
ABSTRACT
Interferon-α (IFNα) has multiple antitumor effects including direct antitumor toxicity and the ability to potently stimulate both innate and adaptive immunity. However, its clinical applications in the treatment of malignancies have been limited because of short half-life and serious adverse reactions when attempting to deliver therapeutically effective doses. To address these issues, we fused IFNα2a to the anti-vascular endothelial growth factor and receptor 2 (VEGFR2) antibody JZA00 with the goal of targeting it to the tumor microenvironment where it can stimulate the antitumor immune response. The fusion protein, JZA01, is effective against colorectal cancer by inhibiting angiogenesis, exhibiting direct cytotoxicity, and activating the antitumor immune response. Although JZA01 exhibited reduced IFNα2 activity in vitro compared with native IFNα2, VEGFR2 targeting permitted efficient antiproliferative, proapoptotic, antiangiogenesis, and immune-stimulating effects against the colorectal tumors HCT-116 and SW620. JZA01 showed in vivo efficacy in NOD-SCID mice-bearing established HCT-116 tumors. In conclusion, this study describes an antitumor immunotherapy that is highly promising for the treatment of colorectal cancer.
Abbreviations
| HIF-1α | = | hypoxia-inducible factor-1alpha |
| MHC | = | major histocompatibility complex |
| CFSE | = | carboxyfluorescein diacetate, succinimidyl ester |
| APCs | = | antigen-presenting cells |
| CTL | = | cytotoxic T lymphocyte |
| ELISA | = | The enzyme-linked immunosorbent assay |
| HRP | = | horseradish peroxidase |
| PBS | = | phosphate buffer saline |
Introduction
The tumor microenvironment (TME) includes surrounding blood vessels, immune cells, fibroblasts, bone marrow-derived inflammatory cells, lymphocytes, signaling molecules, and the extracellular matrix (ECM).Citation1 The tumor and the surrounding microenvironment interact constantly. Tumors can influence the microenvironment by releasing extracellular signals, promoting angiogenesis, and inducing peripheral immune tolerance, whereas the immune cells in the microenvironment can affect the growth and the evolution of cancerous cells by mechanisms such as immune editing and the production of cytokines,Citation2,3 among which interferon-α (IFNα) is effective in regulating the TME.
IFNα is a pleiotropic cytokine, which has been used in the treatment of cancer including hematologic tumors and metastatic solid tumors for over 30 y both alone and in combination with chemotherapeutic agents.Citation4,5 IFNα has several antitumor activities including regulating the immune response through activating dendritic cells, promoting the long-term survival of T cells, and enhancing the killing ability of T cells and NK cells.Citation6-8 Additionally, IFNα exhibits direct cytotoxicity by inhibiting the proliferation and promoting apoptosis of tumor cells.Citation9,10
Although IFNα can exert efficient antitumor effects, because of its short in vivo half-life, it requires frequent administration and at therapeutically effective doses produces toxic side effects including severe depression, fever, headache, joint, and muscle pain leading many patients to abandon IFNα treatment.Citation11 To extend the t1/2, IFNα has been coupled with PEG, albumin, or antibody Fc fragment. These approaches have improved in vivo dynamics but did not eliminate the serious systemic toxicity.Citation12-14 In contrast, fusion proteins with IFNα joined to the tumor targeting anti-CD20 antibody showed both improved pharmacokinetics and tumor targeting in the absence of non-specific toxicity.Citation15,16
Angiogenesis is critical for tumor growth and metastasis with the vascular endothelial growth factor and receptor 2 (VEGF/VEGFR2)-mediated signaling playing an important role.Citation17-19 IFNα can inhibit tumor angiogenesis by downregulating the expression of VEGF and HIF-1α, resulting in a synergistic effect with anti-VEGF.Citation20,21 The combination therapy of the anti-VEGF antibody Bevacizumab with IFNα was approved by the U.S. FDA in 2009 as the first-line treatment of metastatic renal cell carcinoma (mRCC).Citation22 However, it had serious side effects and blocking VEGF by Bevacizumab did not completely inhibit tumor angiogenesis.Citation23-25 In April 2014, the anti-angiogenesis antibody Ramucirumab targeting VEGFR2 was approved by the FDA for the treatment of chemotherapy-resistant gastric cancer.Citation26,27
Thus, the combination of IFNα with antibodies that inhibit angiogenesis can both reactivate the immune state of the TME and block the tumor vascular supply.Citation22,28 As a consequence, VEGF/VEGFR2 signaling pathway inhibitors and IFNα may play a synergistic role in inhibiting proliferation and promoting apoptosis of cancers.Citation10
Based on these observations, we hypothesized that fusing IFNα2 with anti-VEGFR2 cannot only extend the in vivo half-life of IFNα2, but the anti-VEGFR2 will also be able to deliver IFNα2 to the tumor site where it will be active in the absence of systemic toxicity. Hence, IFNα2 carried to the tumor site can activate the immune response in the TME, produce direct toxic effect on tumor cells, and inhibit tumor angiogenesis. Our earlier studies had shown that anti-VEGFR2 exhibits an antitumor effect in breast cancer and leukemia.Citation29,30 We now extend these studies and show that anti-VEGFR2-IFNα2 (JZA01) has therapeutic efficacy both in vivo and in vitro against metastatic colorectal cancer.
Results
Generation and identification of JZA01
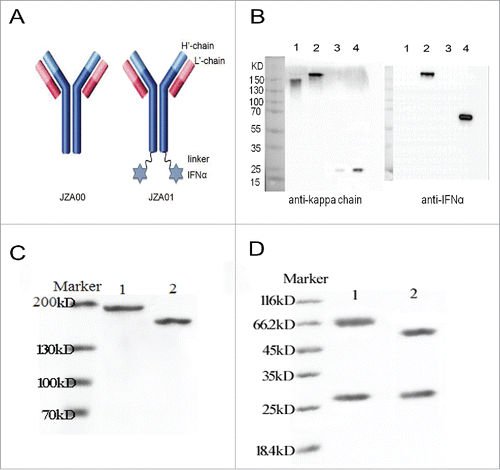
IFNα2 was fused to the carboxy terminus of the H chain of JZA00 through a G4S linker (). Western blot analysis, with anti-human kappa chain and anti-human IFNα following non-reducing and reducing SDS-PAGE, demonstrated that the fully assembled fusion protein JZA01 contained both JZA00 (150 kD) and 2 IFNα (19 kD) resulting in a molecular weight of around 190 kD with the H chain containing IFNα having a molecular weight of about 75 kD (). Similar results were seen with proteins purified by protein A affinity chromatography (unreduced ; reduced ).
Figure 1. Construction and characterization of JZA01. (A) Diagram of JZA01. IFNα2 was linked to the C-terminal of H′-chain of JZA00 with a G4S linker using overlap PCR yielding JZA01. (B) Western blot analysis of purified JZA01 with anti-kappa chain and anti-IFNα shows that it was properly assembled and was of the correct molecular weight. Lane 1: JZA00 non-reducing; Lane 2: JZA01 non-reducing; Lane 3: JZA00 reducing; Lane 4: JZA01 reducing. JZA01 and JZA00 were purified using protein A affinity chromatography and the purity was identified with (C) non-reducing SDS-PAGE and (D) reducing SDS-PAGE; Lane 1: JZA01; Lane 2: JZA00.
JZA01 bounds specifically to VEGFR2
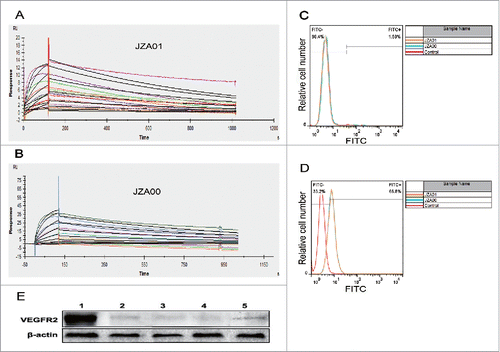
The affinity of human VEGFR2 also known as tyrosine kinase insert domain receptor (KDR) to immobilized JZA01 or JZA00 was determined by the Biacore system with calculations made using the 2:1 binding model. The association increased with the increasing concentration of the KDR3 (from bottom to top), ranging from 0.78125 nM to 100 nM. The complex dissociated when buffer flowed through after 120 s. Both JZA01 and JZA00 showed similar binding affinity: The affinity of JZA01 was KD: 3.72 × 10−9M (kon: 3.15 × 105 M−1 s−1, koff: 1.30 × 10−3 s−1; ), while that of JZA00 was KD: 4.92 × 10−9 M (kon: 2.09 × 105 M−1 s−1, koff: 1.03 × 10−3 s−1; ), indicating that the fusion retains the antigen binding ability of the parental antibody. Using flow cytometry, no binding was seen to human hepatic cell L02 (), while JZA00 and JZA01 bound equivalently to human umbilical vascular endothelial cells (HUVECs) (). To further investigate the expression of VEGFR2 in cancer cells, we detected the expression of VEGFR2 in HCT-116 and SW620 cells using western blot assay and found that there was VEGFR2 expression in HCT-116 and SW620 (). In conclusion, JZA00 and JZA01 are able to specifically bind with KDR and target HUVECs.
Figure 2. JZA01 binds to KDR and to cells expressing VEGFR2. (A) The binding affinity of JZA01 with KDR was determined with the Biacore system. To prepare chips, JZA01 was captured on a CM5 chips with bound anti-human IgG. Increasing concentrations of the KDR (from bottom to top) ranging from 0.78125 nM to 100 nM then flowed over the chip for 120 s. Binding affinity was calculated using the 2:1 model. (B) Binding affinity of JZA00 with KDR was determined as described in (A) with concentrations of KDR ranging from 0.78125 nM to 100 nM. (C) L02 cells or (D) HUVEC were incubated with 100 nM JZA00 or JZA01 for 1 h, washed three times with PBS, and then incubated with FITC labeled anti-human kappa chain and analyzed by flow cytometry assay. (E) Western blot analysis of VEGFR2 expression using either JZA01 or JZA00 as the primary antibody and HRP-conjugated goat-anti-human IgG H+L (Sino Biological Inc., China) as the secondary antibody. Lane 1: JZA01 HUVEC; Lane 2: JZA01 HCT-116; Lane 3: JZA00 HCT-116; Lane 4: JZA01 SW620; Lane 5: JZA00 SW620.
JZA01 inhibits HUVECs proliferation, migration, invasion, and tube formation
To study the anti-angiogenic activity of our fusion antibody, we studied its effects on the proliferation, migration, transwell invasion, and tube formation of HUVECs. Following 72 h of treatment, the growth inhibition of HUVECs by JZA01 exceeded that of JZA00 and was similar to that of IFNα in a dose-dependent manner (). JZA01 also played a role in inhibiting the migration of HUVECs with the inhibitory effect concentration dependent (). All treatments showed inhibition in the transwell invasion assay, with JZA01 being the most effective (). In the tube formation assay, JZA01 showed activity similar to IFNα, and both were superior to JZA00 (). Thus, JZA01 showed superior anti-angiogenic activity in vitro.
Figure 3. JZA01 inhibits the proliferation, migration, transwell invasion, and tube formation of HUVECs. (A) HUVECs were incubated with different concentrations of JZA01, JZA00, or IFNα and proliferation analyzed by MTT assay. Inhibition (%) was calculated as (1 − ODexperiment/ODcontrol) × 100%. (B) and (D) The inhibition of the migration of HUVECs by JZA01 added at different concentrations was analyzed by the scratch assay. The distance of migration at 0 h, 12 h, 16 h, 20 h, 24 h was measured using Image-Pro Plus 6.0 after being photographed by an inverted OLYMPUS microscope at 100× magnification. Inhibition (%) = (1 − lengthexperiment/lengthcontrol) × 100%. (C) and (E) Photomicrographs and quantitative analysis of transwell invasion assay indicated that JZA01 suppressed the invasion of HUVECs better than IFNα or JZA00 in a dose-dependent manner. Inhibition (%) = [(control – experiment)/ control] ×100%. (F) and (G) HUVECs were incubated for 4 h with different concentrations of the JZA00, JZA01, or IFNα in a 96-well plate pretreated with matrigel. The endothelial tubes were photographed by an inverted OLYMPUS microscope at 100× magnification with the number of endothelial tubes counted using Image-Pro Plus 6.0. Inhibition (%) = (1 − numberexperiment/numbercontrol) × 100%. Quantitative analysis were presented as the mean ± SD, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001.
![Figure 3. JZA01 inhibits the proliferation, migration, transwell invasion, and tube formation of HUVECs. (A) HUVECs were incubated with different concentrations of JZA01, JZA00, or IFNα and proliferation analyzed by MTT assay. Inhibition (%) was calculated as (1 − ODexperiment/ODcontrol) × 100%. (B) and (D) The inhibition of the migration of HUVECs by JZA01 added at different concentrations was analyzed by the scratch assay. The distance of migration at 0 h, 12 h, 16 h, 20 h, 24 h was measured using Image-Pro Plus 6.0 after being photographed by an inverted OLYMPUS microscope at 100× magnification. Inhibition (%) = (1 − lengthexperiment/lengthcontrol) × 100%. (C) and (E) Photomicrographs and quantitative analysis of transwell invasion assay indicated that JZA01 suppressed the invasion of HUVECs better than IFNα or JZA00 in a dose-dependent manner. Inhibition (%) = [(control – experiment)/ control] ×100%. (F) and (G) HUVECs were incubated for 4 h with different concentrations of the JZA00, JZA01, or IFNα in a 96-well plate pretreated with matrigel. The endothelial tubes were photographed by an inverted OLYMPUS microscope at 100× magnification with the number of endothelial tubes counted using Image-Pro Plus 6.0. Inhibition (%) = (1 − numberexperiment/numbercontrol) × 100%. Quantitative analysis were presented as the mean ± SD, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001.](/cms/asset/6dd0d322-4898-41ca-a1ee-7432e61f97c7/koni_a_1290038_f0003_oc.gif)
JZA01 inhibits proliferation and promotes apoptosis of colorectal cancer cells
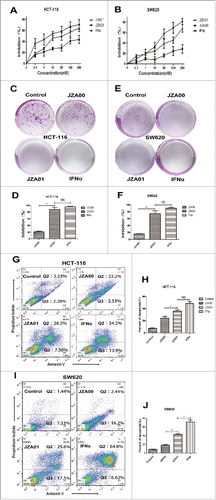
Although SW620 was found to be less sensitive to JZA00 treatment than HCT-116, JZA01 showed a greater growth inhibition of both cell lines than JZA00, resembling IFNα (), while 50 nM JZA00 barely inhibited colony formation of HCT-116 and SW620, few colonies were observed following treatment with 50 nM JZA01 or 100 nM IFNα (). Analysis showed that there was no significant difference between the JZA01- and IFNα-treated groups (HCT-116 p = 0.3525, SW620 p = 0.1245), while there was significant difference between the JZA01- and JZA00-treated groups (HCT-116 p = 0.0066, SW620 p = 0.0097) (). The detection of apoptotic cells using annexin V/PI staining showed significantly more apoptosis following 48 h treatment with 100 nM JZA01 than with 100 nM JZA00 although less apoptosis was seen in the JZA01-treated cells than in cells treated with 200 nM IFNα. (). SW620 cells were more resistant than HCT-116 to JZA00 but both exhibited increased apoptosis following treatment with JZA01 (). Although not as effective as free recombinant IFNα, JZA01 was superior to JZA00 in inhibiting proliferation and promoting apoptosis of both cell lines.
Figure 4. JZA01 inhibits the proliferation, colony formation, and promotes apoptosis of HCT-116 and SW620. (A) HCT-116 or (B) SW620 cells were incubated with different concentrations of JZA00, JZA01, or IFNα for 72 h and proliferation were quantified by MTT assay. Inhibition (%) = (1 − ODexperiment/ODcontrol) × 100%. (C) and (D) HCT-116 or (E) and (F) SW620 cells were diluted to 1000 /mL and incubated in 6-well plates with JZA00, JZA01, or IFNα at 50 nM for 7 d. The numbers of colonies formation was measured by Image-Pro Plus 6.0. Inhibition (%) = (1 − numberexperiment/numbercontrol) × 100%. Quantitative analysis is presented as the mean ± SD, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001. (G) HCT-116 and (I) SW620 cells were incubated with JZA00 100 nM, JZA01 100 nM, or IFNα 200 nM for 48 h and analyzed by flow cytometry following staining with PtdIns and Annexin V. (H) and (J) Annexin V positive cells in Q2 and Q3 were considered apoptotic and are shown quantitatively as the mean ± SD, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001.
JZA01 inhibits migration and invasion of colorectal cancer
HCT-116 () or SW620 () cells were incubated in 12-well plates until adherent. Supernatant was removed and then a straight line was drawn using a 1 mL tip in the middle of each well before adding 100 nM JZA00, JZA01, or 200 nM IFNα. Photographs were taken at 0 h, 12 h, 24 h, 48 h using an OLYMPUS inverted microscope at 100 times magnification. The migration of SW620 was slower than that of HCT-116. JZA01 was more effective than JZA00 in inhibiting the migration of both HCT-116 and SW620 but was not more effective than IFNα (). In the transwell invasion assay (), the inhibition by JZA01 of HCT-116 and SW620 was significantly more than that of JZA00 and slightly more than that of IFNα at the corresponding concentration (). In conclusion, JZA01 was more effective than JZA00 in inhibiting the invasion and migration of the tumor cells HCT-116 and SW620. JZA01 was similar to IFNα in inhibiting invasion, but it was not superior in inhibiting migration.
Figure 5. JZA01 inhibits the migration and invasion of HCT-116 and SW620. For the scratch test, (A) HCT-116 or (C) SW620 cells were incubated in 24-well plates with JZA00, JZA01, or IFNα and photographed at 0 h, 12 h, 24 h, and 48 h. (B) and (D) The migration lengths were measured by Image-Pro Plus 6.0 and data were presented as the mean ± SD, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001. Photomicrographs and quantitative analysis of transwell invasion assay indicated that JZA01 suppressed the invasion of HCT-116 (E) and (F) and SW620 (G) and (H) cells in a dose-dependent manner. % Inhibition = [(control – experiment)/ control] ×100%. Data were presented as the mean ± SD, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001.
![Figure 5. JZA01 inhibits the migration and invasion of HCT-116 and SW620. For the scratch test, (A) HCT-116 or (C) SW620 cells were incubated in 24-well plates with JZA00, JZA01, or IFNα and photographed at 0 h, 12 h, 24 h, and 48 h. (B) and (D) The migration lengths were measured by Image-Pro Plus 6.0 and data were presented as the mean ± SD, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001. Photomicrographs and quantitative analysis of transwell invasion assay indicated that JZA01 suppressed the invasion of HCT-116 (E) and (F) and SW620 (G) and (H) cells in a dose-dependent manner. % Inhibition = [(control – experiment)/ control] ×100%. Data were presented as the mean ± SD, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001.](/cms/asset/bc075fa4-86f2-4825-8dd5-c29a7c73c6c6/koni_a_1290038_f0005_oc.gif)
JZA01 regulates VEGFR2, STAT and apoptosis signaling pathway activation and causes cell cycle arrest of colorectal cancer cells
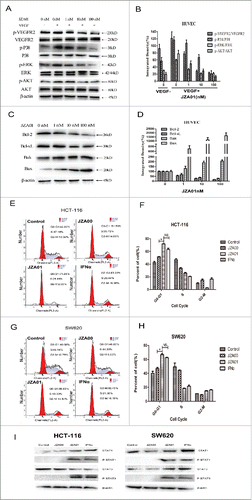
We investigated signaling pathway modulation following JZA01 treatment. With increasing concentrations of JZA01, the amount of p-VEGFR2, p-P38, p-ERK, and p-Akt decreased following VEGF treatment (). JZA01 treatment of 48 h also decreased the expression of Bcl−2 and Bcl−xL, and increased the expression of Bak and Bax () consistent with the fact that Bcl−xL and Bcl−2 inhibit both apoptosis and proliferation, while Bak and Bax promote apoptosis. The cell cycle analysis of HCT-116 and SW620 showed that a greater proportion of HCT-116 and SW620 was arrested at the G0/1 checkpoint following JZA01 treatment than following treatment with JZA00 for 48 h (). Nevertheless, both JZA01 and IFNα showed similar effect at the same concentration (). The activation of p-STAT1 and p-STAT3 was observed in HCT-116 and SW620 cells following 30 min treatment with JZA01 (100 nM) and IFNα (100 nM) but not JZA00 (100 nM) ().
Figure 6. JZA01 downregulates VEGFR2, STAT, and apoptosis signaling pathways and causes cell cycle arrest of colorectal cancer cells. (A) Western blot analysis for p-VEGFR2/VEGFR2, p-P38/P38, p-ERK/ERK, and p-AKT/AKT in HUVECs treated with different concentrations of JZA01 for 48 h. (B) Gray scanning and data statistics of (A) based on the integrated density of β-actin. (C) Western blot analysis of Bcl−2 family proteins in HUVECs treated with different concentrations of JZA01 for 48 h. (D) Gray scanning and data statistics of (C) based on the integrated density of 0 nM. (E) and (F) Cell cycle analysis by flow cytometry of HCT-116 (E) and (F) or SW620 (G) and (H) cells that incubated with JZA00, JZA01, or IFNα for 48 h. Data were presented as the mean ± SD, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001. (I) Western blot analysis of STAT activation in HCT-116 and SW620 following treatment with JZA01 (100 nM) for 30 min.
JZA01 treatment enhances DCs maturation and activity
In exploring JZA01-activated immune function, we found that after 48 h treatment with JZA01, the expression of MHC class I on the surface of HCT-116 increased by 17.2% () in comparison with the control group while that of SW620 increased by 30.6% (). There was a small increase of MHC class II expression after JZA01 or IFNα treatment (Fig. S1A and S1B). JZA01 retained ADCC (Fig. S1D and S1E) and CDC activity but with no enhancement compared with JZA00. Type I IFNs have been reported to potently activate dendritic cells. To determine if JZA01 has similar activating activity, mononuclear cells and immature dendritic cells isolated from human peripheral blood mononuclear cells (huPBMCs) were cultured in AIM-V media containing IL-4 (800 U/mL) and GM-CSF (150 ng/mL) for 4 d. Cells were then incubated with JZA00 (50 nM), JZA01 (50 nM), or IFNα (100 nM) for 48 h. Flow cytometry showed that the expression of CD80 (), CD86 (), MHC class I (), and MHC class II molecules (Fig. S1C) on the surface of DCs increased following JZA01 or IFNα treatment, while no significant increase was observed in the JZA00-treated group. In addition, JZA01 was similar to IFNα alone in promoting DCs maturation. When CD8+ T cells were labeled with CFSE before co-culturing with pretreated DCs at a ratio 10:1, flow cytometry showed a greater proliferation of CD8+ T cells following JZA01 and IFNα treatment than treatment with JZA00 (). The CD8+ T cells treated with JZA01 and IFNα showed greater cytotoxicity () and IFNγ production () than that of JZA00 treated groups. Interestingly, we found that when treated with JZA01, CD8+ T cells exhibited superior cytotoxicity and IFNγ production toward SW620 than HCT-116 cells, possibly reflecting the increased MHC class I expression. In conclusion, following treatment with JZA01, colorectal tumor cells and DCs are more likely to present antigens to CD8+ T cells mainly through the increased expression of MHC class I that may induce the enhanced production of tumor-specific cytotoxic T lymphocytes.
Figure 7. JZA01 treatment enhances DCs maturation and activity. (A) Flow cytometry assay of the expression of MHC class I of HCT-116 under different treatments (JZA00 50 nM, JZA01 50 nM, and IFNα 100 nM) for 48 h. (B) Flow cytometry assay of the expression of MHC class I of SW620 under different treatments (JZA00 50 nM, JZA01 50 nM, and IFNα 100 nM) for 48 h. (C), (D), and (E) Immature dendritic cells were isolated from non-activated huPBMCs and incubated under different treatments of 72 h. The expression of CD80, CD86, and MHC class I were detected by the flow cytometry assay. (F) CD8+ T cells labeled with CFSE were co-cultured for 48 h with DCs (10:1) pretreated with different treatments in (C) and isolated using human CD8+ T cells isolation kit (BD Bioscience;557766) before detected by flow cytometry assay. (G) and (H) CD8+ T cells together with DCs (10:1, in (F)) as effectors, E:T ratio 1:1, 5:1, 10:1, time point of 24 h, Abs 50 nM, IFNα 100 nM, cytotoxicity% were analyzed by LDH release assay. Data were presented as mean of three repeats. % Cytotoxicity = [(experimental − effector spontaneous − target spontaneous)/(target maximum − target spontaneous)]×100. (G) represents the cytotoxicity% of HCT-116, < (H) represented the cytotoxicity % of SW620. (I) and (J) CD8+ T cells together with DCs (10:1, in (F)) as effectors, E:T ratio 10:1, time point of 24 h, Abs 50 nM, IFNα 100 nM, the production of IFNγ was measured by ELISA kit. (I) HCT-116 as target cells; (J) SW620 as target cells.
![Figure 7. JZA01 treatment enhances DCs maturation and activity. (A) Flow cytometry assay of the expression of MHC class I of HCT-116 under different treatments (JZA00 50 nM, JZA01 50 nM, and IFNα 100 nM) for 48 h. (B) Flow cytometry assay of the expression of MHC class I of SW620 under different treatments (JZA00 50 nM, JZA01 50 nM, and IFNα 100 nM) for 48 h. (C), (D), and (E) Immature dendritic cells were isolated from non-activated huPBMCs and incubated under different treatments of 72 h. The expression of CD80, CD86, and MHC class I were detected by the flow cytometry assay. (F) CD8+ T cells labeled with CFSE were co-cultured for 48 h with DCs (10:1) pretreated with different treatments in (C) and isolated using human CD8+ T cells isolation kit (BD Bioscience;557766) before detected by flow cytometry assay. (G) and (H) CD8+ T cells together with DCs (10:1, in (F)) as effectors, E:T ratio 1:1, 5:1, 10:1, time point of 24 h, Abs 50 nM, IFNα 100 nM, cytotoxicity% were analyzed by LDH release assay. Data were presented as mean of three repeats. % Cytotoxicity = [(experimental − effector spontaneous − target spontaneous)/(target maximum − target spontaneous)]×100. (G) represents the cytotoxicity% of HCT-116, < (H) represented the cytotoxicity % of SW620. (I) and (J) CD8+ T cells together with DCs (10:1, in (F)) as effectors, E:T ratio 10:1, time point of 24 h, Abs 50 nM, IFNα 100 nM, the production of IFNγ was measured by ELISA kit. (I) HCT-116 as target cells; (J) SW620 as target cells.](/cms/asset/b70287c9-27be-4642-b86b-5512f027dbdb/koni_a_1290038_f0007_oc.gif)
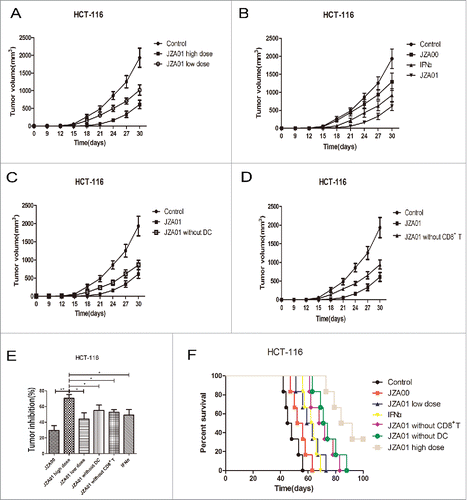
JZA01 suppresses tumor growth and prolongs survival of HCT-116 bearing NOD-SCID mice
Immunodeficient NOD-SCID mice were reconstituted with 1 × 107 huPBMCs isolated from healthy donors together with 5 × 106 HCT-116 cells implanted into the left armpit. The mice with established tumors were treated twice a week with JZA01, JZA00, or IFNα and tumor growth and survival were monitored for 100 d. All the treatments showed a dose-dependent protection against tumor growth with JZA01 demonstrating superior efficacy compared with both JZA00 and IFNα (). To determine whether CD8+ T cells and DCs play key roles in IFNα-mediated immune stimulation, CD8+ T cells or DCs depleted huPBMCs were used (). Tumor growth in CD8+ T cells or DCs depleted groups was more rapid than in mice reconstituted with non-depleted huPBMCs and treated with the same dose JZA01 (1 mg/kg) suggesting that DCs and CD8+ T cells were involved in mediating JZA01-stimulated immune protection (). The low dose of JZA01 (0.5 mg/kg) was more effective in prolonging the survival of HCT-116-bearing NOD-SCID mice than either JZA00 (1 mg/kg) or IFNα (200 μg/kg) at a higher dose (). Inconsistent with the in vitro data, less protection was seen in the IFNα-treated group, which might reflect the altered pharmacokinetics or off-target systemic toxicity. Mice treated with IFNα showed obvious signs of toxicity such as hair loss and anorexia, while no significant toxicity was seen in the JZA00- or JZA01-treated groups. In conclusion, JZA01 that specifically targeted the delivery of IFNα to activate the DC and CD8+ T cell immune response showed better therapeutic efficacy than JZA00 or IFNα in HCT-116-bearing NOD-SCID mice.
Figure 8. JZA01 has superior antitumor efficacy in HCT-116-bearing NOD-SCID mice. (A) and (B) Female NOD-SCID mice 4–6 weeks of age (n = 6 per group) were injected in the left armpit with 5 × 106 HCT-116 cells and 1 × 107 unstimulated huPBMC. (A) Mice was treated with 1 mg/kg (high dose) or 0.5 mg/kg (low dose) JZA01 (i.v.) twice a week with tumor growth followed for a month. (B) Mice were treated with 1 mg/kg of JZA01 or JZA00, or IFNα (200 μg/kg) twice a week and tumor volumes were monitored for a month. (C) and (D) Female NOD-SCID mice 4–6 weeks of age were injected in the left armpit each with 5 × 106 HCT-116 cells and 1 × 107 unstimulated huPBMC from which dendritic cells (C) or CD8+ T cells (D) had been removed. Mice (n = 6 per group) were treated with 1 mg/kg JZA01 twice a week (i.v.) beginning from day 0 for a month with tumor volumes monitored. (E) Tumor inhibition of different dosage groups. JZA01 high dose significantly improved tumor inhibition compared with other groups. Data represent the mean ± SD (n = 6 per group). *p < 0.05, **p < 0.01, ***p < 0.001. (F) The survival of tumor-bearing NOD-SCID mice for 100 d. Data are from the experiments shown in panels A-D. The JZA01-treated group had better survival compared with IFNα-treated group. Log-rank tests: JZA01 high dose versus JZA00, p < 0.001; JZA01 high dose versus JZA01 low dose, p = 0.0012; JZA01 high dose versus IFNα2, p < 0.001; JZA01 high dose versus JZA01 without DCs, p = 0.0409; JZA01 high dose versus JZA01 without CD8+ T, p = 0.0080.
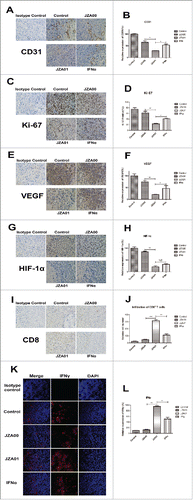
JZA01 influences the tumor microenvironment in HCT-116-bearing NOD-SCID mice
The NOD-SCID mice bearing established HCT-116-tumors were treated with JZA00, JZA01, or IFNα twice a week and killed on day 30 and the alteration of TME was analyzed using immunohistochemistry (IHC) and immunofluorescence (IF). In the tumor sections stained with anti-CD31, tumor neovascularization was found to decrease more in the JZA01-treated group (1 mg/kg), than in the JZA00- (1 mg/kg) or IFNα-treated groups (200 μg/kg) (). There also was a significant decrease in cell proliferation as indicated by Ki-67 expression in JZA01-treated tumors compared with JZA00- and IFNα-treated tumors (). Stress-inducible factors HIF-1α and VEGF are indicators of growing tumor tissue. Tumor sections stained with anti-VEGF () and anti-HIF-1α () showed a greater reduction staining in JZA01-treated group, implying combining anti-VEGFR2 and IFNα2 in JZA01 led to enhanced anti-angiogenic activity (). There was more infiltration of CD8+ T cells in JZA01-treated mice than mice treated with IFNα or JZA00 (). Furthermore, significantly more secretion and expression of IFNγ were observed in the JZA01-treated group than in JZA00- or IFNα-treated groups (), indicating that IFNα targeting by anti-VEGFR2 enhanced IFNα-stimulated immune responses and potentiated its influence on the TME.
Figure 9. JZA01 inhibits the HCT-116 tumor growth at least in part through influencing the tumor microenvironment in vivo. (A) CD31+ blood vessels were identified with an anti-CD31 antibody (brown staining). Photomicrographs show representative pictures from three independent tumor samples. (B) Semiquantitative and statistical analysis of CD31+ blood vessels. The CD31 density was calculated with Image Pro Plus. Relative expression of CD31 (%) = (Experiment/Control) ×100%. (C) Expression of Ki-67 in paraffin sections of xenografted tumor identified with an anti-Ki-67 antibody (brown staining). (D) The tumor cells expressing Ki-67 were counted with Image-Pro-Plus. Ki-67 positive (%) = (cell expressing Ki-67/total cells)×100%. (E) Staining of VEGF using anti-VEGF (brown staining). (F) Semiquantitative and statistical analysis of VEGF. (G) Expression of HIF-1α identified by anti-HIF-1α (brown staining). JZA01 was more effective in downregulating the expression of HIF-1α and VEGF than IFNα or JZA00. (H) Semiquantitative and statistical analysis of HIF-1α. VEGF and HIF-1α density were quantified with Image-Pro-Plus. Relative expression (%) = (Experiment/Control) ×100%. (I) The infiltration of CD8+ T cells was identified with an anti-CD8+ antibody (brown staining). (J) Semiquantitative and statistical analysis of the infiltration of CD8+ T cells. The relative number of CD8+ T cells was calculated with Image-Pro-Plus. (K) Expression of IFNγ in the tumor microenvironment was identified by staining with anti-IFNγ (red fluorescence) following different treatments. Expression of IFNγ in JZA01 group is the highest. All pictures were photographed by an inverted OLYMPUS microscope at 400× magnification. (L) Semiquantitative and statistical analysis of IFNγ expression made with Image-Pro-Plus. Relative expression (%) = (Experiment/Control) ×100%.
Discussion
Current treatment of colorectal cancer is surgery combined with chemotherapy or radiotherapy.Citation31 In recent years, drugs targeting cell receptors have significantly prolonged patients overall survival and improved the quality of life.Citation32,33 For example, Bevacizumab with 5-fluorine (5-FU) or capecitabine-based combination chemotherapy has become the first-line and second-line treatment of metastatic colorectal cancer. In clinical trials, Bevacizumab combined with IFNα can effectively prolong the survival of patients with mRCC. However, serious adverse reactions including hypertension, proteinuria, fatigue, and anorexia nervosa have limited its clinical application.Citation34,35 Therefore, it is urgent to reduce the toxic side effects and enhance therapeutic efficacy for the treatment of metastatic colorectal cancer.
In our study, the anti-VEGFR2 antibody JZA00 was fused with IFNα2. We found that our fusion antibody, JZA01, was more effective in inhibiting angiogenesis than JZA00 both in vivo and in vitro. It showed anti-angiogenic activity similar to IFNα in vitro but was more effective in vivo. JZA01 can regulate the VEGFR2 signaling pathway, downregulate the expression of VEGFR2-related downstream signaling proteins and reduce the expression of HIF-1α and VEGF to further inhibit tumor angiogenesis.Citation36 In addition, JZA01 was able to inhibit the invasion and migration of HUVECs and colorectal cancer cells demonstrating its potential for inhibiting tumor metastasis. Our data revealed that both SW620 and HCT-116 were more sensitive to direct killing, inhibition of proliferation, and induction of apoptosis by JZA01 than by JZA00. Although data showed that fusing IFNα to anti-VEGFR2 decreased the direct cytotoxicity of IFNα, anti-VEGFR2 and IFNα can play a synergistic role in suppressing tumor angiogenesis and metastasis. In HCT-116 tumor-bearing NOD-SCID mice, JZA01 was more effective in inhibiting the tumor growth and prolonging the survival than IFNα alone and was able to do so without toxic side effects.Citation37,38 Subsequent IHC and IF analyses showed that JZA01 was more effective than IFNα and JZA00 in stimulating the release of cytokines, suggesting it has improved efficacy on regulating the TME in vivo.
Most current strategies to improve the therapeutic effect of antibodies focus on increasing the direct tumor cell cytotoxicity of antibodies and cytotoxic drug conjugates.Citation39,40 In contrast, JZA01 cannot only inhibit proliferation, arrest cell cycle, and produce apoptosis of tumor cells, but can also inhibit tumor angiogenesis. More importantly, it can target the TME and activate the immune response. In our study, we found that JZA01 can more effectively upregulate the expression of MHC class I on the colorectal cancer cell lines HCT-116 and SW620 than JZA00, thereby facilitating the direct presentation of tumor cell antigens. Analysis of the expression of CD80, CD86, MHC class I, and MHC class II on the surface of DCs showed that JZA01 can promote the maturation of DCs, so that DCs will be better able to present antigens to CD8+ T cells and promote proliferation and IFNγ release of CD8+ T cells. DCs are the most important APCs for CTL inductionCitation41 Thus, activating DCs with JZA01 confers better immune recognition and killing of tumor cells. In vivo experiments showed that the antitumor effect of JZA01 decreased in mice depleted of DCs or CD8+ T cells suggesting that DCs and CD8+ T cells played a key role in the antitumor effect of JZA01. The CD8+ T cell infiltration and IFNγ released by mice treated with JZA01 were much greater than in mice treated with JZA00 or IFNα. Taken together, the IFNα in JZA01 retains the ability to activate the immune response. Furthermore, JZA01 is more effective than JZA00 or IFNα alone in activating the immune response that can probably be attributed to the TME targeting ability of anti-VEGFR2.
Overall, this study has some important implications for the field of antitumor immune therapy. We fused hIFNα2 with the anti-VEGFR2, to deliver IFNα2 to the TME, thereby inhibiting tumor angiogenesis, directly killing tumor cells, and activating the immune response. We found that anti-VEGFR2-IFNα2 was also effective against tumor cells such as SW620 that had not been previously shown to be sensitive to anti-VEGFR2 treatment. Antibody-cytokine fusions such as Ab-IFNα fusion proteins provide a new strategy for promoting the immunotherapy of cancer. Collectively, this study provides a new approach for the treatment of colorectal cancer and opens up several novel avenues for optimizing targeted immune therapy that may impact clinical cancer therapy.
Material and methods
Mice and cell lines
All animal experiments were conducted under protocols approved by the Ministry of Health of the People's Republic of China (Document No. 55, 2001) and the Guidelines for the Care and in accordance with the Provision and General Recommendation of Chinese Experimental Animals Administration Legislation and approved by the Science and Technology Department of Jiangsu Province (SYXK (SU) 2016–0011). All PBMC healthy donors were given informed consent for this study and approved by the ethical committee of China Pharmaceutical University. The NOD/SCID mice were purchased from Model Animal Research Center of Nanjing University (MARC). Animals used in experiments were between 4 and 8 weeks of age. The Chinese hamster ovary cell line CHO-pro (Amprotein Co., Ltd., Hangzhou, China) was cultured in IMDM medium (Gibco, Grand Island, USA), supplemented with 5% (v/v) fetal bovine serum (FBS, Gibco, Auckland, NZ). HUVECs were purchased from American Type Culture Collection and were maintained in endothelial culture medium supplemented with 5% (v/v) FBS and 1% (v/v) endothelial cell growth supplement (ECGS, ScienCell, San Diego, CA). The human colorectal cancer cell lines HCT-116 and SW620 cultured in 1640 medium (Gibco, Life, America) were obtained from the cell bank of the Chinese Academy of Science (Shanghai, China).
Construction, expression and purification of JZA00 and JZA01
The construction of anti-VEGFR2-IFNα2 was conducted as we previously reported. In brief, the variable region of Ramucirumab was used to replace the variable region of anti-CD138.Citation42 IFNα2a was fused to the carboxy terminus of the heavy chain with a flexible pentapeptide (Gly-Gly-GlyGly-Ser). Subsequently, the recombinant plasmid was introduced into CHO-pro cell through lipofectin-mediated transfection. Stable clones were selected using 1 mg/mL G418, 1 mM Histidinol (Sigma-Aldrich), and screened by ELISA (HRP-conjugated goat-anti-human IgG Fc, Sino Biological Inc., China) and western blot analysis (HRP-conjugated goat-anti-human IgG H+L, Sina Biological Inc., China). The fusion protein was purified from culture supernatants using protein A affinity chromatography (GE Healthcare, UPPsala, Sweden) and characterized by reducing and non-reducing SDS-PAGE (10%).
Binding affinity analysis
The binding affinity of JZA01 to tyrosine KDR (human VEGFR2) was measured by the Biacore X100 (GE Healthcare, Sweden). First, anti-human IgG (Fc) antibody was fixed on the CM5 chip with the Human Antibody Capture Kit (GE Healthcare, Sweden). Then, JZA01 was captured, and KDR was added into running buffer (HBS-EP, PH 7.4) with different concentrations ranging from 0.78125 nM to 100 nM. One flow cell of the sensor chip was set as a control. The captured antibody-analyte complex was removed by regeneration solution (3 M magnesium chloride). The association (kon) and dissociation (koff) rate constants were calculated and analyzed using the bivalent analytemodel and the equilibrium dissociation constant (KD) was calculated (KD = kon/koff).
Cell proliferation assay
Cells were plated into 96-well plates at 4 × 103 cells/well. After incubation with different concentrations of treatments at 37°C for 72 h, cell proliferation was quantified by MTT assay. Inhibition (%) was calculated as (1 − ODexperiment/ODcontrol) × 100%.
Colony formation
One × 103 cells were incubated with different treatments (JZA00 50nM, JZA01 50nM, and IFNα 100 nM) in 1 mL RPMI 1640 medium supplemented with 10% (v/v) FBS at 37°C for 7 d. Supernatant was removed, cells were fixed with 4% (v/v) paraformaldehyde for 20 min at room temperature, and stained with 1% (w/v) crystalline violet. Images were taken by Canon camera and the number of cell clones was counted with the Image-Pro-Plus program. Inhibition (%) = (1 − numberexperiment/numbercontrol) × 100%.
Tube formation
Matrigel (BD, Bioscience, USA) diluted, 1:1, with serum-free ECM was plated in 96-well plates, 50 μL/well and allowed to polymerize at 37°C for 1 h. Then, 2 × 104 HUVECs incubated with different concentrations of treatments in 100 µL of ECM supplemented with 2% (v/v) FBS, 1% (v/v) ECGS was added into each well. After 2–16 h incubation, endothelial tubes were photographed with an OLYMPUS inverted microscope under 100-time magnification and counted with the Image-Pro-Plus program. Inhibition (%) = (1 − numberexperiment/numbercontrol) × 100%.
Transwell invasion
One × 104 HUVECs suspended in ECM supplemented with 1% (v/v) FBS and various concentrations of the treatments were incubated in the upper well of 24-well transwell chambers (Millipore, Billerica, USA), which were pretreated with matrigel (BD, Bioscience, USA). The lower chambers were filled with 600 µL of ECM containing 5% (v/v) FBS and 1% (v/v) ECGS. After a 12–24 h incubation, the upper cells were removed, while the lower cells were fixed with 4% polyoxymethylene for 20 min and then stained with 1% (w/v) crystalline violet before being photographed under an OLYMPUS inverted microscope at 100-time magnification. The number of the invasive cells was determined with the Image-Pro-Plus program. Inhibition (%) = [(controlnumber – experimentnumber)/ controlnumber] ×100%.
Scratch assay
Two × 105/well HUVECs suspended in ECM containing 1% (v/v) ECGS and 5% (v/v) FBS were incubated in a 12-well plate until adherent. Supernatant was removed and then a straight line was drawn in the middle of each well with a 1 mL tip. ECM supplemented with different concentrations of the treatments was then added into the 12-well plate. Images were taken using an OLYMPUS inverted microscope at 100× magnification. The length of migration was analyzed using the Image-Pro-Plus program. Inhibition (%) = (1 − lengthexperiment/lengthcontrol) × 100%.
Apoptosis assay
HCT-116 or SW620 cells were incubated in 6-well plates with various treatments (JZA00 100 nM, JZA01 100 nM, and IFNα 200 nM) at 37°C for 48 h and the supernatant was removed and cells were washed three times with PBS followed by staining with annexinV-FITC and propidium iodide (PtdIns) for 30 min. After being washed with PBS for three times, the samples were analyzed by flow cytometry to distinguish populations of early apoptotic (annexin V+/PtdIns−), late apoptotic (annexin V+/PtdIns+), and necrotic (annexin V−/PtdIns+) using annexin V/PtdIns Apoptosis Assay Kit (Sangon Biotech, Shanghai, China). The percentage of apoptotic cells (annexin V+) was calculated as the sum of both early apoptotic (annexin V+/PtdIns−) and late apoptotic (annexin V+/PtdIns+).
Cell cycle assay
Three × 105 HCT-116 cells and SW620 cells were incubated in 6-well plates with various treatments (50 nM) at 37°C for 48 h. After that, the supernatant was removed and cells were washed three times with PBS and then fixed in 75% ethanol overnight. Subsequently, samples were stained with PtdIns (Beyotime Biotech, Shanghai, China) for 30 min and washed with PBS for three times before flow cytometry assay. All the assays were performed using a BD FACS flow cytometer and PtdIns fluorescence intensity represents the different cell cycles. The proportion of cells from each cell cycle was calculated with the MFLT32 software.
MHC class I and MHC class II expression on the tumor surface
Three × 105 HCT-116 or SW620 cells were cultured in 6-well plates at 37°C with different treatments (JZA00 50 nM, JZA01 50 nM, and IFNα 100 nM) for 48 h. After that, cells were, respectively, incubated with the anti-MHC class I (Bioscience, 560965) and anti-MHC class II (Bioscience, 560943) for 1 h before being analyzed by flow cytometry.
Maturation of dendritic cells
Mononuclear cells and immature dendritic cells were isolated from huPBMCs and incubated in AIM-V medium (Gibco, #087–0112DK) containing IL-4 (800 U/mL) and GM-CSF (150 ng/mL) for 4 d. Half of the medium was changed every 2 d. Different treatments (JZA00 50 nM, JZA01 50 nM, and IFNα 100 nM) were given on the fourth day for 48 h. After that, cells were, respectively, incubated with different antibodies (anti-CD80, Biosciences 560925; anti-CD86, Bioscience 560956; anti-MHC class I, Bioscience 560965; anti-MHC class II, Bioscience 560943) for 1 h in PBS containing 2% FBS and detected by flow cytometry.
CD8+ T cells activation and cytotoxicity assay
CD8+ T cells isolated from the huPBMCs (from five healthy donors) using a human CD8+ T isolation kit (BD Bioscience 557766) were labeled with CFSE (BD Bioscience 565082) and then co-cultured for 48 h with tumor cells and DCs (T:DC = 10:1) that had been pretreated as we described previously in the maturation of dendritic cells. Flow cytometry analysis was then performed and the expression of IFNγ was detected by the ELISA kit (Arigo, AGR80117). Data were analyzed with the FlowJo 7.6 software and GraphPad Prism 5. The ADCC and CDC assays were performed as previously reported.Citation29 Cytotoxicity was detected by the CytoTox 96 Nonradioactive Cytotoxicity kit (Promega, Madison, USA). LDH analysis was performed according to the manufacture's protocol. % Cytotoxicity = [(experimental − effector spontaneous − target spontaneous) / (target maximum − target spontaneous)]×100%.
HCT-116-transplanted NOD-SCID mice model and in vivo efficacy
Five × 106 HCT-116 cells mixed with 1 × 107 non-activated human PBMCs from five healthy donors were implanted into the left armpit of 6-week-old NOD/SCID mice (MARC of Nanjing University). CD8+ T cells were removed with human CD8+ T isolation kit (BD Bioscience, 557766) and DCs were removed with DC immunomagnetic beads (BD Bioscience, 558420). There were eight groups (six per group): control (without huPBMCs), control (with huPBMCs), JZA00 (1 mg/kg), JZA01 (1 mg/kg), JZA01 (0.5 mg/kg), JZA01 (1 mg/kg, without DCs), JZA01 (1 mg/kg, without CD8+ T cells), IFNα2 (200 μg/kg). All treatments were administered twice a week by tail vein injection from day 0 to a month. Growth of the tumor was measured every day and the tumor volume was determined using the formula V = (length × widthCitation2)/2. The survival of the NOD-SCID mice with xenografts was monitored until 100 d.
Immunohistochemistry and immunofluorescence analysis
The mice were killed and the tumors were made into paraffin tissue sections. Paraffin sections were cut into 5 μm sections and fixed in 4% paraformaldehyde. For IHC staining, the sections were incubated with CD31-, Ki67-, VEGF-, HIF-1α-, or CD8+-specific antibody (Cell Signaling Technology, Boston, USA), and horseradish peroxidase-labeled secondary antibody and analyzed with the Vectastain ABC Kit (Dako, Copenhagen, Denmark). For IF staining, the sections were incubated with anti-IFNγ (Abcam, Cambridge, UK) and slides were mounted with Vectashield mounting media containing DAPI before analyzed under fluorescence microscope.
Statistical analysis
All data are indicated as mean ± SD. Data were compared using the student's t-test and p < 0.05 was considered statistically significant. Analyses were performed with the GraphPad Prism software (San Diego, CA).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1290038_supplementary_data.zip
Download Zip (1.4 MB)Funding
This study was supported by the National Natural Science Foundation of China (NSFC81473125), the Natural Science Foundation of Jiangsu Province (BK20161459), Jiangsu Province Qinglan Project (2014), and a project Funded by the Priority Academic Program Development Innovation Program of Jiangsu Higher Education Institutions.
References
- Spill F, Reynolds DS, Kamm RD, Zaman MH. Impact of the physical microenvironment on tumor progression and metastasis. Curr Opin Biotechnol 2016; 40:41-8; PMID:26938687; http://dx.doi.org/10.1016/j.copbio.2016.02.007
- Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015; 348(6230):74-80; PMID:25838376; http://dx.doi.org/10.1126/science.aaa6204
- Pittet MJ. Behavior of immune players in the tumor microenvironment. Curr Opin Oncol 2009; 21(1):53-9; PMID:19125019; http://dx.doi.org/10.1097/CCO.0b013e32831bc38a
- Buchbinder EI, McDermott DF. Interferon, interleukin-2, and other cytokines. Hematol Oncol Clin North Am 2014; 28(3):571-83; PMID:24880948; http://dx.doi.org/10.1016/j.hoc.2014.02.001
- Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov 2007; 6(12):975-90; PMID:18049472; http://dx.doi.org/10.1038/nrd2422
- Farkas A, Kemeny L. Interferon-alpha in the generation of monocyte-derived dendritic cells: recent advances and implications for dermatology. Br J Dermatol 2011; 165(2):247-54; PMID:21410666; http://dx.doi.org/10.1111/j.1365-2133.2011.10301.x
- Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med 2011; 208(10):2005-16; PMID:21930765; http://dx.doi.org/10.1084/jem.20101159
- Paolini R, Bernardini G, Molfetta R, Santoni A. NK cells and interferons. Cytokine Growth Factor Rev 2015; 26(2):113-20; PMID:25443799; http://dx.doi.org/10.1016/j.cytogfr.2014.11.003
- Rizza P, Moretti F, Capone I, Belardelli F. Role of type I interferon in inducing a protective immune response: perspectives for clinical applications. Cytokine Growth Factor Rev 2015; 26(2):195-201; PMID:25466627; http://dx.doi.org/10.1016/j.cytogfr.2014.10.002
- Parmar S, Platanias LC. Interferons: mechanisms of action and clinical applications. Curr Opin Oncol 2003; 15(6):431-9; PMID:14624225; http://dx.doi.org/10.1097/00001622-200311000-00005
- Gresser I. Antitumour effects of interferons: past, present and future. Br J Haematol 1991; 79 Suppl 1:1-5; PMID:1718388; http://dx.doi.org/10.1556/OH.2013.29718
- Jabbour E, Kantarjian H, Cortes J, Thomas D, Garcia-Manero G, Ferrajoli A. PEG-IFN-alpha-2b therapy in BCR-ABL-negative myeloproliferative disorders: final result of a phase 2 study. Cancer 2007; 110(9):2012-8; PMID:17849460; http://dx.doi.org/10.1002/cncr.23018
- Flisiak R, Flisiak I. Albinterferon-alpha 2b: a new treatment option for hepatitis C. Expert Opin Biol Ther 2010; 10(10):1509-15; PMID:20828335; http://dx.doi.org/10.1517/14712598.2010.521494
- Jones TD, Hanlon M, Smith BJ, Heise CT, Nayee PD, Sanders DA, Hamilton A, Sweet C, Unitt E, Alexander G et al. The development of a modified human IFN-alpha2b linked to the Fc portion of human IgG1 as a novel potential therapeutic for the treatment of hepatitis C virus infection. J Interferon Cytokine Res 2004; 24(9):560-72; PMID:15450132; http://dx.doi.org/10.1089/jir.2004.24.560
- Xuan C, Steward KK, Timmerman JM, Morrison SL. Targeted delivery of interferon-alpha via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma. Blood 2010; 115(14):2864-71; PMID:20139095; http://dx.doi.org/10.1182/blood-2009-10-250555
- Trinh KR, Vasuthasawat A, Steward KK, Yamada RE, Timmerman JM, Morrison SL. Anti-CD20-interferon-beta fusion protein therapy of murine B-cell lymphomas. J Immunother 2013; 36(5):305-18; PMID:23719241; https://doi.org/10.1097/CJI.0b013e3182993eb9
- Kiselyov A, Balakin KV, Tkachenko SE. VEGF/VEGFR signalling as a target for inhibiting angiogenesis. Expert Opin Investig Drugs 2007; 16(1):83-107; PMID:17155856; http://dx.doi.org/10.1517/13543784.16.1.83
- Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2011; 2(12):1097-105; PMID:22866201; http://dx.doi.org/10.1177/1947601911423031
- Saharinen P, Eklund L, Pulkki K, Bono P, Alitalo K. VEGF and angiopoietin signaling in tumor angiogenesis and metastasis. Trends Mol Med 2011; 17(7):347-62; PMID:21481637; http://dx.doi.org/10.1016/j.molmed.2011.01.015
- Wu WZ, Sun HC, Shen YF, Chen J, Wang L, Tang ZY, Iliakis G, Liu KD. Interferon alpha 2a down-regulates VEGF expression through PI3 kinase and MAP kinase signaling pathways. J Cancer Res Clin Oncol 2005; 131(3):169-78; PMID:15662525; http://dx.doi.org/10.1007/s00432-004-0615-2
- Wang J, Li G, Wang Y, Tang S, Sun X, Feng X, Li Y, Bao G, Li P, Mao X et al. Suppression of tumor angiogenesis by metformin treatment via a mechanism linked to targeting of HER2/HIF-1alpha/VEGF secretion axis. Oncotarget 2015; 6(42):44579-92; PMID:26625311; http://dx.doi.org/10.18632/oncotarget.6373
- Summers J, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab plus interferon for advanced renal cell carcinoma. Oncologist 2010; 15(1):104-11; PMID:20061402; http://dx.doi.org/10.1634/theoncologist.2009-0250
- Smith NR, Baker D, Farren M, Pommier A, Swann R, Wang X, Mistry S, McDaid K, Kendrew J, Womack C et al. Tumor stromal architecture can define the intrinsic tumor response to VEGF-targeted therapy. Clin Cancer Res 2013; 19(24):6943-56; PMID:24030704; http://dx.doi.org/10.1158/1078-0432.CCR-13-1637
- Grignol VP, Olencki T, Relekar K, Taylor C, Kibler A, Kefauver C, Wei L, Walker MJ, Chen HX, Kendra K et al. A phase 2 trial of bevacizumab and high-dose interferon alpha 2B in metastatic melanoma. J Immunother 2011; 34(6):509-15; PMID:21654521; http://dx.doi.org/10.1097/CJI.0b013e31821dcefd
- Rini BI, Halabi S, Rosenberg JE, Stadler WM, Vaena DA, Ou SS, Archer L, Atkins JN, Picus J, Czaykowski P et al. Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206. J Clin Oncol 2008; 26(33):5422-8;PMID:18936475; http://dx.doi.org/10.1200/JCO.2008.16.9847
- Poole RM, Vaidya A. Ramucirumab: first global approval. Drugs 2014; 74(9):1047-58; PMID:24916147; http://dx.doi.org/10.1007/s40265-014-0244-2
- Casak SJ, Fashoyin-Aje I, Lemery SJ, Zhang L, Jin R, Li H, Zhao L, Zhao H, Zhang H, Chen H et al. FDA approval summary: ramucirumab for gastric cancer. Clin Cancer Res 2015; 21(15):3372-6; PMID:26048277; http://dx.doi.org/10.1158/1078-0432.CCR-15-0600
- Burkholder B, Huang RY, Burgess R, Luo S, Jones VS, Zhang W, Lv ZQ, Gao CY, Wang BL, Zhang YM et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim Biophys Acta 2014; 1845(2):182-201; PMID:24440852; http://dx.doi.org/10.1016/j.bbcan.2014.01.004
- Xie W, Liu F, Wang Y, Ren X, Wang T, Chen Z, Tang M, Sun F, Li Z, Wang M et al. VEGFR2 targeted antibody fused with MICA stimulates NKG2D mediated immunosurveillance and exhibits potent anti-tumor activity against breast cancer. Oncotarget 2016; 7(13):16445-61; PMID:26909862; http://dx.doi.org/10.18632/oncotarget.7501
- Ren X, Xie W, Wang Y, Xu M, Liu F, Tang M, Li C, Wang M, Zhang J. VEGFR2-targeted fusion antibody improved NK cell-mediated immunosurveillance against K562 cells. Immunol Res 2016; 64(4):1060-70; PMID:27154226; http://dx.doi.org/10.1007/s12026-016-8800-3
- McGuire S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv Nutr 2016; 7(2):418-9; PMID:26980827; http://dx.doi.org/10.3945/an.116.012211
- Moriarity A, O'Sullivan J, Kennedy J, Mehigan B, McCormick P. Current targeted therapies in the treatment of advanced colorectal cancer: a review. Ther Adv Med Oncol 2016; 8(4):276-93; PMID:27482287; http://dx.doi.org/10.1177/1758834016646734
- Board RE, Valle JW. Metastatic colorectal cancer: current systemic treatment options. Drugs 2007; 67(13):1851-67; PMID:17722954; http://dx.doi.org/10.2165/00003495-200767130-00004
- Deng T, Zhang L, Liu XJ, Xu JM, Bai YX, Wang Y, Han Y, Li YH, Ba Y. Bevacizumab plus irinotecan, 5-fluorouracil, and leucovorin (FOLFIRI) as the second-line therapy for patients with metastatic colorectal cancer, a multicenter study. Med Oncol 2013; 30(4):752; PMID:24174316; http://dx.doi.org/10.1007/s12032-013-0752-z
- Gruenberger B, Tamandl D, Schueller J, Scheithauer W, Zielinski C, Herbst F, Gruenberger T. Bevacizumab, capecitabine, and oxaliplatin as neoadjuvant therapy for patients with potentially curable metastatic colorectal cancer. J Clin Oncol 2008; 26(11):1830-5; PMID:18398148; http://dx.doi.org/10.1200/JCO.2007.13.7679
- Ban HS, Uto Y, Won M, Nakamura H. Hypoxia-inducible factor (HIF) inhibitors: a patent survey (2011-2015). Expert Opin Ther Pat 2016; 26(3):309-22; PMID:26882240; http://dx.doi.org/10.1517/13543776.2016.1146252
- Rossi EA, Rossi DL, Cardillo TM, Stein R, Goldenberg DM, Chang CH. Preclinical studies on targeted delivery of multiple IFNalpha2b to HLA-DR in diverse hematologic cancers. Blood 2011; 118(7):1877-84; PMID:21680794; http://dx.doi.org/10.1182/blood-2011-03-343145
- Levin D, Harari D, Schreiber G. Stochastic receptor expression determines cell fate upon interferon treatment. Mol Cell Biol 2011; 31(16):3252-66; PMID:21690295; http://dx.doi.org/10.1128/MCB.05251-11
- Fayad L, Offner F, Smith MR, Verhoef G, Johnson P, Kaufman JL, Rohatiner A, Advani A, Foran J, Hess G et al. Safety and clinical activity of a combination therapy comprising two antibody-based targeting agents for the treatment of non-Hodgkin lymphoma: results of a phase I/II study evaluating the immunoconjugate inotuzumab ozogamicin with rituximab. J Clin Oncol 2013; 31(5):573-83; PMID:23295790; http://dx.doi.org/10.1200/JCO.2012.42.7211
- Hurvitz SA, Dirix L, Kocsis J, Bianchi GV, Lu J, Vinholes J, Guardino E, Song C, Tong B, Ng V et al. Phase II randomized study of trastuzumab emtansine versus trastuzumab plus docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol 2013; 31(9):1157-63; PMID:23382472; http://dx.doi.org/10.1200/JCO.2012.44.9694
- Steinman RM. Decisions about dendritic cells: past, present, and future. Ann Rev Immunol 2012; 30:1-22; PMID:22136168; http://dx.doi.org/10.1146/annurev-immunol-100311-102839
- Yoo EM, Trinh KR, Tran D, Vasuthasawat A, Zhang J, Hoang B, Lichtenstein A, Morrison SL. Anti-CD138-targeted interferon is a potent therapeutic against multiple myeloma. J Interf Cytokine Res 2015; 35(4):281-91; PMID:25353626; http://dx.doi.org/10.1089/jir.2014.0125