
ABSTRACT
VXM01 is a first-in-kind orally applied tumor vaccine based on live attenuated Salmonella typhi carrying an expression plasmid encoding VEGFR2, an antigen expressed on tumor vasculature and a stable and accessible target for anti-angiogenic intervention. A recent randomized, placebo-controlled, phase I dose-escalation trial in advanced pancreatic cancer patients demonstrated safety, immunogenicity and transient, T-cell response-related anti-angiogenic activity of four priming vaccinations applied within one week. We here evaluated whether monthly boost vaccinations are safe and can sustain increased frequencies of vaccine-specific T cells.
Patients with advanced pancreatic cancer were randomly assigned at a ratio of 2:1 to priming with VXM01 followed by up to six monthly boost vaccinations, or placebo treatment. Vaccinations were applied orally at two alternative doses of either 106 colony-forming units (CFU) or 107 CFU, and concomitant treatment with standard-of-care gemcitabine during the priming phase, and any treatment thereafter, was allowed in the study. Immunomonitoring involved interferon-gamma (IFNγ) ELIspot analysis with long overlapping peptides spanning the entire VEGFR2 sequence.
A total of 26 patients were treated. Treatment-related adverse events preferentially associated with VXM01 were decreases in lymphocyte numbers in the blood, increased frequencies of neutrophils and diarrhea. Eight out of 16 patients who received at least one boosting vaccination responded with pronounced, i.e. at least 3-fold, increase in VEGFR2-specific T cell response over baseline levels. In the VXM01 vaccination group, VEGFR2-specific T cells peaked preferentially during the boosting phase with an average 4-fold increase over baseline levels.
In conclusion, prime/boost vaccination with VXM01 was safe and immunogenic and increased vaccine specific T cell responses compared with placebo treatment.
Introduction
In the past decade, advances in immuno-oncology and the unprecedented clinical success of immune checkpoint inhibition have paved the way for the development of adoptive transfer of genetically modified lymphocytes and cancer vaccination.Citation1-6
Pancreatic cancer is expected to become the third leading cause of cancer-related deaths in the United State.Citation7 More than 85% of the cases are pancreatic ductal adenocarcinomas (PDAC). The poor 5-y survival rate of 8% is largely due to the mostly advanced, non-resectable situation at clinical presentation, and to marked resistance to chemotherapy.Citation8-11 Treatment options are limited compared with other cancers, and since 2011, only one drug has been approved by the FDA.Citation12 Several approaches to immunotherapy for pancreatic cancer have been explored with little success in early clinical trials including immune checkpoint inhibitors,Citation13,14 therapeutic vaccines,Citation15-19 adoptive T cell therapy including chimeric antigen receptor transduced T cells,Citation20 monoclonal antibodies, and antibody-drug conjugates directed against various antigens, as well as adjuvant and cytokine therapies.Citation21 Clinical success of pancreatic cancer immunotherapy is limited by the peculiar microenvironment, which is characterized by the presence of a low number of tumor-specific CD8+ T cells, predominance of TH-2 polarized CD4+ T helper cells, impaired NK cell activity and increased frequencies of immune suppressive regulatory T cells, myeloid-derived suppressor cells, and M2 macrophages.Citation22 The dense and complex pancreatic tumor stroma, involving an anergic tumor vasculature not only supports tumor growth and promotes angiogenesis and metastasis, but also serves as a physical barrier that is not readily permeable to immune effector cells and drugs.Citation23 Thus, targeting such barriers will be instrumental for efficient pancreatic cancer immunotherapy. Current efforts focus on combination therapies using the immunogenic response generated by chemotherapy or radiation therapy,Citation24 and immune checkpoint blockade, to profoundly improve clinical responses and survival benefits in patients with pancreatic cancer.
Oral vaccination is a novel approach in cancer immunotherapy that exploits the large surface area and the immunologic contexture of the intestine for immune stimulation. Oral Salmonella typhimurium-mediated DNA vaccination has shown consistent anti-angiogenic and antitumor activity in different murine tumor models.Citation25-27 Moreover, an analogous human-specific carrier strain Salmonella typhi Ty21a has been thoroughly studied and is widely used as a vaccine for the prevention of typhoid fever.Citation28 The oral T cell vaccine VXM01 consists of live, attenuated Salmonella typhi carrying a eukaryotic expression plasmid that encodes for full length vascular endothelial growth factor receptor 2 (VEGFR2).Citation29 VEGFR2 is a high-affinity receptor for vascular endothelial growth factor-A (VEGF-A) and is the main mediator of the VEGF-A-related endothelial growth and survival signals.Citation30,31 VEGFR2 is highly expressed on tumor vasculature as well as on certain tumorCitation32 and immune cells.Citation33,34 Moreover, the modulation of tumor growth with anti-VEGF-A and VEGFR2 antibodies and small-molecule VEGFR2 inhibitors in cancer patients has established VEGFR2 as a therapeutic target in several cancer indications including pancreatic cancer.Citation35-40 We have recently reported safety, tolerability, immune responses, and preliminary efficacy data of the first-in-human oral antitumor vaccine VXM01 during a dose-escalation study. We demonstrated that four priming vaccinations with VXM01 applied throughout one initial week resulted in overall increased VEGFR2-specific, polyfunctional mixed CD8+ and CD4+ effector T cell responses and correlated with reduced tumor perfusion.Citation41 VEGFR2-specific T cell responses peaked after 21 d but rapidly declined within 3 mo to levels comparable to those before vaccination. We therefore conducted an extension trial to assess the safety and tolerability of a continuous boosting treatment with VXM01, and to explore whether this regimen can maintain elevated VEGFR2-specific T cell responses after the initial prime vaccination.
Results
Patients and treatment
Between August 2013 and February 2014, 27 patients with locally advanced, inoperable, or stage IV pancreatic cancer were enrolled and randomly assigned. One patient did not receive treatment due to serious pre-treatment adverse event (AE) leading to withdrawal from the study. Therefore, the analysis included 18 patients treated with VXM01, and 8 patients treated with placebo (isotonic sodium chloride). There were no statistical differences in the demographic baseline disease characteristics of the patients between the two groups, except for the gender distribution (). Moreover, the placebo group had a lower proportion of patients with metastatic disease, a Karnofsky performance status <90 or increased CA19-9 levels (both elevated and >1,000 U/mL). The only notable difference between the two dose groups of patients treated with VXM01 was the frequency of patients with CA19-9 levels higher than 1,000 U/mL (50% and 0% in the 106 and 107 CFU group, respectively).
Table 1. Patient demographics and clinical characteristics
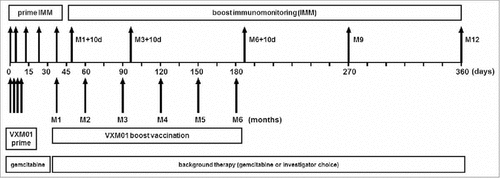
Treatment consisted of four priming vaccinations on days 1, 3, 5, and 7, followed by six monthly boost vaccinations beginning 1 mo after the last priming vaccination (). Vaccinations were orally given as a drink solution buffered with bicarbonate at two alternative doses of either 106 colony-forming unit (CFU, (N = 12)) or 107 CFU (N = 6) of VXM01. Concomitant treatment with standard-of-care gemcitabine up to day 38, and any treatment thereafter, was allowed in the study (Fig. S1). All the 26 enrolled patients entered the priming part and received all four priming doses of VXM01 (n = 18) or placebo treatment (n = 8). In total, 24 patients entered the boosting part and 22 patients received at least one boosting dose of VXM01 (n = 15) or placebo (n = 7). Finally, 11 out of 15 patients having entered the VXM01 boosting phase received all six boosting doses. The study was prematurely discontinued by 11 patients (n = 7 in the VXM01 arms, and n = 4 in the placebo arm), 3 patients during the priming part, and 8 patients during the boosting part due to death (n = 5), AEs (n = 1), deterioration of state of health (n = 1), or consent withdrawal (n = 4) as detailed in Fig. S2. The doses for boosting were identical to the doses administered for initial priming except for three patients in the 107-dose group who received a reduced dose, i.e., 106 CFU, due to the fecal excretion of VXM01 (see below). One patient in the VXM01 106 CFU arm was not treated with boosting administrations due to the fecal excretion of VXM01 and discontinued the study due to the withdrawal of consent.
Figure 1. Study design including vaccination schedule and immunomonitoring time points. During the prime phase of the trial, patients received four prime doses of VXM01 or placebo as add-on to their standard chemotherapy with gemcitabine on days 1, 3, 5, and 7. Boosting vaccination with VXM01 or placebo was started 1 mo after the last prime administration and performed following a 1× per month administration schedule, as add-on to their background therapy, i.e., gemcitabine or investigator choice on days 38 (Month 1; M1), 60 (M2), 90 (M3), 120 (M4), 150 (M5), and 180 (M6). T cell response was measured during the priming phase on days 0, 4, 14, 21, and 38, and during the boosting phase on days 48 (M1+10 d), 100 (M3+10d), 190 (M6+10d), 270 (M9), and 360 (M12). IMM, immunomonitoring.
Adverse events (AE)
In total, 576 treatment-emergent adverse events (TEAEs) were reported, 369 TEAEs (160 after prime and 209 after boosting doses) in 18 patients after VXM01 administration, and 207 TEAEs (95 after prime and 112 after boosting doses) in 8 patients after placebo administration. Most of the TEAEs were of mild and moderate severity (). Severe AEs were reported in six patients after prime (n = 3 in the VXM01 arms, n = 3 in the placebo arm) and 14 patients after boost vaccination (n = 10 in the VXM01 arms, n = 4 in the placebo arm). The most frequent TEAEs of any grade skewed toward the VXM01 treatment group after the prime vaccination were decreases in platelets (44.4% vs. 12.5%) and in lymphocytes (27.8% vs. 12.5%), nausea (16.7% vs. 0%), and diarrhea (22.2% vs. 12.5%) (), confirming the findings of the previous study.Citation41 Treatment-related TEAEs preferentially associated with boosting doses of VXM01 were decreased in lymphocytes (22% vs. 0%), and increased in neutrophil count (16.7% vs. 0%) and diarrhea (22% vs. 0%). There were no marked differences in nature, frequency, and severity of TEAEs between the two VXM01 doses tested (data not shown).
Table 2. Incidence and cumulative number of adverse events in priming and boosting vaccination phases
Excretion in fecal samples was observed up to day 38 in 2 out of 12 patients (16.7%) in the lowest dose group and in 4 out of 6 patients (66.7%) in the highest dose group after the administration of VXM01. Both patients who excreted VXM01 at a dose of 106 CFU had a gastric bypass. No such anatomic changes were present in patients who excreted VXM01 after administration of 107 CFU. Subsequent stool cultures were negative without any antibiotic intervention. Neither VXM01 nor plasmid DNA was found or excreted in any other body fluids in any patient throughout the study (data not shown).
Two patients, both from the highest dose VXM01 treatment arm, were screened positive, i.e., with anti-LPS antibody indexes >1.1, for IgG antibodies against the carrier bacterium at day 0 and 38, but none of the patients were screened positive for anti-LPS IgM (Fig. S3). Seroconversion was not detected in any of the patients up to 9 mo after the first administration, except for one patient in the lower dose VXM01 treatment arm who experienced two episodes of diarrhea between months 3 and 6, presumably due to food-borne Salmonella infection, with a positive result at months 6 and 7. Moreover, we did not observe any seroconversion upon further follow-up of the patients up to 24 mo.
T cell responses
Assessment of VEGFR2-specific T cell responses was conducted by interferon gamma (IFNγ) enzyme-linked immunosorbent spot (ELIspot) analysis.Citation41 T cells were stimulated by autologous dendritic cells (DC) (T cell:DC ratio 5:1) that were pulsed with (i) a pool of long overlapping synthetic peptides spanning the entire VEGFR2 sequence, (ii) equal amounts of human IgG that served as negative control antigen, or (iii) with a mixture of viral recall antigens derived from CMV and adenovirus as positive control. The respective original mean IFNγ spot counts from all patients are shown in Table S1. A few patients in both treatment arms (6 out of 26) showed the presence of preexisting VEGFR2-specific T cells already before the vaccination, i.e., a significant (p < 0.05) difference between the VEGFR2 and control spot count mean values and a mean absolute spot count at D0 1.5 times higher in VEGFR2-containing wells than in control wells (data not shown). The course of VEGFR2-specific T cell responses in each individual patient was recorded as either (i) the difference between mean IFN-gamma spot counts in VEGFR2-containing wells and in negative control wells (), (ii) the fold increase of VEGFR2-specific spots over d0 values (Fig. S4A) or (iii) the fold increase of mean spot counts in VEGFR2-containing wells over those in negative control wells (Fig. S4B). In total, 66.7% (8 out of 12) of the patients in the lower dose group and 75% (3 out of 4) of the patients in the higher dose group showed a clear increase, i.e., >1.8-fold, of VEGFR2-specific T cell responses after priming or boosting. The magnitude and time course of the IFNγ T cell response displayed distinct response profiles encompassing immune responses (a) remaining at low baseline levels throughout the study, (b) peaking rapidly after the priming phase and returning to baseline levels, (c) peaking early after the initiation of the boosting phase and declining before the end of the boosting phase, and (d) rising gradually during and after the boosting phase. In the higher dose group, T cell responses in responding patients consistently peaked after one to three boosting vaccinations, and in all cases vaccine-specific T cells declined after the last vaccination, reaching baseline levels after 1 y in the remaining two patients.
Figure 2. Analysis of the VEGFR2-specific T cell response by IFNγ ELISpot. (A) The individual and (B) mean absolute specific IFNγ T cell response to DC loaded with long synthetic peptides derived from VEGFR2, defined as the negative control subtracted from the absolute SFU count per 105 cells and per well (means of triplicates), (C) adjusted p-values obtained after multiple comparison (ANOVA) between the indicated groups at the indicated time point, and (D) fold change of the specific IFNγ T cell response over baseline level (day 0) are plotted over time. Each symbol represents the mean of the absolute specific IFNγ response (B) or the fold change over baseline level (D) at the indicated time point for all patients in the placebo (•, blue curves; N = 8), VXM01 106 CFU (■, yellow curves; N = 12), and VXM01 107 CFU (▴, red curves; N = 6) groups. Error bars represent the SEM. (E) The area under the curve was computed for the specific IFNγ T cell responses over the duration of the study in each patient. The red bar represents the mean of area under curve values for all patients in the corresponding arm. (F) The strength of treatment-associated VEGFR2-specific T cell responses was categorized for each evaluable patient and time point according to a pre-defined grading strategy, where grade 0 represents a non-significant difference between test wells and negative control wells (unpaired t-test) or no increased difference of absolute spot numbers in test and negative control wells compared with d0 and grades 1–3 represent a significant difference between test and control wells and increased difference of absolute spot numbers in VEGFR2 and negative control wells compared with day 0 < 3-fold (grade 1), ≥ 3-fold and < 5-fold (grade 2), and ≥ 5-fold (grade 3). These response grade values are plotted staggered individually over time into different panels corresponding to the indicated treatment arm. The red (VXM01 107 CFU), yellow (VXM01 106 CFU), and blue (placebo) staircase curves represent the medians of the grade values for all patients in the corresponding arm at the indicated time point. In all panels, the green tick marks and the light gray rectangles represent the vaccination, i.e., priming and boosting, time points and periods, respectively. ns: not significant, and #: statistically significant difference (p < 0.05) as determined by two-tailed t-test. CFU, colony-forming unit; SFU, spot-forming unit.
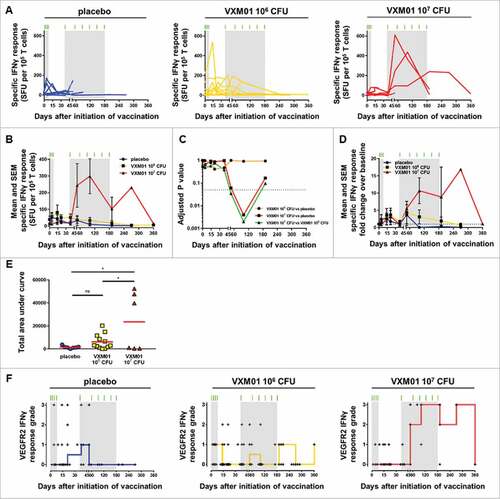
Altogether, mean VEGFR2 T cell responses in vaccinated patients exceeded those in placebo patients (). Due to the large heterogeneity in individual response patterns including non-responding patients, the mean difference between the placebo group and the lower VXM01 dose group was small and statistically not significant. However, VEGFR2 responses were significantly increased in the higher VXM01 dose group compared with both the placebo group and the lower dose group (). Adjusted p values for group-wise comparisons of mean VEGFR2-specific IFN-gamma spots conducted for all time points revealed respective statistical significance for increased spot counts in the high-dose group at day 90 (). A statistical trend toward an increased T cell response was further detectable in this group compared with the placebo group and the lower dose group at day 60 and in comparison to the lower dose group also at day 120.
In the lower dose group, the mean VEGFR2-specific IFNγ T cell response reached a peak already after the priming phase (81 ± 155 spot-forming units (SFU) per 105 T cells at day 15). Subsequently, vaccine-specific T cells declined slowly over time because only a minority of the patients in this group showed a clear response to the boosting vaccination (). In contrast, patients receiving the high-dose VXM01 vaccination showed an overall strong anti-VEGFR2 response particularly after the first boosting vaccination, reaching a peak value of 299 ± 180 SFU per 105 T cells at day 100, and remaining higher than 100 SFU per 105 T cells up to day 270. This strong increase of the vaccine-specific T cell response was observed in three patients out of four (75%) that entered the boosting phase (Fig. S4A). In the placebo group, VEGFR2-specific T cell responses declined over time and were below the baseline levels beyond day 100. In contrast in both VXM01 arms vaccine-specific T cells remained elevated at 1.9 and 4.4 times the baseline level up to day 180 (Fig. S4B) and 1.1- and 16.9-times the baseline level up to day 360 in patients vaccinated with 106 CFU and 107 CFU VXM01, respectively (). Due to these differences in T cell response kinetics, we compared the overall strength of VEGFR2-specific T cell response between the groups not only in regard to individual time points, but also by integrating response values at all time points as the “area under the curve.” Again, respective cumulative data shown in demonstrate significantly increased VEGFR2-specific T cell responses in the high-dose group. Despite increased T cell responses in several patients there was no overall significant increase in VEGFR2-specific T cell responsiveness in the lower dose group.
For a more systematic comparison of T cell responses in vaccinated and placebo-treated patients, we graded the increase of VEGFR2-specific T cell responses throughout the observation period according to the study protocol using prefixed criteria, which had been previously determined in the frame of the previously reported VXM01 trial,Citation41 as detailed in the Patients and Methods section. We detected increases in VEGFR2-specific T cell response grade throughout and early after the priming phase in both placebo and vaccinated patients (). These were also observed in placebo-treated patients of the previous clinical trial and thus potentially related to concomitant treatment regimen applied to both patient groups. For example, gemcitabine has previously been shown to exert immune stimulatory effects in cancer patients.Citation42,43 VEGFR2-specific T cell responses vanished over time in the placebo group, with median VEGFR2 T cell response below grade 1 after day 48 (). In contrast, strong immune responses persisted in several of VXM01-vaccinated patients and the median VEGFR2-T cell response grades remained higher or equal to 1, and higher or equal to 2 up to day 270, in the low and high-dose VXM01 groups, respectively (). Altogether, we observed in the boosting period pronounced, i.e., grade 2 and 3, T cell responses in 5 out of 11 patients who received at least one boosting vaccination and who had evaluable ELIspot results.
Regarding clinical outcome, we did not detect statistically significant differences between placebo and VXM01-treated patients. No clear trend was observed for VEGFA or collagen IV serum levels, and no significant difference was observed when comparing the tumor marker CA19-9 between the different treatment arms during the study. The tumor perfusion, as evaluated with DCE-MRI,Citation44,45 only increased slightly in patients treated with 106 CFU VXM01 after the boosting period. No alteration in the blood pressure levels was observed in the different treatment groups in this study (data not shown).
Discussion
We show here that a prime-boost regimen with VXM01, a first-in-kind, oral T cell vaccine, based on recombinant live attenuated Salmonella typhi vector targeting VEGFR2-expressing cells, can be safely administered and can elicit strong and sustained T cell responses against VEGFR2. Side effects were mostly mild and in accordance to those reported previously for a VXM01 priming-only regimen.Citation41 Importantly, the frequency of drug-related TEAEs were comparable after prime and boost doses indicating that further dosing with VXM01 did not increase the nature, severity, and frequency of TEAEs.
The aim of the study was not to characterize the polyfunctional nature of induced vaccine-specific T cells, nor the correlation with anti-angiogenic and clinical activities, because these issues had been already addressed in detail in the preceding VXM01 main study.Citation41 The main objective of this study was rather to explore whether repetitive administration of VXM01 after an initial priming period could maintain higher frequencies of VEGFR2-specific T cells. We were also interested in exploring a potential dose-effect of VXM01, although the cohort size of the highest dose group was kept small because we had not observed such an effect in the VXM01 main study.Citation41
In contrast to the main study, the results obtained from the extension study suggested an improved T cell response in the highest dose group. This might be due to the fact that the study cohorts of both trials differed from each other in regard to the levels of pre-existing VEGFR2-specific T cells that were frequent in the main study but rare in the extension study. Spontaneous T cell responses against tumor cell-associated antigens have been previously detected by us and others in a broad variety of different tumor entities, including pancreatic cancer.Citation46-51 It appears possible that the reactivation of pre-existing T cells in the main study was efficiently induced already by lower doses of the vaccine, while higher doses may be particularly effective in eliciting and maintaining new T cell responses against the vaccination antigen. Nevertheless, data from the extension trial correspond very well to the subgroup of patients in the main study who presented with low numbers of pre-existing VEGFR2-specific T cells.Citation41 Likewise, in both studies, the placebo groups revealed a transient early increase of VEGFR2-specific T cells during the first 2 weeks, possibly caused by concomitant immunogenic chemotherapies.Citation52-54
Vaccination of pancreatic cancer patients against tumor-stroma-associated antigens such as VEGFR2 is hampered by central thymic and peripheral tolerance against auto-antigens and by the well-established strong immunosuppressive tumor microenvironment in this entity.Citation49-51 Our study is further limited by the overall small study group, high inter-individual variations in T cell response patterns, and tumor burden among vaccinated and placebo patients and by the potentially confounding effect of the preceding gemcitabine treatment. Nevertheless, our cumulative data provide evidence for the capacity of VXM01 prime-boost regimens to induce, boost, and maintain throughout the vaccination period VEGFR2-specific T cells at elevated levels in highly advanced pancreatic cancer patients. This was particularly and consistently apparent in the higher dose group. It may be important to note that after peaking during the boosting period, VEGFR2-specific T cell responses tend to decline in most patients despite continued vaccination although their levels remained elevated as compared with baseline levels. T cell responses fully declined to baseline levels only after discontinuation of the boosting regimen. Thus, the continuation of the vaccinations might be suitable to maintain VEGFR2-specific T cells at elevated levels even in advanced stage pancreatic cancer patients. We assume that the reduction of VEGFR2-specific T cells after the peak was either due to reduced immunogenicity of the vaccine, to tolerogenic mechanisms or exhaustion, and/or may have been be related to a mobilization of induced T cells into VEGFR2-expressing target tissuesCitation41
The intestinal mucosa represents a highly immunologically active, large, and orally accessible interface, and thus represents an attractive organ for vaccination. Not only can high numbers of T cells be activated in this mucosal compartment and the related mesenteric lymph nodes,Citation55,56 but also are those T cells prone to acquire the cellular machinery for homing, accumulation, and proper cytotoxic activation within the gastrointestinal tract, governing gastrointestinal and mucosal tumors such as pancreatic, gastric, or colonic cancer or, potentially, lung cancer.Citation57
In conclusion, this study in patients with highly advanced pancreatic cancer demonstrated safety and excellent tolerability of a prime-boost regimen for VXM01 as well as promising specific T cell activity, in particularly when administered at the highest doses of 107 CFU. Based on the observed immunological response and the favorable safety profile, VXM01 is being further explored in colorectal cancer and in glioblastoma, and its combination with anti-PD-L1 immune checkpoint inhibition blockade is under development for the treatment of colorectal cancer and squamous cell carcinoma of the head and neck.
Patients and methods
Patients
Eligible patients had inoperable, locally advanced, or stage IV pancreatic cancer, completed at least one cycle of gemcitabine treatment, were chemotherapy naïve within 60 d before the screening visit except gemcitabine treatment, had a Karnofsky index above 70 and an anticipated life expectancy of > 3 mo, and an adequate bone marrow reserve, hepatic, and renal function at the start of the trial. Exclusion criteria included autoimmune disease; major surgery or recent significant trauma; uncontrolled hypertension or arterial thromboembolic events in the prior 6 mo, congestive heart failure ≥grade III, serious ventricular arrhythmia, peripheral arterial occlusive disease >grade 2b, hemoptysis, or gastrointestinal bleeding or other relevant conditions; and concomitant treatment or treatment in the prior 2 weeks with antibiotics, anti-angiogenic anticancer therapy, systemic corticosteroids, or any other immunosuppressive agents.
Study design
The monocentric study (EudraCT No 2015-003068-34) was a randomized, placebo-controlled, phase I trial consisting of a double-blind priming period (four oral doses) and an open-label boosting period (up to six single monthly doses). This is an extension and bridging study to the phase I dose escalation study completed previously.Citation41 Patients were randomly assigned to VXM01 or placebo at a ratio of 2:1 with a computer-generated random list, such that in both dose groups (106 and 107 CFU), patients were also assigned to placebo. Randomization was concealed so that neither patients, nor the investigator, nor the sponsor knew which agent was being administered. To maintain masking, a sodium bicarbonate-containing drinking solution with either VXM01 or isotonic sodium chloride (placebo) was prepared by personnel not otherwise involved in the study. The data safety monitoring board (DSMB) had access to the random list. All patients were included in the safety analysis. Only minor protocol violations were recorded.
The primary objective of the study was to assess safety and immunologic responses of the prime-boost regimen. The study was reviewed by the responsible institutional review board (Ethics Committee of the Medical Faculty of Heidelberg University) and approved by the federal authority (Paul-Ehrlich-Institut, Langen, Germany). The trial was conducted according to the Declaration of Helsinki and the Good Clinical Practice guidelines of the International Conference of Harmonisation. All participants provided written informed consent before entering the study.
Treatment
During the first part of the extension study, eligible patients were hospitalized from day 0 to day 10. The patients received the prime doses of VXM01 (N = 18) or placebo (N = 6) as add-on to their standard chemotherapy gemcitabine on days 1, 3, 5, and 7 (). The tested doses were 106 CFU for the first dose group, and 107 CFU for the second dose group. Boosting re-vaccination with up to six single oral doses of VXM01 or placebo was started 1 mo after the last prime administration and performed following a once-per-month administration schedule, as add-on to their background therapy, i.e., gemcitabine or investigator choice after day 38 (Fig. S1) on days 38 (Month 1; M1), 60 (M2), 90 (M3), 120 (M4), 150 (M5), and 180 (M6) (). The boosting doses were identical to the doses administered for initial vaccination (priming) as long as no reduction of dose was required due to positive fecal excretion results. Patients treated with 107 CFU during prime treatment with positive excretion results received a reduced dose of 106 CFU in the boosting part. Detailed activities during the prime and boosting parts of the study are provided in the flowchart (Fig. S2).
Clinical laboratory and safety evaluation
Hematology, clinical chemistry, coagulation, and urinalysis were measured up to M18. Medical and surgical history, cancer history, status and anticancer treatment, physical examination, vital signs, electrocardiogram, and blood pressure were also documented. AEs and TEAEs were graded according to the National Cancer Institute's CTCAE criteria (version 4.03). In addition, VXM01 distribution and shedding was investigated in blood, saliva, tears, urine, and stool samples on days 0, 2, 4, and 8.
Anticarrier antibodies (anti-LPS IgG and IgM) as well as anti-VEGFR2 antibodies (IgG and IgM) were analyzed by a validated enzyme-linked immunosorbent assay (ELISA) according to GLP regulations on days 0, 38 and months 3, 6, and 9, by Aurigon (Tutzing, Germany) and ATRC (Dunakeszi, Hungary) after assay transfer.
Serum levels of the angiostatic biomarkers collagen IV and VEGF-A were analyzed by commercial ELISA assays (Human Collagen IV ELISA, Serum, KT-035; Kamiya Biomedical Company, Seattle, US, and Quantikine Human VEGF-A Immunoassay; DVE00, R&D Systems GmbH, Wiesbaden, Germany, respectively) according to GLP regulations on days 0, 38 and months 3, 6, and 7 by Aurigon (Tutzing, Germany) and ATRC (Dunakeszi, Hungary) after assay transfer. The level of the tumor marker CA19-9 was measured in serum samples on days 0, 38 and months 6 and 7.
Tumor perfusion was analyzed by dynamic contrast enhanced magnetic resonance imaging (DCE-MRI) on a 1.5 Tesla System (Siemens Avanto, Erlangen, Germany) at day 0, day 38, and months 3, 6, and 7.
Immunologic assessment of the T cell response
Immunomonitoring of VEGFR2-specific T cells was performed on days 0, 4, 14, 21, 38, 48 (M1+10 d), 100 (M3+10 d), 190 (M6+10 d), 270 (M9), and 360 (M12) () in peripheral-blood lymphocytes isolated from cryopreserved peripheral-blood mononuclear cells (PBMCs) from all patients included in this study () by IFNγ ELIspot assay. PBMCs were isolated by the NCT laboratory, Heidelberg, Germany.
Whole blood was obtained by venipuncture using lithium heparin blood collection tubes (Sarstedt). Transportation of blood was done under ambient conditions to the Monitoring Laboratory. Processing of PBMCs was performed using density gradient centrifugation (Biochrome, Berlin, Germany) in 50 mL Falcon tubes (with frit) filled with ca. 18 mL of blood and ca. 15 mL of RPMI medium. PBMCs were processed and cryopreserved within a median time of 4 h after blood draw. The average cell yield, determined with trypan blue manual count after processing, was 1.1×106 PBMCs per mL of whole blood.
For cryopreservation, slow freezing method was performed using freezing medium composed of 70% fetal calf serum (Biochrom), 20% X-VIVO 20 medium (Lonza). and 10% DMSO (Sigma Aldrich). Aliquots of 1.0 to 1.2 × 107 cells/mL/vial were stored in slow freezing containers (Nalgene) at −80°C for up to 3 d, and then transferred to liquid nitrogen for long-term storage.
After thawing, the average cell viability was 90% and the mean cell yield of viable cells was 70%, as determined by Trypan blue (Sigma Aldrich) viability test. As cells underwent a 1-week cell culture before being plated for ELISpot, no additional cell count was performed.
Autologous dendritic cells were used as antigen-presenting cells. PBMCs were thawed and cultured for 4 h, and non-adherent cells were washed off and cultured in X-VIVO 20 medium (Lonza) supplemented with 100 U/mL IL-2 and 60 U/mL IL-4 (Miltenyi Biotec, Bergisch Gladbach, Germany). For dendritic cell generation, adherent cells were cultured with 560 U/mL of recombinant human GM-CSF and 500 U/mL IL-4 (Miltenyi). After 7 d of cell culture, cells were cultured in cytokine free media (X-VIVO 20; Lonza) for 24 h and purified. Dendritic cells were enriched by negative selection using the Pan Mouse IgG Dynabeads® (Invitrogen, Darmstadt, Germany), coupled to mouse anti-human CD3, CD19 (both LifeTechnology, Oslo, Norway), and CD56 (clone: C218, Beckman Coulter, Krefeld, Germany) antibodies. T cell purification was performed using the Untouched™ Human T Cells system (Invitrogen).
The quantitative evaluation of antigen-specific T cells secreting IFNγ was conducted as described previouslyCitation41 ELIspot plates (MAHA S4510; Millipore, Ireland) were coated with 1 µg of anti-IFNγ antibody (clone 1-D1K; Mabtech, Nackastrand, Sweden) per well, incubated overnight at 4 °C, washed and blocked with complete RPMI medium (10% human AB serum). 2 × 104 dendritic cells were plated in 100 µL per well in triplicate, and loaded with 10 µg/mL of antigenic peptides. A pool of 20-mer synthetic peptides #1-135 derived from human VEGFR2 with 10 amino acids overlap (omitting #21+22+76+77) (Proimmune, Oxford, UK) was used as test peptides. A pool of 15-mer synthetic peptides covering the complete sequence of pp65 protein (CMV pp56 PepTivator) with 11 amino acids overlap, mixed with a pool of 15-mer synthetic peptides covering the complete sequence of the hexon protein of human adenovirus 5 with 11 amino acids overlap (AdV5 Hexon PepTivator), as well as SEB (Staphylococcus enterotoxin B), was used as a positive control. Human Normal Immunoglobulin (KIOVIG, Baxter) was used as a negative control. After 14 h of antigen pulsing, 1 × 105 purified T cells (DynaBeads) were added to each well for an additional 40-h co-culture. IFNγ spots were developed using an enzyme-coupled detection antibody system using an anti-IFNγ antibody coupled to biotin (clone 7_B6-1 diluted 1:1000, Mabtech), streptavidin ALP (diluted 1:1000, Mabtech), and NCT/BCIP substrate kit (BioRad, Munich, Germany). Plates were analyzed using an automated ELIspot reader and ImmunoSpot V 5.0.9 Smart Count Software (CTL, Bonn, Germany) using the following spot definition settings: Size: 100%; Size Count: normal; Maximum Spot Size: 9.6250 mm2; Minimum Spot size 0.0051 mm2; Spot Separation: 3; Diffuse Processing: normal; Background Balance: 0–80.
The strength of the VEGFR2-specific T cell response after vaccination was graded for each time point in patients matching the acceptance criterion of a minimum mean of 10 SFU at day 0 according to the following rules: (a) grade 0: non-significant difference between test wells and negative control wells, as evaluated with t-test, or no increased difference of absolute spot numbers in test and negative control wells compared with day 0; (b) grade 1–3: significant difference between test and control wells, as evaluated using unpaired t-test, and increased ratio of absolute spot numbers in VEGFR2 to negative control wells compared with day 0 lower than three-fold (grade 1), higher than or equal three-fold and less than five-fold (grade 2), and higher than or equal five-fold (grade 3). Grading definition criteria were pre-defined in the statistical analysis plan.
The laboratory has not fully established good clinical laboratory practice conditions. Sample reception, PBMC processing, PBMC slow freezing for long-term storage, thawing of PBMCs, and cell counting were performed according to established laboratory protocols that were then approved as standard operation procedures. ELIspot was performed according to established laboratory protocols. The study was performed using general research investigative but pretested assays. For all experiments, raw data can be provided upon request.
Statistical analysis
Data were analyzed by two-tailed t-test (ELIspot), and one-way ANOVA with Tukey's multiple comparison tests. All statistical analyses, including the area under curve analysis, were performed using GraphPad Prism v.7.01 (GraphPad Software, Inc., LaJolla, CA, USA).
Abbreviations
| AE | = | adverse event |
| CFU | = | colony-forming unit |
| DC | = | dendritic cell |
| DSMB | = | data safety monitoring board |
| IFNγ | = | interferon gamma |
| IMM | = | immunomonitoring |
| PDAC | = | pancreatic ductal adenocarcinoma |
| SFU | = | spot-forming unit |
| TEAE | = | treatment-emergent adverse event |
| VEGFR2 | = | vascular endothelial growth factor receptor 2 |
Disclosure of potential conflicts of interest
FHSW, MWB, WEH and PB are advisors to VAXIMM AG. HL is an officer of VAXIMM GmbH, and HL and MS own stock options of VAXIMM AG. SW and AVK are employees of VAXIMM. KB is a director of VAXIMM AG. All other authors and MWB declare that they have no conflicts of interest.
Author contributions
Conception and design: FHSW, TS, LP, NH, TF, HL, MS, KB, GM, MWB, AVK, RK, CS, PK, MB, LG, WEH, and PB
Provision of study materials or patients: FHSW, TS, NH, TF, GM, MWB, RK, CS, PK, LG, WEH, and PB
Collection and assembly of data: FHSW, TS, LP, NH, TF, HL, MS, SW, KB, GM, MWB, AVK, RK, CS, PK, MB, LG, WEH, and PB
Data analysis and interpretation: FHSW, TS, LP, NH, TF, HL, MS, SW, KB, GM, MWB, AVK, RK, CS, PK, MB, LG, WEH, and PB
KONI_A_1303584_supplementary_data.zip
Download Zip (818.8 KB)Acknowledgments
DSMB Members: Andreas Engert, Department for Internal Medicine, Cologne, Germany; Christoph Huber, Immunology Cluster of Excellence, Mainz, Germany; Meinhard Kieser, Medical Biometry and Informatics, Heidelberg, Germany. VAXIMM AG provided the study drug and funding for the conduct of the study. SAE and trial management: The Coordination Center for Clinical Trials (KKS) Heidelberg. Preliminary results of this trial were presented at the American Society of Clinical Oncology annual meeting 2016.
Additional information
Funding
References
- Hodi FSS, O'Day SJJ, McDermott DFF, Weber RWW, Sosman JAA, Haanen JBB, Gonzalez R, Robert C, Schadendorf D, Hassel JCC et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711-23; PMID:20525992; https://doi.org/10.1056/NEJMoa1003466
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al. Safety, activity, and immune correlates of anti-pd-1 antibody in cancer. N Engl J Med 2012; 366:2443-54; PMID:22658127; https://doi.org/10.1056/NEJMoa1200690
- Morrissey K, Yuraszeck T, Li CC, Zhang Y, Kasichayanula S. Immunotherapy and novel combinations in oncology: current landscape, challenges, and opportunities. Clin Transl Sci 2016; 9:89-104; PMID:26924066; https://doi.org/10.1111/cts.12391
- Palucka AK, Coussens LM. The basis of oncoimmunology. Cell 2016; 164:1233-47; PMID:26967289; https://doi.org/10.1016/j.cell.2016.01.049
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015; 348:62-8; PMID:25838374; https://doi.org/10.1126/science.aaa4967
- Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol 2016; 13:273-90; PMID:26977780; https://doi.org/10.1038/nrclinonc.2016.25
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: Cancer J Clin 2016; 66:7-30; PMID:26742998; https://doi.org/10.3322/caac.21332
- Garrido-Laguna I, Hidalgo M. Pancreatic cancer: from state-of-the-art treatments to promising novel therapies. Nat Rev Clin Oncol 2015; 12:319-34; PMID:25824606; https://doi.org/10.1038/nrclinonc.2015.53
- Rishi A, Goggins M, Wood LD, Hruban RH. Pathological and molecular evaluation of pancreatic neoplasms. Semin Oncol 2015; 42:28-39; PMID:25726050; https://doi.org/10.1053/j.seminoncol.2014.12.004
- Waddell N, Pajic M, Patch A, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015; 518:495-501; PMID:25719666; https://doi.org/10.1038/nature14169
- Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014; 74:2913-21; PMID:24840647; https://doi.org/10.1158/0008-5472.CAN-14-0155
- Wang-Gillam A, Li CP, Bodoky G, Dean A, Shan YS, Jameson G, MacArulla T, Lee KH, Cunningham D, Blanc JF et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet 2016; 387:545-57; PMID:26615328; https://doi.org/10.1016/S0140-6736(15)00986-1
- Foley K, Kim V, Jaffee E, Zheng L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016 Oct 10; 348(1):244-51; https://doi.org/10.1016/j.canlet.2015.12.020
- Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 2010; 33:828-33; PMID:20842054; https://doi.org/10.1097/CJI.0b013e3181eec14c
- Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, Wamwea A, Bigelow E, Lutz E, Liu L et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother 2015; 38:1-11; PMID:25415283; https://doi.org/10.1097/CJI.0000000000000062
- Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, Springett G, Morse M, Zeh H, Cohen D, Fine RL et al. Safety and survival with GVAX pancreas prime and Listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol 2015; 33:1325-33; PMID:25584002; https://doi.org/10.1200/JCO.2014.57.4244
- Salman B, Zhou D, Jaffee EM, Edil BH, Zheng L. Vaccine therapy for pancreatic cancer. Oncoimmunology 2013; 2:e26662; PMID:24498551; https://doi.org/10.4161/onci.26662
- Yamamoto K, Ueno T, Kawaoka T, Hazama S, Fukui M, Suehiro Y, Hamanaka Y, Ikematsu Y, Imai K, Oka M et al. MUC1 peptide vaccination in patients with advanced pancreas or biliary tract cancer. Anticancer Res 2005; 25:3575-9; PMID:16101182
- Bernhardt SL, Gjertsen MK, Trachsel S, Moller M, Eriksen J A, Meo M, Buanes T, Gaudernack G. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: A dose escalating phase I/II study. Br J Cancer 2006; 95:1474-82; PMID:17060934; https://doi.org/10.1038/sj.bjc.6603437
- Abate-Daga D, Rosenberg S a, Morgan R a. Pancreatic cancer. Oncoimmunology 2014; 3:e29194; PMID:25083334; https://doi.org/10.4161/onci.29194
- Kunk PR, Bauer TW, Slingluff CL, Rahma OE. From bench to bedside a comprehensive review of pancreatic cancer immunotherapy. J Immunother cancer 2016; 4:14; PMID:26981244; https://doi.org/10.1186/s40425-016-0119-z
- Wachsmann M, Pop L, Vitetta E. Pancreatic ductal adenocarcinoma: A review of immunologic aspects. J Investig Med. 2012; 60:643-63; PMID:22406516; https://doi.org/10.2310/JIM.0b013e31824a4d79
- Kleeff J, Beckhove P, Esposito I, Herzig S, Huber PE, Löhr JM, Friess H. Pancreatic cancer microenvironment. Int J Cancer 2007; 121:699-705; PMID:17534898; https://doi.org/10.1002/ijc.22871
- Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, Pfirschke C, Voss R, Timke C, Umansky L et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013; 24:589-602; PMID:24209604; https://doi.org/10.1016/j.ccr.2013.09.014
- Xiang R, Luo Y, Niethammer AG, Reisfeld RA. Oral DNA vaccines target the tumor vasculature and microenvironment and suppress tumor growth and metastasis. Immunol Rev 2008; 222:117-28; PMID:18363997; https://doi.org/10.1111/j.1600-065X.2008.00613.x
- Niethammer AG, Xiang R, Becker JC, Wodrich H, Pertl U, Karsten G, Eliceiri BP, Reisfeld RA. A DNA vaccine against VEGF receptor 2 prevents effective angiogenesis and inhibits tumor growth. Nat Med 2002; 8:1369-75; PMID:12415261; https://doi.org/10.1038/nm1202-794
- Reisfeld RA, Niethammer AG, Luo Y, Xiang R. DNA vaccines designed to inhibit tumor growth by suppression of angiogenesis. Int Arch Allergy Immunol 2004; 133:295-304; PMID:14988601; https://doi.org/10.1159/000077009
- Fraser A, Paul M, Goldberg E, Acosta CJ, Leibovici L. Typhoid fever vaccines: Systematic review and meta-analysis of randomised controlled trials. Vaccine 2007; 25:7848-57; PMID:17928109; https://doi.org/10.1016/j.vaccine.2007.08.027
- Niethammer AG, Lubenau H, Mikus G, Knebel P, Hohmann N, Leowardi C, Beckhove P, Akhisaroglu M, Ge Y, Springer M et al. Double-blind, placebo-controlled first in human study to investigate an oral vaccine aimed to elicit an immune reaction against the VEGF-Receptor 2 in patients with stage IV and locally advanced pancreatic cancer. BMC Cancer 2012; 12:361; PMID:22906006; https://doi.org/10.1186/1471-2407-12-361
- Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: A crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2011; 2:1097-105; PMID:22866201; https://doi.org/10.1177/1947601911423031
- Shibuya M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem 2013; 153:13-9; PMID:23172303; https://doi.org/10.1093/jb/mvs136
- Bruce D, Tan PH. Vascular endothelial growth factor receptors and the therapeutic targeting of angiogenesis in cancer: Where do we go from here? Cell Commun Adhes 2011; 18:85-103; PMID:22017472; https://doi.org/10.3109/15419061.2011.619673
- Suzuki H, Onishi H, Wada J, Yamasaki A, Tanaka H, Nakano K, Morisaki T, Katano M. VEGFR2 is selectively expressed by FOXP3high CD4+ Treg. Eur J Immunol 2010; 40:197-203; PMID:19902430; https://doi.org/10.1002/eji.200939887
- Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet A-L, Latreche S, Bergaya S, Benhamouda N, Tanchot C et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med 2015; 212:139-48; PMID:25601652; https://doi.org/10.1084/jem.20140559
- Sennino B, McDonald DM. Controlling escape from angiogenesis inhibitors. Nat Rev Cancer 2012; 12:699-709; PMID:23001349; https://doi.org/10.1038/nrc3366
- Spratlin JL, Cohen RB, Eadens M, Gore L, Camidge DR, Diab S, Leong S, O'Bryant C, Chow LQM, Serkova NJ et al. Phase I pharmacologic and biologic study of ramucirumab (imc-1121b), a fully human immunoglobulin G1 monoclonal antibody targeting the vascular endothelial growth factor receptor-2. J Clin Oncol 2010; 28:780-7; PMID:20048182; https://doi.org/10.1200/JCO.2009.23.7537
- Spratlin J. Ramucirumab (IMC-1121B): Monoclonal antibody inhibition of vascular endothelial growth factor receptor-2. Curr. Oncol. Rep. 2011; 13:97-102; PMID:21222245; https://doi.org/10.1007/s11912-010-0149-5
- Okamoto I, Arao T, Miyazaki M, Satoh T, Okamoto K, Tsunoda T, Nishio K, Nakagawa K. Clinical phase I study of elpamotide, a peptide vaccine for vascular endothelial growth factor receptor 2, in patients with advanced solid tumors. Cancer Sci 2012; 103:2135-8; PMID:22957712; https://doi.org/10.1111/cas.12014
- Miyazawa M, Ohsawa R, Tsunoda T, Hirono S, Kawai M, Tani M, Nakamura Y, Yamaue H. Phase I clinical trial using peptide vaccine for human vascular endothelial growth factor receptor 2 in combination with gemcitabine for patients with advanced pancreatic cancer. Cancer Sci 2010; 101:433-9; PMID:19930156; https://doi.org/10.1111/j.1349-7006.2009.01416.x
- Yamaue H, Tsunoda T, Tani M, Miyazawa M, Yamao K, Mizuno N, Okusaka T, Ueno H, Boku N, Fukutomi A et al. Randomized phase II/III clinical trial of elpamotide for patients with advanced pancreatic cancer: PEGASUS-PC Study. Cancer Sci 2015; 106:883-90; PMID:25867139; https://doi.org/10.1111/cas.12674
- Schmitz-Winnenthal FH, Hohmann N, Niethammer AG, Friedrich T, Lubenau H, Springer M, Breiner KM, Mikus G, Weitz J, Ulrich A et al. Anti-angiogenic activity of VXM01, an oral T-cell vaccine against VEGF receptor 2, in patients with advanced pancreatic cancer: A randomized, placebo-controlled, phase 1 trial. Oncoimmunology 2015; 4:e1001217; PMID:26137397; https://doi.org/10.1080/2162402X.2014.1001217
- Lesterhuis WJ, Salmons J, Nowak AK, Rozali EN, Khong A, Dick IM, Harken JA, Robinson BW, Lake RA. Synergistic effect of CTLA-4 blockade and cancer chemotherapy in the induction of anti-tumor immunity. PLoS One 2013; 8:1-8; PMID:23626745; https://doi.org/10.1371/journal.pone.0061895
- Ding ZC, Huang L, Blazar BR, Yagita H, Mellor AL, Munn DH, Zhou G. Polyfunctional CD4+ T cells are essential for eradicating advanced B-cell lymphoma after chemotherapy. Blood 2012; 120:2229-39; PMID:22859605; https://doi.org/10.1182/blood-2011-12-398321
- Kim SH, Lee JM, Gupta SN, Han JK, Choi BI. Dynamic contrast-enhanced MRI to evaluate the therapeutic response to neoadjuvant chemoradiation therapy in locally advanced rectal cancer. J Magn Reson Imag 2014; 40:730-7; PMID:24307571; https://doi.org/10.1002/jmri.24387
- Stieltjes B, Meissner M, Ing D, Zimmer F, Burkholder I, Kroll A, Combs SE, Vogt-schaden M, Giesel FL, Zoubaa S et al. Biopsy targeting gliomas. Invest Radiol 2010; 45:755-68; PMID:20829706; https://doi.org/10.1097/RLI.0b013e3181ec9db0
- Sommerfeldt N, Beckhove P, Ge Y, Schütz F, Choi C, Bucur M, Domschke C, Sohn C, Schneeweis A, Rom J et al. Heparanase: A new metastasis-associated antigen recognized in breast cancer patients by spontaneously induced memory T lymphocytes. Cancer Res 2006; 66:7716-23; PMID:16885374; https://doi.org/10.1158/0008-5472.CAN-05-2363
- Schmitz-Winnenthal FH, Volk C, Z'graggen K, Galindo L, Nummer D, Ziouta Y, Bucur M, Weitz J, Schirrmacher V, Buchler MW et al. High frequencies of functional tumor-reactive T cells in bone marrow and blood of pancreatic cancer patients. Cancer Res 2005; 65:10079-87; PMID:16267034; https://doi.org/10.1172/JCI39608
- Sommerfeldt N, Schütz F, Sohn C, Förster J, Schirrmacher V, Beckhove P. The shaping of a polyvalent and highly individual T-cell repertoire in the bone marrow of breast cancer patients. Cancer Res 2006; 66:8258-65; PMID:16912206; https://doi.org/10.1158/0008-5472.CAN-05-4201
- Bonertz A, Weitz J, Pietsch DH, Rahbari NN, Schlude C, Ge Y, Juenger S, Vlodavsky I, Khazaie K, Jaeger D et al. Antigen-specific Tregs control T cell responses against a limited repertoire of tumor antigens in patients with colorectal carcinoma. J Clin Invest 2009; 119:3311-21; PMID:19809157; https://doi.org/10.1172/JCI39608
- Jandus C, Bioley G, Dojcinovic D, Derré L, Baitsch L, Wieckowski S, Rufer N, Kwok WW, Tiercy JM, Luescher IF et al. Tumor antigen-specific FOXP3+ CD4 T cells identified in human metastatic melanoma: Peptide vaccination results in selective expansion of Th1-like counterparts. Cancer Res 2009; 69:8085-93; PMID:19808957; https://doi.org/10.1158/0008-5472.CAN-09-2226
- Baumgaertner P, Rufer N, Devevre E, Derre L, Rimoldi D, Geldhof C, Voelter V, Liénard D, Romero P, Speiser DE. Ex vivo detectable human CD8 T-cell responses to cancer-testis antigens. Cancer Res 2006; 66:1912-6; PMID:16488988; https://doi.org/10.1158/0008-5472.CAN-05-3793
- Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, Martin F, Apetoh L, Rébé C, Ghiringhelli F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res 2010; 70:3052-61; PMID:20388795; https://doi.org/10.1158/0008-5472.CAN-09-3690
- Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: Implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ 2014; 21:15-25; PMID:23787994; https://doi.org/10.1038/cdd.2013.67
- Zitvogel L, Kepp O, Kroemer G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol 2011; 8:151-60; PMID:21364688; https://doi.org/10.1038/nrclinonc.2010.223
- Macpherson AJ, Smith K. Mesenteric lymph nodes at the center of immune anatomy. J Exp Med 2006; 203:497-500; PMID:16533891; https://doi.org/10.1084/jem.20060227
- Perez-Lopez A, Behnsen J, Nuccio S-P, Raffatellu M. Mucosal immunity to pathogenic intestinal bacteria. Nat Rev Immunol 2016; 16:135-48; PMID:26898110; https://doi.org/10.1038/nri.2015.17
- Sandoval F, Terme M, Nizard M, Badoual C, Bureau M-F, Freyburger L, Clement O, Marcheteau E, Gey A, Fraisse G et al. Mucosal imprinting of vaccine-induced CD8+ T cells is crucial to inhibit the growth of mucosal tumors. Sci Transl Med 2013; 5:172ra20-172ra20; PMID:23408053; https://doi.org/10.1126/scitranslmed.3004888