as CD137 or CD28.Citation4 First-generation CARs carry only the CD3ζ intracellular sequence. First-generation CAR T cells however do not display potent antitumoral responses in vivo. Second-generation CAR T cells have besides CD3ζ also a co-stimulatory domain that prevents the development of anergy and increases interleukin-2 (IL-2) production upon antigenic stimulation.Citation5 All clinical trials that reported high rates of clinical responses involved second-generation CAR T cells with either CD28 or CD137 as co-stimulation.
TCR transgenic T cells directed to tumor antigens such as NY-ESO-1 and MART1 have also entered clinical trial to study their efficacy in melanoma, sarcoma and multiple myeloma.Citation6,7 Clinical TCR gene therapy studies may be challenged by (1) low affinity of the receptor (directed towards autoantigens), (2) interference of the endogenous TCR with the expression of the transgenic TCR either by cross-pairing of the TCRα and TCRβ chains or by competition for CD3,Citation8-13 (3) the generation of graft versus host disease (GvHD)Citation13 and/or (4) the lack of co-stimulation.Citation14 Several strategies have been reported to address these challenges such as increasing affinity of the TCR, preventing cross-pairing by introducing cysteine residues in the transgenic TCR chains, by gene editing the endogenous TCR loci or, similar to CARs, by inclusion of CD3ζ and CD28 sequences in the TCRα and TCRβ construct (TCR with built-in co-stimulation).Citation8,9,15-20
In some malignancies, CAR T cells seem to be less potent to eradicate the tumor than in others. For instance, CD19 CAR T cells seem to be less efficient in non-Hodgkin lymphoma than in acute lymphoblastic leukemia.Citation21 In addition, the effectiveness seems to be dependent on the intensity of the conditioning regimen, suggesting that concomitant chemotherapy acts synergistically due to the added antitumoral effect and/or due to the immune suppressive effect that inhibits anti-CAR T-cell immune responses by the patient.Citation1,2,22 These data suggest that for some malignancies, CAR T-cell therapy should be combined with other therapeutic regimens to increase cure rate. One possibility is to combine antigen receptor (AR) T-cell therapy with allogeneic stem cell transplantation and/or chemotherapy. Directing allogeneic T cell toward defined cancer antigens, while at the same time avoiding the toxicity of these T lymphocytes caused by alloreactivity of the polyclonal TCR repertoire, may provide a very potent and safe therapy in combination with chemotherapy. For these reasons, it could be beneficial to generate AR+ T cells from allogeneic sources such as banked cord blood or from adult allogeneic donors.Citation23-25 To prevent GvHD, expression of the endogenous TCRs by the peripheral blood T cells should be suppressed to generate mono-specific CAR or TCR-transgenic T cells. Various methods have been described to accomplish this, mostly based on the targeting of the Cα of the TCR locus either by gene editing or siRNAs.Citation15,26
We previously reported that mono-specific TCR-transgenic cells lacking endogenous rearrangements can be generated from cord blood hematopoietic progenitor cells (HPCs).Citation27 This method is based on allelic exclusion, a phenomenon that occurs physiologically in the thymus and that ensures that each T cell successfully rearranges only a single TCRβ locus. Because of allelic exclusion, T cells generated from TCR-transgenic HPCs do not rearrange endogenous TCR loci and T cells that are generated are mono-specific and express only the transgenic TCR. This strategy involves transduction of HPCs to express a tumor-specific TCR and subsequent differentiation of the HPCs to T cells on OP9 feeder cells in the presence of cytokines and Notch stimulation.
Here, we have investigated whether CARs and TCRs with or without built-in CD28 co-stimulatory domain could inhibit rearrangements of endogenous TCR loci while at the same time allow the generation of functional transgenic AR+, tumor-specific T cells. In addition, we assessed whether cells expressing ARs with built-in CD28 co-stimulation acquired more potent T-cell functional capacity. We observed that very potent functional T cells can be generated using this strategy and that these cells express the transgenic CAR or TCR in the virtual absence of rearranged endogenous TCR loci. These cells are therefore assumed to be devoid of alloreactivity.
Results
Antigen receptor transgenic CD34+ HPCs differentiate to mature T cells on OP9-DL1 feeder cells
Human T-lineage committed CD34+ HPCs either isolated directly from thymus or generated in vitro from cord blood CD34+ cells were transduced to express a “second-generation” carcino-embryonic antigen (CEA)-specific CAR carrying an intracellular CD3 ζ-chain signaling sequence and the transmembrane and co-stimulatory CD28 intracellular signaling sequence (CAR:28ζ) (Fig. S1). Twenty to sixty percent of the cells expressed the CAR and the co-transduced GFP after transduction. Transduced GFP+ and untransduced GFP− cells were subsequently cultured together on OP9-DL1 feeder cells for 25 d in the presence of growth factors to obtain CAR+ T cells (). Compared to untransduced cells, the percentages of immature CD4+ (7.2% vs 13.6%) and CD4+CD8+ double positive (DP) (51.9% vs 64.2%) cells were consistently reduced in the GFP+ CAR transgenic population due to a prominent population of mature CD27+ CD1a− cells, which were virtually all double negative (DN) or CD8+ (not shown), in the CAR transgenic cells whereas only few mature CD27+CD1a− cells were present in untransduced cultures (45.3% vs 2.6%).
Figure 1. Differentiation of T-lineage committed CD34 cells after transduction with various antigen receptor constructs. Thymus-derived T-lineage CD34+ precursor cells were transduced to express a transgenic AR and subsequently cultured on OP9-DL1 feeder cells to induce terminal T cell maturation. (A) Flow cytometric analysis of transduced GFP+ and untransduced GFP− cells 25 d after transduction of the cells to express the CAR:28ζ specific for CEA and subsequent culture on OP9-DL1 feeder cells. (B) T-lineage CD34+ precursor cells transduced to express the CAR:ζ or the CAR:28ζ. GFP+ cells are shown after 14 d and 25 d of culture (N = 5). (C) T-lineage CD34+ precursor cells transduced to express the CAR:ζ or the CAR:28ζ 25 d after the initiation of culture on OP9-DL1 feeder cells. The transgenic CAR was recorded using an anti-human IgG1 antibody. The percentages of CD4+ CD8β co-expressing cells and of mature CD27+CD1a− cells were determined within a gate for cells with low CAR expression and a gate for cells with high CAR expression for CAR:ζ and CAR:28ζ transgenic cultures (N = 3).(D) T-lineage CD34+ precursor cells transduced to express the HLA-A2 restricted, gp100 specific wtTCR, TCR:ζ or the TCR:28ζ. Vβ14β+ cells are shown after 20 d of culture on OP9-DL1 feeder cells (N = 5), and (E) after an additional 7 d culture in the presence of the specific peptide (N = 2).
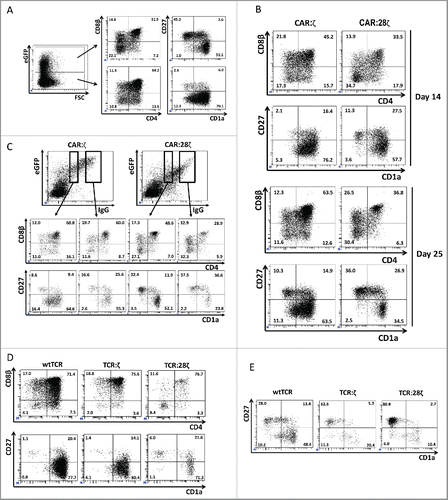
We have shown previously that, in untransduced OP9-DL1 cultures, mature T cells are mainly TCRγδ+ cells.Citation28 In addition, we have shown that in cultures initiated with HPCs transduced to express a TCRαβ, mature CD27+CD1a− T cells are virtually absent, but addition of the agonist peptide in the presence of the restricting HLA antigen induces maturation.Citation27 Here, antigen-dependent maturation is unlikely as CEA expression analysis on these cultures with qPCR was consistently negative (data not shown). Subsequently, we investigated whether CD28 co-stimulatory signals may be inducing terminal maturation in the absence of ligand. Cultures transgenic for a “first-generation” CAR containing only the transmembrane and intracellular CD3ζ-chain signaling sequence (further referred as CAR:ζ) and cultures transgenic for the second-generation CAR containing the transmembrane and intracellular CD28 signaling sequence as well as the intracellular CD3ζ-chain signaling sequence (CAR:28ζ), both specific for CEA, were compared side by side (). The percentage of DPs of the GFP+ CAR transgenic cells was higher in the CAR:ζ transduced cells compared to the CAR:28ζ transduced cells (45.2% vs 33.5% at day 14, 63.5% vs 36.8% at day 25). At the same time, the level of antigen-independent maturation as evidenced by the presence of mature CD27+CD1a− cells was lower in the CAR:ζ transgenic cells compared to the CAR:28ζ transgenic cultures (2.1% vs 11.3% at day 14, 10.3% vs 36.0% at day 25). These differences were not caused by a co-stimulator-induced acceleration of T-cell differentiation as this phenomenon was observed in early (day 14) as well as in late (day 25) cultures. We hypothesized that the number of CAR molecules per cell may impact the signaling strength. Accordingly, the CAR:ζ was expressed to a lower degree than the CAR:28ζ, while the transduced cells expressed the same GFP levels (). To exclude that differences in antigen-independent maturation of the different transgenic cells was caused by differential expression levels of the CAR rather than the co-stimulatory properties and/or the transmembrane domain of the CAR, we stained the different CARs directly with anti-human IgG1 antibody, which binds to the extracellular spacer of the CAR, and compared percentages of DPs and of mature cells for both transgenic cells. For cells with the same expression levels of the AR, a decreased percentage of DPs (48.6% vs 60.8% and 28.9% vs 60.0%) and an increased percentage of antigen-independent maturation (32.4% vs 8.6% and 37.5% vs 16.6%) were noted in CAR:28ζ+ cells vs CAR:ζ+ cells. AR expression levels also contributed, with high AR-expressing cells maturing at higher frequencies than low AR-expressing cells (16.6% vs 8.6% and 37.5% vs 32.4%). We conclude that phenotypically mature T cells are generated from CAR transgenic cells in a ligand-independent manner and that the efficiency of maturation is dependent on the levels of AR expression and on the presence of co-stimulatory domain and/or the nature of the transmembrane domain in the AR.
In addition, the dimerized CD3ζ intracellular domain of the first-generation CAR:ζ itself seems to induce a prominent antigen-independent maturation (). To further substantiate this, we generated modular receptors from a cloned TCR similar to CARs. Both the α and the β chains of a wildtype (wt) HLA-A2 restricted TCR specific for gp100 were linked with the intracellular domain of the CD3ζ only (TCR:ζ) or in addition to the CD28 co-stimulatory domain (TCR:28ζ) (Fig. S1). HPCs transgenic for the wtTCR, for the TCR:ζ and the TCR:28ζ were identified by positive staining for Vβ (). wtTCR transgenic cells generated only a low percentage of mature T cells in the absence of the TCR ligand, as expected (1.1%). In contrast to first-generation CARs, we did not observe increased ligand-independent maturation of the TCR:ζ transgenic cultures (1.4%) compared to wtTCR. However, when a co-stimulatory CD28 domain was included in the TCR, we did observe an increased efficiency of ligand-independent maturation (6.0%). As previously described,Citation27 addition of the TCR cognate peptide further increased maturation of the wtTCR transgenic cells to mature CD27+CD1a− T cells (28.0%). This was also the case for the TCR:ζ and TCR:ζ:28 transgenic cells (62.6% and 80.9%, respectively) (). In conclusion, wtTCR and TCR with incorporated CD3ζ required specific ligand to drive maturation of the T cells, whereas TCR with incorporated CD28 transmembrane and co-stimulatory domain induced antigen-independent maturation of T-cell precursors which can be further enhanced by addition of ligand.
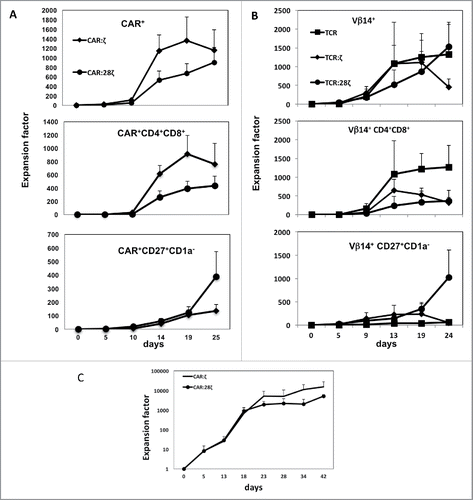
Since DP cells are highly proliferative cells in culture, we asked whether premature ligand-independent maturation of the precursor cells impacts cell expansion in OP9-DL1 cultures of cells transgenic for ARs with built-in CD28 sequence. In , expansion after CAR transduction is presented. Consistent with the lower levels of DP cells observed in CAR:28ζ transgenic cells, total expansion of these cells was lower than that of first-generation CAR:ζ transgenic cells (670- vs 1361-fold increase for CAR:28ζ and CAR:ζ AR-transgenic cultures, respectively, on day 19). Although the total number of cells and the number of DPs that were obtained for TCR:28ζ-transgenic cells were significantly lower, the number of CD27+CD1a− mature T cells obtained from these cultures tended to be higher mainly due to late generation and/or expansion of mature T cells (388- vs 134-fold CAR:28ζ and CAR:ζ AR-transgenic cultures, respectively, on day 25). Similar expansion rates were observed for the TCR constructs (). T-lineage committed HPCs transgenic for wtTCR expanded early together with the generation of DP cells, but a few mature cells are generated without agonist stimulation. TCR:ζ transgenic cells generated comparable cell numbers but more CD27+CD1a− mature T cells were generated. Cultures transgenic for TCR:28ζ displayed reduced early proliferation due to defective DP generation; however, at later stage more CD27+CD1a− mature T cells were generated, suggesting that co-stimulatory signaling induces longer survival or proliferation of mature T cells in vitro. In all cases, total cell expansion starting from fresh CD34+ HPCs is substantial (). After initial culture on OP9-DL1 for 10–14 d, the T-lineage committed cells are transduced and further cultured on OP9-DL1 for additional 28–35 d. Total expansion rates were 1.56×104 times for first-generation CAR:ζ transgenic cells compared to 5.23×103 for cultures with second-generation CAR:28ζ. In conclusion, AR-transgenic HPCs from cord blood can be expanded more than 1000-fold and differentiated in OP9-DL1 cultures to mature cells. Unlike wtTCR transgenic HPCs, AR transgenic cells with build-in CD28 co-stimulator sequence did not require antigen for expansion.
Figure 2. Expansion of T-lineage committed and fresh CD34+ HPC-derived transgenic AR+ T cells. Expansion and differentiation of the various AR-transgenic cell populations. (A) Progeny of one CD34+ cell obtained from thymus after transduction with the CAR:ζ or CAR:28ζ specific for CEA. Error bars represent SD, N = 3. (B) Progeny of one CD34+ cell obtained from thymus after transduction with the gp100-specific, HLA-A2 restricted wtTCR, the TCR:ζ or the TCR:28ζ construct with the same specificity. Error bars represent SD, N = 5. (C) Progeny of fresh CD34+ cord blood cells expanded and differentiated on OP9DL1 feeder cells for 13 d to CD5+CD7+ T-lineage restricted precursors and subsequent transduction to express the CAR:ζ or CAR:28ζ specific for CEA. Error bars represent SD, N = 3.
AR-transgenic CD34+ HPCs generate AR+ T cells that lack CD3 membrane expression
Since CARs do not require CD3 for membrane expression, we checked whether the cells generated in these cultures had TCR/CD3 complexes expressed on the membrane. In , it is shown that >7% of the untransduced GFP− cells express CD3 in combination with either a TCRαβ or a TCRγδ receptor. In contrast, the GFP+ CAR transgenic cells in the same cultures are largely CD3 negative, despite the presence of a higher percentage of mature CD27+ CD1a− cells (). This was the case for both CAR:ζ and CAR:28ζ transgenic cultures. A similar phenomenon was observed in TCR-transgenic cultures (). wtTCR transgenic cells expressed CD3 as well as the transgenic TCR, as evidenced by the Vβ14 expression. In contrast, Vβ14-expressing TCR:ζ and TCR:28ζ transgenic cells expressed the AR in the absence of CD3 membrane expressionCitation16,20. These transgenic cells expressed no CD3. A small population of Vβ14+ cells expressed CD3, but these are most likely untransduced cells that have rearranged the endogenous TCR at the Vβ14 gene segment. To exclude that the CD3/TCR complex was not expressed on the membrane of CAR-transgenic T cells due to the absence of CD3 protein expression, we performed cytoplasmic CD3ϵ staining. shows that cCD3 was present at high levels in the cytoplasm, but not on the cell surface (sCD3), which is likely due to the absence of rearranged endogenous TCRs rather than due to the non-T-cell nature of the cells. Cytoplasmic expression of CD3 suggests that the CAR+, TCR/CD3− cells are bona fide T cells. However, natural killer (NK) cells generated from cord blood on OP9-DL1 feeder cells may also express cytoplasmic CD3.Citation29 We therefore analyzed the mature CAR+ cells for other T and NK-lineage markers. The CAR transgenic cells were positive for CD5, CD7 and CD2 (). The combination of these three markers is exclusively expressed by T cells, whereas NK cells are consistently CD5−. While NK cells are defined as CD3− CD56+ cells, the CAR+ cells generated in vitro were to a large degree negative for the NK cell marker CD56. Only a minor population of the cells was CD56+, a marker that is also present on activated T cells. NKG2D, a marker for both NK cells and mature CD8+ single positive T cells, was expressed on these cells.
Figure 3. Phenotype and endogenous TCR expression of CD34+ HPC-derived transgenic AR+ T cells. Flow cytometric analysis of the AR-transgenic T cells. (A) CAR-transgenic GFP+ cells of cultures transduced to express either the CAR:ζ or the CAR:28ζ were analyzed on day 26 of OP9-DL1 culture for CD3 and TCRαβ expression. As a control, GFP− cells are shown from the OP9-DL1 culture transduced to express the CAR:ζ (N = 5). (B) Dot plots show CD3 expression of cells from the OP9-DL1 cultures transgenic for the wtTCR, TCR:ζ and TCR:28ζ. Vβ14 staining is used to mark transgene expression, as no GFP is expressed by the transgenic cells (N = 5). (C) Surface and cytoplasmic staining for CD3 of in vitro generated mature T cells that were expanded for one cycle on feeder cells in the presence of cytokines. (D) Expression of various membrane markers by the CD27+CD1a− mature T cells at the end of OP9-DL1 culture (46 d) (N = 2). (E) Day 0: fresh cord blood after MACS CD34 enrichment sorted using the sorting window shown. Day 13: cord blood cells cultured on OP9-DL1 were sorted for CD5 CD7 double positive cells, using the indicated sorting window. The cells were then transduced to express CAR:28ζ and further differentiated on OP9-DL1 feeder layer. Day 21: analysis of the transgenic GFP+ cultured cells for DP cells and CD27+CD1a− mature cells. (F) Flow cytometric analysis of GFP+ CAR:28ζ-transgenic cultures, gated on GFP+ CD27+ CD1a− mature AR+ cells (N = 2).
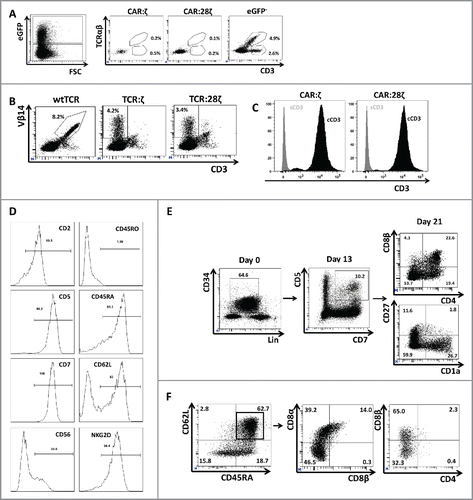
To further substantiate the T-cell nature of the CAR+ CD3− cells, we initiated an OP9-DL1 culture with a pure population (>99%) of CD34+ cord blood cells (). Thirteen days later, the T-cell lineage committed CD5+CD7+ cells were again sorted to homogeneity (>99% purity), transduced and put back in culture to obtain mature T cells. Another 8 d later, GFP+ CAR+ DP cells as well as GFP+CAR+ CD27+CD1a− mature cells were generated from these cultures, indicating that the CAR+ mature cells were efficiently generated from CD5+CD7+ T-lineage precursors and therefore are T lineage cells despite the absence of membrane CD3 expression. We therefore conclude that the CAR+CD3− cells represent mature T cells.
The CAR+ mature T cells were heterogeneous with respect to CD45RA/CD45RO expression (). However, most cultures generated a large population of CD62L+ CD45RA+ CD45RO− cells, indicating that the generated T cells have a naive T-cell phenotype (). As expected, the CAR T-cell populations did not contain CD4+ T cells. Most CAR+ cells were either double negative (DN) or expressed CD8α and CD8β, similar to the reported phenotype of TCRαβ transgenic T cells generated on OP9-DL1 feeder cells.
CD34+ HPC derived transgenic AR+ T cells lack endogenous TCRα and TCRβ rearrangements
We previously reported that transgenic expression of a wtTCRαβ in HPCs completely suppresses rearrangements of the endogenous TCRβ locus and that agonist selection by TCR stimulation suppresses rearrangements of the endogenous TCRα locus, resulting in mature T cells that express only the transgenic TCR.Citation27. In line with this, we showed evidence in the previous section that the CAR:ζ, CAR:28ζ, TCR:ζ and TCR:28ζ expressing cells were CD3 negative, suggesting the absence of endogenous rearrangements of the TCR loci. To further substantiate the absence of endogenous rearrangements, we measured mRNA levels coding for the different components of the CD3/TCR complex in the (TCR-negative) CAR transgenic HPC-derived cell lines. Transcripts encoding CD3γ, CD3δ, CD3ϵ and CD3ζ were detected, whereas TCRα and TCRβ transcripts were selectively lacking in the HPC-derived cell lines (). Next, we analyzed whether a second transduction to express a transgenic (CMV-specific) wtTCRα or a wtTCRβ chain or both could induce membrane expression of the CD3 complex in TCR:ζ and TCR:28ζ transgenic HPC-derived cell lines. As expected, when both wtTCR chains were introduced, membrane CD3 expression was observed in both cell lines, indicating that all components required for TCR expression were present. However, introduction of only the TCRα chain failed to induce membrane CD3 expression, indicating that no endogenous TCRβ chains were expressed. About 0.9–1.2% of the cells expressed CD3 upon transgenic TCRβ introduction, indicating, as previously reported, that low levels of early TCRα rearrangements may occur during the DP differentiation stage.Citation27
Figure 4. TCRα and TCRβ rearrangements in CD34+ HPC derived transgenic AR+ T cells. (A) Expression of the various components of the CD3/TCRαβ complex at the mRNA level. RT-PCR was performed on the JY B cell line as negative control, on a CAR:28ζ transgenic PBMC-derived T cell line (PBMC) as a positive control and on CAR transgenic HPC-derived cell lines of OP9 cultures transduced to express either the CAR:ζ (CAR:ζ) or the CAR:28ζ (CAR:28ζ). (B) TCR:ζ and TCR:28ζ transgenic CD3-negative HPC-derived T-cell lines were transduced to express the TCRα chain of a CMV-specific TCR and GFP as marker, the TCRβ chain with truncated NGFR as marker or with both TCR chains. Three days later, cells were gated for GFP+, NGFR+ or double positive cells and the CD3/TCRαβ expression was measured. Note that the TCRαβ antibody does not bind CD3-negative TCR:ζ nor TCR:28ζ complex although it binds to wtTCR/CD3 complexes. Percentage CD3/TCR positive cells is indicated in the upper right quadrant. (C) Histograms of read counts per CDR3 nucleotide length. CDR3α and CDR3β histograms are shown for wtTCR, TCR:ζ, TCR:28ζ, CAR:ζ and CAR:28ζ transgenic HPC-derived cell lines and as a control CAR:28ζ transgenic PBMC-derived T-cell line. All samples were spiked with Jurkat T cell line mRNA and CDR3α and CDR3β sequences of each transgenic cell line were determined by next-gen sequencing. Asterisk denotes the CDR3 length of the transgenic reads: CDR3α of 48 nucleotides encoding CAASTSGGTSYGKLTF and CDR3β of 39 nucleotides encoding CASSLGSSYEQYF. Arrow points at the CDR3 length of spiked Jurkat CDR reads: CDR3α of 51 nucleotides encoding CAVSDLEPNSSASKIIF and CDR3β of 48 nucleotides encoding CASSFSTCSANYGYTF).
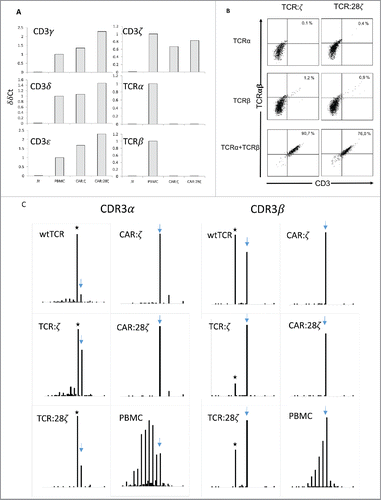
Finally, we studied endogenous rearrangements by high-throughput sequencing of PCR-amplified complementary determining region (CDR3) RNA. Whereas CAR:28ζ transgenic PBMC derived cell line showed a normal distribution of in frame CDR3 lengths, the HPC-derived AR+ cell lines show virtually no endogenous TCRβ rearrangements and severely reduced TCRα rearrangements (). These data show that expression of a CAR, similar to a TCRαβ, suppresses endogenous rearrangements to a large degree leading to single receptor AR+ CD3/TCR− cells.
CD34+ HPC-derived transgenic AR+ T cells display specific antitumor activity
We analyzed whether the in vitro generated AR+ CD8+ and double DN T cells displayed the characteristic functional capacities of T cells, i.e., cytokine release and specific cytotoxicity towards cognate target cells. The in vitro generated mature T cells were expanded for one cycle on feeder cells in the presence of IL-7 and IL-15 and subsequently assessed for specific activity. To exclude the effects of the few CD3+ cells present in these cultures, cells were sorted for GFP+CD3− cells. Specific killing by the CAR+ T cells was tested using the CEA− Colo320 and the CEA+ LS174T tumor cell lines as targets. As shown in , in vitro generated T-cell lines transgenic for CAR:ζ and CAR:28ζ constructs killed the CEA+ tumor cells specifically to a similar degree. Killing was specific and largely mediated by the CAR since CEA+ tumor cell lines cells were killed much more efficiently than CEA− tumor cell lines (Fig. S2).
Figure 5. Antitumor activity of CD34+ HPC-derived transgenic AR+ T cells. Functional analysis of the in vitro generated AR+ cells after one round of expansion on feeder cells. (A) Killing activity at different E:T ratios against the CEA− COLO320 and the CEA+ LST174T tumor cell line. HPC-derived cells were sorted for GFP+CD3− cells before expansion on feeder cells. Error bars represent SD of duplicate determinations (N = 3). (B) Killing activity of CAR:28ζ transgenic peripheral blood CD4+ and CD8+ (PB-derived) and in vitro generated, CD34+ HPC-derived CAR:28ζ transgenic cells. Error bars represent SD of duplicate determinations (N = 2). (C) Killing activity of wtTCR, TCR:ζ and TCR:28ζ transgenic in vitro generated cells toward gp-100+ FM3 and gp100− COLO320 tumor cell lines. TCR:ζ and TCR:28ζ transgenic cultures were sorted for Vβ14+CD3−, wtTCR transgenic cultures were sorted for Vβ14+CD3+ cells. Error bars represent SD of duplicate determinations (N = 5). (D) Cytokine production by CAR:ζ and CAR:28ζ transgenic T-cell lines, either unstimulated or stimulated by PMA and ionomycin, plate-bound anti-IgG1, CEA− COLO320 or CEA+ LST174T cells. All dot plots were gated on GFP+ cells (N >3 for all conditions). (E) Cytokine production by wtTCR, TCR:ζ and TCR:28ζ transgenic T-cell lines, sorted for Vβ14+ cells, after addition of T2 cells pre-incubated with an influenza peptide (T2-INF) or a gp100 peptide (T2-gp100), gp100− COLO320 or gp100+ FM3 tumor cell lines (N >3 for all conditions).
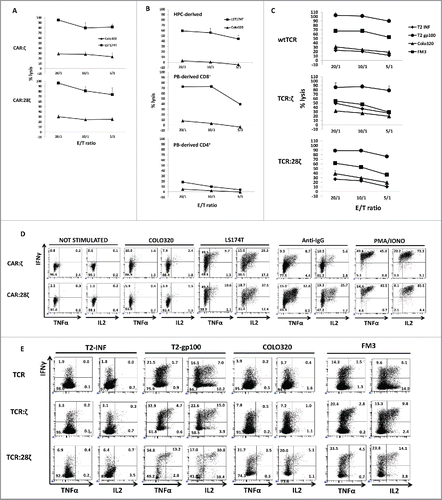
To compare killing activity to peripheral blood derived T cells, killing by the CAR:28ζ transgenic in vitro generated T cells was assessed side by side with CAR:28ζ transgenic sorted CD4+ and CD8+ T cells. shows that whereas peripheral blood derived CD4+ T cells kill tumor cells only marginally, peripheral blood derived CD8+ T cells and in vitro generated T cells vigorously kill CEA+ LST174T cells, indicating that the in vitro generated CD8+ and DN T cells have similar cytotoxic activity as have conventional CD8+ T cells. Finally, we compared killing by wtTCR-transgenic, single AR+ T cells to TCR:ζ and TCR:28ζ transgenic, single AR+ T cells: again killing activity was similar, independent of the nature of the TCR that was expressed, on T2 targets loaded with the gp100 peptide as well as on the gp100 expressing tumor cells ( and S3).
To test for cytokine production, the cells were stimulated and subsequently analyzed for intracellular IFNγ, TNFα and IL-2 production by flow cytometry. We tested for IFNγ and TNFα, cytokines that are produced by NK cells as well as CD4+ and CD8+ T cells and for IL-2, a cytokine produced at high levels by peripheral blood CD4+ T cells, to a lesser extent by peripheral blood CD8+ T cells and not at all by NK cells. Maximal stimulation with PMA and ionomycin of the CAR-transgenic T cells demonstrated that virtually all cells of both T-cell lines were able to secrete simultaneously all three cytokines (). When stimulated by plate bound anti-IgG1 antibody, CAR:ζ-transgenic cells produced consistently lower levels of cytokines and a consistently lower fraction of cells co-produced two cytokines (8.7 and 5.6% vs 32.8% and 25.7%) compared to CAR:28ζ-transgenic cells, which is in line with the previous reports.Citation5 Specific cytokine production was assessed after stimulation with the CEA− Colo320 and the CEA+ LS174T tumor cells. Although there was some background IFNγ secretion that is AR-independent by 5–10% of the cells, upon specific CAR engagement, T cells secreting all three cytokines are observed only after stimulation with the CEA+ cell line LST174T cells. This is especially the case for the CAR:28ζ-transgenic cells.
Finally, we compared specific cytokines secretion by in vitro generated wtTCR transgenic T cells with TCR:ζ and TCR:28ζ-transgenic T cells (). After specific gp100 peptide addition to T2 HLA-A2+ antigen presenting cells, the levels of all three cytokines were increased. Similar to the CAR transgenic cells, the TCR:28ζ transgenic T cells with built-in CD28 co-stimulatory domain demonstrated higher numbers of cells that produced two cytokines compared to wt or TCR:ζ transgenic cells (13.2% and 30.8% for TCR:28ζ vs 1.7% and 7.0% for wtTCR and 4.7% and 15.0% for TCR:ζ). This was the case upon stimulation with T2-gp100 peptide as well as upon co-incubation with gp100+ cell lines FM3. In conclusion, in vitro CD34+ derived T cells, in particular when transgenic for an AR with build-in CD28 co-stimulatory domain, are highly cytotoxic and cytokine producing cells despite their CD8+ or DN phenotype. These cells are CD3 negative, mono-specific T cells that produce high levels of IL-2 and IFNγ upon specific stimulation discriminating these cells from NK cells.
Discussion
We demonstrated that CAR or TCR expressing T cells can be generated from cord blood HPCs in vitro. Importantly, CARs as well as TCRs block the endogenous rearrangements of the TCRα and TCRβ locus resulting in mono-specific T cells since they express only the transgenic AR and lack the endogenous TCR. In addition, we showed that these T cells are very potent in cytokine production as well as cytotoxicity especially when CAR or TCR-constructs are linked to the CD28 co-stimulation signal. Importantly, the CD28 co-stimulation signal does not compromise T-lineage differentiation and the development of functional T cells from HPC.
By transducing CD34+ HPCs, which we subsequently differentiated upon OP9-DL1 feeder cells, we took advantage of the phenomenon of allelic exclusion to generate mono-specific, transgenic AR+ T-cell populations that do not express a polyclonal, endogenous TCR repertoire as is the case with transgenic peripheral blood T cells. It has been shown in TCR-transgenic mice, that rearrangement at the TCRβ locus are completely blocked, whereas the TCRα rearrangements are reduced.Citation30 TCRα rearrangements are operative during the DP stage in the thymus and are terminated by positive selection. In humans, TCRβ locus rearrangements are suppressed in TCR-transduced HPCs which were transplanted in humanized mice, as well as in T cell derived from iPSC generated from a T-cell clone.Citation31-34 We have shown that also in OP9-DL1 cultures TCRβ rearrangements are blocked and that TCRα rearrangements can be largely prevented by early agonist selection, thereby reducing the length of the DP stage.Citation27 Since TCR:28ζ-transgenic cells already mature spontaneously, it is not surprising that these cells rapidly pass through the DP stage and that TCRα rearrangements are severely reduced. As shown in this report, also CARs potently block TCR locus rearrangements to the extent that virtually no TCR/CD3 expressing cells are generated. The observation is in line with a previous report that CAR-transgenic iPSC-derived T cells have a reduced TCR repertoire, however these iPSC cells were derived from a T-cell clone.Citation35 As the T-cell clone has rearranged TCRβ and TCRα locus, it is not unexpected that de novo rearrangements were inhibited and the issue whether this was caused by the CAR or by the pre-TCR was not addressed. A report by Zakrzewski et al. using transfer of murine CD19-CAR-transgenic T-cell precursors in mice reported no effect of the first generation CAR on the development of the TCRβ repertoire, nor on the expression of CD4+, CD8+ and CD3.Citation36 However, in a recent study, HIV-specific CAR-transduced human HPCs injected in BLT mice resulted in a reduced number of CD3 positive cells in the thymus and the TRECs per cell were decreased, indicating that TCR rearrangements were blocked.Citation37 The latter data in concert with the data presented here strongly suggest that also CARs block rearrangements during T-cell development in vivo as well as in vitro with the result that mono-specific T cells are generated. Discrepant results could be explained by the expression level of the CAR in the thymus as we have shown that low expression affects T-cell differentiation to a lesser degree. As expression of certain promoters such as CMV promoters are shut down in the thymus this may explain the discrepant results in the in vivo studies.
Alternative methods to generate mono-specific T cells without expression of the endogenous TCR, using gene editing of peripheral blood T cells, have limitations. Gene editing methods can be applied to CAR T cells as one TCR-chain can be edited and by subsequent screening for CD3− CAR+ cells, CD3/TCR negative CAR T cells can be isolated also from peripheral blood. However, for TCR transgenic T cells, this method is more cumbersome, as both chains have to be eliminated to prevent cross-pairing. In addition, this requires lengthy screening, which may not be practical in a clinical setting.Citation15 Furthermore, gene editing has its own pitfalls, in particular off-target mutations which may prove to be oncogenic depending on the targeted sequence involved.
The absence of endogenous TCRs has several advantages for adoptive cell therapy. Interference with expression of the tumor-specific TCR is minimized due to the expected absence of cross-pairing. Cross-pairing may induce unexpected autoreactivity and GvHD as well as reduce the expression levels of the transgenic TCR.Citation13,16 In the approach described here, additional measures were taken to prevent interference with the tumor-specific TCR expression: The linked CD3ζ and CD28 signaling sequences force both transgenic chains to preferentially pair with each other and not with endogenous chains.Citation17,20 In addition, as these TCRs are surface-expressed independent of CD3, there is no competition for endogenous CD3 as the cytoplasm brightly stains for superfluous CD3 that cannot incorporate into the membrane due to lack of a TCR.
The CD28 co-stimulatory signal may not only prevent the development of anergy, but may also significantly increase survival and proliferation of these cells upon antigen stimulation.Citation17 Here, we show that the mature CD27+CD1a− cells survive and/or proliferate much better in the OP9-DL1 cultures compared to AR+ cells without co-stimulatory signal and we show that these cells produce higher levels of cytokines including IL-2. The high levels of cytotoxic activity together with polycytokine secreting capabilities, despite their DN or CD8+ phenotype, make these cells good candidates for a redirected cell therapy of cancer.
A limitation of this study is the lack of proof of in vivo functionality. Preliminary in vivo data suggest that these cells require human IL-15 for survival and experiments with human IL-15 transgenic mice are now being set up.
Two groups reported rejuvenation of T cells by generating iPSCs from T-cell clones and subsequent differentiation to T cells.Citation33,34,38 These authors report that they obtain CD62L+ CD45RA+ T cells and that these T cells may have again become naive, conventional T cells. As the cells they describe are in many respects similar to the T cells derived here from HPCs, data underline their potential value in cell therapy.
The mono-specific AR+ T cells can be applied in several ways in cancer therapy. Similar to allogeneic virus-specific T cells that are administered to patients after bone marrow transplantation, one can generate a bank of common HLA-expressing, cord blood HPC-derived allogeneic CAR T cells. In this way, partially matched CAR T cells can be administered to post-transplant patient. Using this approach, CMV-specific T cells induce remission of CMV disease.Citation39 Whether such an approach would also work for CAR T cells in treatment of malignancies is unclear, as it has been shown that survival of CAR T cells for at least 6 mo is required for cure.Citation3 Alternatively, allogeneic CAR therapy could be combined with heavy immunosuppressive conditioning. This conditioning will work in synergy with CAR T cells on the tumor and at the same time will prevent anti-CAR T cell alloresponses and anti-CAR responses, thus allowing the cells to survive longer. In the most intensive therapy, the latter could be combined with T-cell depleted stem cell therapy, either using HLA matched cord blood or adult mobilized blood cells. In these cases, stem cell chimerism will induce lifelong tolerance sustaining survival of the donor CAR T cells. In all these instances, CAR T cells can be generated from healthy donor material, under optimal conditions and sufficiently in numbers in advance for a broad cohort of patients.
Materials and methods
Isolation of human cells
Postnatal thymus (0–12 y of age) and cord blood were obtained and used following guidelines of the Medical Ethical Committee of the Ghent University Hospital (Belgium). Informed consent was obtained in accordance with the Declaration of Helsinki (Ethics Committee UZ Ghent decision B670201215078). Human T-lineage committed CD34+ cells were isolated from postnatal thymus and CD34+ HPCs were isolated from cord blood by magnetic activated cell separation (MACS, Miltenyi, cat# 130-046-703). Cord blood CD34+ cells were subsequently T-lineage committed by culture on OP9-DL1 in the presence of 50 ng/mL SCF, 20 ng/mL Flt3L and 10 ng/mL IL7 for 12–14 d. Purities were usually > 90%.
LST174T (ATCC CCL 188) and H508 (ATCC CCL 253) are CEA-expressing carcinoma cell lines, Colo320 (ATCC CCL 220), Colo201 (ATCC CCL 224) and H716 (ATCC CCL251) are CEA− colon carcinoma cell lines, FM3, MEL624 and MEL526 are HLA-A2+ melanoma cell lines expressing gp100 obtained from R Debets. JY is a human HLA-A2+ B cell line and Jurkat is a CD3+ leukemic line. These cell lines were maintained under standard culture conditions.
Production of retroviral vectors
The different construct used are represented in Fig. S1. The carcino-embryonic antigen (CEA)-specific CAR:ζ and CAR:28ζ constructsCitation5,40 were transferred into the LZRS IRES eGFP (LIE) vector using XhoI and BglII and viral particles were produced using the Phoenix packaging cell line. Retroviral supernatans were collected at day 14 after transfection and hygromycin selection and frozen until use. Transduced cells were detected either by eGFP expression or by an anti-IgG antibody directed against the human IgG1 spacer domain present in the extracellular domain of both CARs.
The TCR constructs were generated from a gp100-specific (epitope: YLEPGPVTA) HLA-A2 restricted receptor using TRBV14s1.Citation16,17,41 The TCR:ζ was constructed by linking the extracellular part of both the wt TCRα and TCRβ chain to the full length transmembrane and intracellular parts of CD3ζ.Citation16,42 The TCR:28ζ was constructed by linking the extracellular part of both the wt TCRα and TCRβ chain to the CD28-CD3ζ tail described above (kindly provided by H. Abken, Cologne, Germany) (Fig. S1).Citation20 wtTCR, the TCR:ζ and the TCR:28ζ cDNAs were cloned into pBullet retroviral vectors. VSV-G envelope-pseudotyped Moloney murine leukemia virus retroviral particles that contain TCR RNAs were freshly produced by a co-culture of the packaging cells 293T and Phoenix-A following calcium phosphate transfections and were used fresh. wtTCR, the TCR:ζ and the TCR:28ζ were detected by a Vβ14-specific antibody (Beckman Coulter, cat# PN IM2047).
Retroviral transduction
Fresh thymic CD34+ were pre-activated for 1 d in the presence of cytokines (5 ng/mL SCF and 10 ng/mL IL7). CD34+ CB cells were transduced after T-lineage committment (see above) in the presence of SCF (5 ng/mL, PeproTech, 300–07)), Flt-3L (10 ng/mL, PeproTech 300–19) and IL-7 (10 ng/mL, R&D Systems, 207-IL). Transduction efficiencies varied but were usually between 20% and 60% for the CAR-transgenic cells, and between 2% and 8% for the TCR transgenic cells. Unless stated otherwise, the mixture of transduced and untransduced cells was cultured without prior Fluorescent Activated Cell Sorting (FACS) on OP9-DL1 cells.
OP9-DL1 co-cultures
Transduced cells were co-cultured on a subconfluent OP9-DL1 cell layer in a-MEM medium (Gibco, 22561–021) supplemented with 20% FBS (Bovogen, SFBS), 2 mM L-glutamine, 100 IU/mL penicillin, 100 IU/mL streptomycin, 5 ng/mL SCF, 10 ng/mL Flt3L and 10 ng/mL Il-7, as described previously.Citation43
Agonist peptide stimulation
Cells were harvested from OP9-DL1 and were, either as such or after FACS sorting for immature DP cells, seeded in tissue culture plates (BD Biosciences) in complete IMDM in the presence of 10 ng/mL IL-7 for 7 d. The gp100 YLEPGPVTA peptide and, as irrelevant control, influenza matrix protein M158–66 peptide were used (Anaspec by Eurogentec). For the CAR experiment, a CEA-expressing adherent cell line (LST174T) was added to the cultures.
T-cell expansion
Cultures containing mature CD27+CD1a− cells were expanded on irradiated allogenic feeder cells, consisting of a mixture of 40-Gy irradiated peripheral blood mononuclear cells and 50-Gy irradiated JY cells. Cells were cultured in IMDM (Gibco) with 10% FBS, supplemented with 2 μg/mL PHA (Oxoid, R30852801) or 10 ng of IL-7 and IL-15 (R&D Systems 247-ILB-025).
Flow cytometry and antibodies
Surface marker staining was performed in DPBS with 1% FBS using antibody concentration recommended by the supplier. Intracellular staining was performed following the supplier's protocol using Fix & Perm (BD Biosciences). Flow cytometric analysis was performed on the LSR II and cell sorting on the ARIA II (both BD Biosciences), both equipped with fur lasers. All populations studied were devoid of dead cells based on propidium iodide negativity and of doublets based on FSC-A FSC-W ratios. The following anti-human monoclonal antibodies are CD5 (BD Biosciences, cat# 345782), IgG-Fc (eBioscience, cat# 12-4998-82), Vβ14 (Beckman Coulter, cat# PN IM2047); APC-conjugated: CD8α (BD Biosciences, cat# 345775), TCRαβ (Miltenyi, cat# 130-091-237), CD45 (Miltenyi, cat# 130-091-230), CD34 (BD Biosciences, cat# 345804); Amcyan-conjugated: CD45 (BD Biosciences, cat# 339192); V450-conjugated: CD7 (BD Biosciences, cat# 642916), CD3 (eBioscience, cat# 48-0038-42); APC-Cy7-conjugated: CD27 (eBioscience, cat# 47-0279-42); PE-Cy7-conjugated: CD8β (eBioscience, cat# 25-5273-42); AF-700-conjugated: CD4+ (BD Biosciences, cat# 557922); BV510-conjugated: CD45 (BD Biosciences, cat# 563204); biotin-conjugated: CD1a (ATCC); PerCPCy5.5-conjugated: Streptavidin (eBioscience, cat# 45-4317-82).
Flowcytometric determination of cytokine production
One hundred thousand culture expanded cells were stimulated by co-incubation with a cell line expressing the relevant antigen (LS174T, FM3 or Colo320) at 105 cells in 96-well flat-bottom plates. After 1 h, GolgiSTOP (BD Biosciences, cat# 51-2092KZ)) was added. After an additional 16 h of stimulation, the cells were harvested, permeabilized, labeled and analyzed for cytokine expression using TNF-PE-Cy7 (BD Biosciences, cat#557647), IFNγ-PE (BD Biosciences, cat# 340452) and IL2-APC (BD Biosciences, cat# 554567).
51Chromium release assay
Target cells were labeled with51 Chromium, washed and used at different effector to target ratios. T2 cells were loaded for 2 h with the indicated peptide at a concentration of 10 μg/mL unless otherwise indicated before radioactive labeling. Effector cells consisted of culture expanded peripheral blood mononuclear cells or OP9-DL1 cultures. After a 4 h of co-incubation, supernatant was harvested and measured in a 1450 LSC & Luminescence Counter (Perkin Elmer). Specific lysis is calculated as followed: (experimental release − spontaneous release)/(maximal release − spontaneous release) × 100%.
Real-time quantitative PCR (RT-qPCR)
Total RNA was prepared using the miRNeasy minikit (Qiagen). cDNA synthesis was performed by the Superscript First Strand Synthesis System for RT-PCR kit (Invitrogen). RT-qPCR with the SYBR Green I technology was performed using the LightCycler 480 SYBR Green I Master kit on a LightCycler 480 II (both Roche) according to the manufacturer's protocol. Used primers (Biolegio, Nijmegen, Netherlands) are listed in Table S1. Results were analyzed using the δδCt method using actin as a reference gene.
CDR3α and CDR3β high-throughput sequencing
RNA was isolated from culture expanded HPC-derived wtTCR, TCR:ζ, TCR:28ζ, CAR:ζ, CAR:28ζ transgenic cell lines and from a PBMC-derived CAR:28ζ transgenic T cell line (105–106 cells) with the RNeasy microkit (Qiagen) followed by template-switch anchored RT-PCR.Citation44 To each sample, Jurkat RNA was added for a total of 10% of the amount of RNA as an internal standard. A template-switch adaptor, AAGCAGTGGTATCAACGCAGAGTACATrGr GrG, was ligated at the 5′ end of mRNA during cDNA generation using the Superscript II RT enzyme (Invitrogen). The cDNA product was purified using AMPure XP Beads (Agencourt). Then, PCR amplification (Lightcycler, Roche) was performed using a Cα specific primer (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG TCTCAGCT GGTACACGGCAGGGTCAGGGT-3′, adapter in italic) or a Cβ specific primer (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG TGCTTCTGATGGCTCAAACACAGCGACCT-3′, adapter in italic) containing an adapter used in subsequent sequencing, and a primer complementary to the template-switch adapter (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG AAGCAGTGGTATCAACGCAG-3′, adapter in italic) with the KAPA Real-Time Library Amplification Kit (Kapa Biosystems). After purification with AMPure XP Beads, an index PCR with Illumina sequencing adapters was performed using the Nextera XT Index Kit. This second PCR product was again purified with AMPure XP beads. High-throughput sequencing of the generated amplicon products containing the CDR3α or CDR3β sequences was performed on an Illumina MiSeq platform using the V2 300 kit, with 200 bp at the 3′ end (read 2) and 100 bp at the 5′ end (read 1) (at the GIGA center, University of Liège, Belgium). CDR3 sequences were obtained by aligning the fastq files (read 2) with the MiXCR software version 1.7.1Citation45 with specified “-loci” parameter “TRA” and “TRB” for the alignment of CDR3α and CDR3β, respectively. Then, CDR3 sequences were assembled and exported. Spectratyping plots were generated via the routine PlotFancySpectratype of VDJTools version 1.0.7.Citation46
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_materials.zip
Download Zip (209.4 KB)Acknowledgments
The authors would like to thank Dr. Katrien Francois, Department of Cardiac Surgery and Dr. Conny Matthys, Cord Blood Bank of Ghent University Hospital for providing thymus and cord blood samples. Finally, we would like to thank Dr. Tom Boterberg for irradiation of the feeder cells and Sophie Vermaut for help with flow cytometry and cell sorting.
Funding
This work was supported by the Kinderkankerfonds, Research Foundation - Flanders (Fonds voor Wetenschappelijk Onderzoek Vlaanderen, FWO), Stichting tegen Kanker, the “Interuniversity Attraction Poles” (IAP) Program of the Belgian Science Policy Office (BELSPO) and the Fonds National de la Recherche Scientifique (FNRS). SV, YVC and GV are supported by the Instituut voor de Aanmoediging van Innovatie door Wetenschap en Technologie in Vlaanderen (IWT). SD and TK are supported by the FWO and PT is supported by the FNRS (Télévie).
References
- Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 2016; 126(6):2123-38; PMID:27111235; http://dx.doi.org/10.1172/JCI85309
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015; 385(9967):517-28; PMID:25319501; http://dx.doi.org/10.1016/S0140-6736(14)61403-3
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371(16):1507-17; PMID:25317870; http://dx.doi.org/10.1056/NEJMoa1407222
- Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev 2014; 257(1):83-90; PMID:24329791; http://dx.doi.org/10.1111/imr.12125
- Hombach A, Sent D, Schneider C, Heuser C, Koch D, Pohl C, Seliger B, Abken H. T-cell activation by recombinant receptors: CD28 costimulation is required for interleukin 2 secretion and receptor-mediated T-cell proliferation but does not affect receptor-mediated target cell lysis. Cancer Res 2001; 61(5):1976-82; PMID:11280755
- Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, Badros AZ, Garfall A, Weiss B, Finklestein J et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 2015; 21(8):914-21; PMID:26193344; http://dx.doi.org/10.1038/nm.3910
- Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 2011; 29(7):917-24; PMID:21282551; http://dx.doi.org/10.1200/JCO.2010.32.2537
- Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res 2007; 67(8):3898-903; PMID:17440104; http://dx.doi.org/10.1158/0008-5472.CAN-06-3986
- Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res 2006; 66(17):8878-86; PMID:16951205; http://dx.doi.org/10.1158/0008-5472.CAN-06-1450
- Ahmadi M, King JW, Xue SA, Voisine C, Holler A, Wright GP, Waxman J, Morris E, Stauss HJ. CD3 limits the efficacy of TCR gene therapy in vivo. Blood 2011; 118(13):3528-37; PMID:21750319; http://dx.doi.org/10.1182/blood-2011-04-346338
- Heemskerk MH, Hagedoorn RS, van der Hoorn MA, van der Veken LT, Hoogeboom M, Kester MG, Willemze R, Falkenburg JH. Efficiency of T-cell receptor expression in dual-specific T cells is controlled by the intrinsic qualities of the TCR chains within the TCR-CD3 complex. Blood 2007; 109(1):235-43; PMID:16968899; http://dx.doi.org/10.1182/blood-2006-03-013318
- Kuball J, Dossett ML, Wolfl M, Ho WY, Voss RH, Fowler C, Greenberg PD. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood 2007; 109(6):2331-8; PMID:17082316; http://dx.doi.org/10.1182/blood-2006-05-023069
- Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, Kaiser AD, Pouw N, Debets R, Kieback E et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med 2010; 16(5):565-70, 1p following 70; PMID:20400962; http://dx.doi.org/10.1038/nm.2128
- Ghorashian S, Velica P, Chua I, McNicol AM, Carpenter B, Holler A, Nicholson E, Ahmadi M, Zech M, Xue SA et al. CD8 T cell tolerance to a tumor-associated self-antigen is reversed by CD4 T cells engineered to express the same T cell receptor. J Immunol 2015; 194(3):1080-9; PMID:25539815; http://dx.doi.org/10.4049/jimmunol.1401703
- Provasi E, Genovese P, Lombardo A, Magnani Z, Liu PQ, Reik A, Chu V, Paschon DE, Zhang L, Kuball J et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med 2012; 18(5):807-15; PMID:22466705; http://dx.doi.org/10.1038/nm.2700
- Sebestyen Z, Schooten E, Sals T, Zaldivar I, San Jose E, Alarcon B, Bobisse S, Rosato A, Szöllosi J, Gratama JW et al. Human TCR that incorporate CD3zeta induce highly preferred pairing between TCRalpha and beta chains following gene transfer. J Immunol 2008; 180(11):7736-46; PMID:18490778; http://dx.doi.org/10.4049/jimmunol.180.11.7736
- Govers C, Sebestyen Z, Roszik J, van Brakel M, Berrevoets C, Szoor A, Panoutsopoulou K, Broertjes M, Van T, Vereb G et al. TCRs genetically linked to CD28 and CD3epsilon do not mispair with endogenous TCR chains and mediate enhanced T cell persistence and anti-melanoma activity. J Immunol 2014; 193(10):5315-26; PMID: 25320284; http://dx.doi.org/10.4049/jimmunol.1302074
- Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 2013; 36(2):133-51; PMID:23377668; http://dx.doi.org/10.1097/CJI.0b013e3182829903
- Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013; 122(6):863-71; PMID:23770775; http://dx.doi.org/10.1182/blood-2013-03-490565
- Schaft N, Lankiewicz B, Drexhage J, Berrevoets C, Moss DJ, Levitsky V, Bonneville M, Lee SP, McMichael AJ, Gratama JW et al. T cell re-targeting to EBV antigens following TCR gene transfer: CD28-containing receptors mediate enhanced antigen-specific IFNgamma production. Int Immunol 2006; 18(4):591-601; PMID:16507598; http://dx.doi.org/10.1093/intimm/dxh401
- Maude SL, Shpall EJ, Grupp SA. Chimeric antigen receptor T-cell therapy for ALL. Hematol: Am Soc Hematol Educ Prog 2014; 2014(1):559-64; http://dx.doi.org/10.1182/asheducation-2014.1.559
- Lamers CH, Klaver Y, Gratama JW, Sleijfer S, Debets R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem Soc Trans 2016; 44(3):951-9; PMID:27284065; http://dx.doi.org/10.1042/BST20160037
- Cruz CR, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, Diouf O, Liu E, Barrett AJ, Ito S et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood 2013; 122(17):2965-73; PMID:24030379; http://dx.doi.org/10.1182/blood-2013-06-506741
- Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, Hakim FT, Halverson DC, Fowler DH, Hardy NM et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013; 122(25):4129-39; PMID:24055823; http://dx.doi.org/10.1182/blood-2013-08-519413
- Brudno JN, Somerville RP, Shi V, Rose JJ, Halverson DC, Fowler DH, Gea-Banacloche JC, Pavletic SZ, Hickstein DD, Lu TL et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-Cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J Clin Oncol 2016; 34(10):1112-21; PMID:26811520; http://dx.doi.org/10.1200/JCO.2015.64.5929
- Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, Butler K, Rivat C, Wright G, Somana K, Ghorashian S, Pinner D, Ahsan G, Gilmour K, Lucchini G, Inglott S, Mifsud W, Chiesa R, Peggs KS, Chan L, Farzeneh F, Thrasher AJ, Vora A, Pule M, Veys P et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. 2017; 9(374)pii:eaaj2013; PMID:28123068; http://dx.doi.org/10.1126/scitranslmed.aaj2013
- Snauwaert S, Verstichel G, Bonte S, Goetgeluk G, Vanhee S, Van Caeneghem Y et al. In vitro generation of mature, naive antigen-specific CD8(+) T cells with a single T-cell receptor by agonist selection. Leukemia 2014; 28(4):830-41; PMID:24091848; http://dx.doi.org/10.1038/leu.2013.285
- Van Coppernolle S, Verstichel G, Timmermans F, Velghe I, Vermijlen D, De Smedt M, Leclercq G, Plum J, Taghon T, Vandekerckhove B et al. Functionally mature CD4 and CD8 TCRalphabeta cells are generated in OP9-DL1 cultures from human CD34+ hematopoietic cells. J Immunol 2009; 183(8):4859-70; PMID:19801512; http://dx.doi.org/10.4049/jimmunol.0900714
- De Smedt M, Taghon T, Van de Walle I, De Smet G, Leclercq G, Plum J. Notch signaling induces cytoplasmic CD3 epsilon expression in human differentiating NK cells. Blood 2007; 110(7):2696-703; PMID:17630354; http://dx.doi.org/10.1182/blood-2007-03-082206
- Kisielow P, Teh HS, Bluthmann H, von Boehmer H. Positive selection of antigen-specific T cells in thymus by restricting MHC molecules. Nature 1988; 335(6192):730-3; PMID:3262831; http://dx.doi.org/10.1038/335730a0
- Vatakis DN, Arumugam B, Kim SG, Bristol G, Yang O, Zack JA. Introduction of exogenous T-cell receptors into human hematopoietic progenitors results in exclusion of endogenous T-cell receptor expression. Mol Ther 2013; 21(5):1055-63; PMID:23481324; http://dx.doi.org/10.1038/mt.2013.28
- Giannoni F, Hardee CL, Wherley J, Gschweng E, Senadheera S, Kaufman ML, Kaufman ML, Chan R, Bahner I, Gersuk V, Wang X et al. Allelic exclusion and peripheral reconstitution by TCR transgenic T cells arising from transduced human hematopoietic stem/progenitor cells. Mol Ther 2013; 21(5):1044-54; PMID:23380815; http://dx.doi.org/10.1038/mt.2013.8
- Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D, Nakayama-Hosoya K, Iriguchi S, Uemura Y, Shimizu T. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013; 12(1):114-26; PMID:23290140; http://dx.doi.org/10.1016/j.stem.2012.11.002
- Vizcardo R, Masuda K, Yamada D, Ikawa T, Shimizu K, Fujii S, Koseki H, Kawamoto H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell 2013; 12(1):31-6; PMID:23290135; http://dx.doi.org/10.1016/j.stem.2012.12.006
- Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, Sadelain M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol 2013; 31(10):928-33; PMID:23934177; http://dx.doi.org/10.1038/nbt.2678
- Zakrzewski JL, Suh D, Markley JC, Smith OM, King C, Goldberg GL, Jenq R, Holland AM, Grubin J, Cabrera-Perez J et al. Tumor immunotherapy across MHC barriers using allogeneic T-cell precursors. Nat Biotechnol 2008; 26(4):453-61; PMID:18376399; http://dx.doi.org/10.1038/nbt1395
- Zhen A, Kamata M, Rezek V, Rick J, Levin B, Kasparian S, Chen IS, Yang OO, Zack JA, Kitchen SG. HIV-specific immunity derived from chimeric antigen receptor-engineered Stem Cells. Mol Ther 2015; 23(8):1358-67; PMID:26050990; http://dx.doi.org/10.1038/mt.2015.102
- Crompton JG, Rao M, Restifo NP. Memoirs of a reincarnated T cell. Cell Stem Cell 2013; 12(1):6-8; PMID:23290132; http://dx.doi.org/10.1016/j.stem.2012.12.009
- Clancy LE, Blyth E, Simms RM, Micklethwaite KP, Ma CK, Burgess JS, Antonenas V, Shaw PJ, Gottlieb DJ. Cytomegalovirus-specific cytotoxic T lymphocytes can be efficiently expanded from granulocyte colony-stimulating factor-mobilized hemopoietic progenitor cell products ex vivo and safely transferred to stem cell transplantation recipients to facilitate immune reconstitution. Biol Blood Marrow Transplant 2013; 19(5):725-34; PMID:23380344; http://dx.doi.org/10.1016/j.bbmt.2013.01.021
- Hombach A, Wieczarkowiecz A, Marquardt T, Heuser C, Usai L, Pohl C, Seliger B, Abken H. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J Immunol 2001; 167(11):6123-31; PMID:11714771; http://dx.doi.org/10.4049/jimmunol.167.11.6123
- Schaft N, Willemsen RA, de Vries J, Lankiewicz B, Essers BW, Gratama JW, Figdor CG, Bolhuis RL, Debets R, Adema GJ. Peptide fine specificity of anti-glycoprotein 100 CTL is preserved following transfer of engineered TCR alpha beta genes into primary human T lymphocytes. J Immunol 2003; 170(4):2186-94; PMID:12574392; http://dx.doi.org/10.4049/jimmunol.170.4.2186
- Willemsen RA, Weijtens ME, Ronteltap C, Eshhar Z, Gratama JW, Chames P, Bolhuis RL. Grafting primary human T lymphocytes with cancer-specific chimeric single chain and two chain TCR. Gene Therapy 2000; 7(16):1369-77; PMID:10981663; http://dx.doi.org/10.1038/sj.gt.3301253
- De Smedt M, Hoebeke I, Plum J. Human bone marrow CD34+ progenitor cells mature to T cells on OP9-DL1 stromal cell line without thymus microenvironment. Blood Cells Mol Dis 2004; 33(3):227-32; PMID:15528136; http://dx.doi.org/10.1016/j.bcmd.2004.08.007
- Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest 2014; 124(5):2246-59; PMID:24667641; http://dx.doi.org/10.1172/JCI73639
- Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, Chudakov DM. MiXCR: Software for comprehensive adaptive immunity profiling. Nat Methods 2015; 12(5):380-1; PMID:25924071; http://dx.doi.org/10.1038/nmeth.3364
- Shugay M, Bagaev DV, Turchaninova MA, Bolotin DA, Britanova OV, Putintseva EV, Pogorelyy MV, Nazarov VI, Zvyagin IV, Kirgizova VI et al. VDJtools: unifying post-analysis of T cell receptor repertoires. PLoS Comput Biol 2015; 11(11):e1004503; PMID:26606115; http://dx.doi.org/10.1371/journal.pcbi.1004503