
ABSTRACT
The CXCR3 ligands CXCL9, 10, and 11 play critical roles in the amplification of immune responses by recruiting CXCR3+ immune effector cells to the tumor site. Taking advantage of this property of CXCR3 ligands, we aimed to establish a novel approach to identify immunogenic mutated-antigens. We examined the feasibility of using CXCR3 ligand mRNAs as sensors for detection of specific immune responses in human and murine systems. We further investigated whether this approach is applicable for the identification of immunogenic mutated-antigens by using murine sarcoma lines. Rapid synthesis of CXCR3 ligand mRNAs occurred shortly after specific immune responses in both human and murine immune systems. Particularly, in CMS5 tumor-bearing mice, we detected specific immune responses to mutated mitogen-activated protein kinase 2 (ERK2), which has previously been identified as an immunogenic mutated-antigen. Furthermore, by combining this approach with whole-exome and transcriptome sequencing analyses, we identified an immunogenic neo-epitope derived from mutated staphylococcal nuclease domain-containing protein 1 (Snd1) in CMS7 tumor-bearing mice. Most importantly, we successfully detected the specific immune response to this neo-epitope even without co-administration of anti-cytotoxic T-lymphocyte protein-4 (CTLA-4), anti-programmed cell death-1 (PD-1) and anti-glucocorticoid-induced TNFR-related protein (GITR) antibodies, which vigorously augmented the immune response and consequently enabled us to detect the specific immune response to this neo-epitope by conventional IFNγ intracellular staining method.
Our data indicate the potential usefulness of this strategy for the identification of immunogenic mutated-antigens. We propose that this approach would be of great help for the development of personalized cancer vaccine therapies in future.
Introduction
Cellular immune responses against mutated antigens originating from somatic genomic alterations occurring in an individual's tumor play critical roles in controlling and eradicating tumors.Citation1-3 One line of evidence supporting this notion comes from recent advances in immune checkpoint blockade therapies, which utilize monoclonal antibodies (mAbs) targeting CTLA-4 and PD-1.Citation1,4-6 Although the precise mechanism by which these antibodies exert their effect is still not fully understood, animal and human clinical studies indicate that neo-antigen specific autologous T cells, activated by these checkpoint inhibitors, play critical roles in killing tumor cells and impeding tumor growth. This concept is further supported by the fact that tumors with a high load of somatic mutations are more likely to respond to this therapy.Citation5,7,8
A second line of evidence supporting the role of immune responses against mutated tumor antigens for cancer treatment comes from adoptive tumor-infiltrating lymphocyte (TIL) therapy that has been used for treating patients with melanoma and a patient with cholangiocarcinoma.Citation9,10 These serial studies have also emphasized the significance of mutated antigen-specific T cells by showing that the frequency and number of these T cells among the expanded TILs were closely correlated with the clinical efficacy of the treatment.
Collectively, these results indicate that mutated antigens are key target antigens recognized by autologous T cells, which can cause a substantial reduction in tumor burden. Given that immune responses against mutated antigens directly correlate with a positive clinical outcome, it is important to better understand the biologic interactions between the host immune system and the mutated antigens to develop more effective immunotherapies; hence, it has become increasingly important to analyze cellular immune responses against the mutated antigens and identify the immunogenic neo-epitopes that could be potentially used as ideal targets for immunotherapies. Indeed, advanced techniques in next-generation sequencing have enabled us to rapidly identify genomic mutations. However, it has been laborious to identify populations of very rare mutated antigens, which are able to elicit immune responses in vivo, from among a numerous number of candidate antigens. Although several screening approaches, such as using synthetic mRNAs simultaneously expressing multiple antigens,Citation11 mass-spectrometry-based techniquesCitation12 or tetramer-based conditional UV-mediated peptide exchange technology,Citation13 have been explored to overcome this obstacle, identifying immunogenic epitopes remains challenging.
In this study, we aimed at establishing an alternative strategy for the identification of immunogenic neo-epitopes by using CXCR3 ligand mRNAs as rapid indicators of specific immune responses. Although the methodology using CXCR3 ligand mRNA as an indicator of immune responses has been established by Chakera et al.,Citation14 we hypothesized that improvement of this method would enable us to simultaneously analyze immune responses against several synthetic neo-epitope peptides in a short time of period. We determined the feasibility of this approach in multiple models of human and murine immune response. By combining this approach with whole-exome and transcriptome sequencing analyses, we were able to identify immunogenic neo-epitopes encoded by the murine fibro-sarcoma cell line CMS7.
Results
CXCR3 ligand mRNAs are sensitive indicators of specific immune responses
The first aim of our study was to determine whether CXCR3 ligands are appropriate molecular indicators of specific immune response. For this purpose, we first used a model of the human cellular immune response against the immunogenic cancer/testis antigen NY-ESO-1.Citation15 After retroviral transduction with an α/β TCR (T cell receptor) that can recognize the NY-ESO-1p157–165 epitope (SLLMWITQC) in an HLA-A0201 restricted manner (),Citation16 human CD8+ T cells were incubated overnight with the NY-ESO-1 expressing melanoma cell line 397mel or with 397melA0201, a cell line derived from 397mel that stably expresses HLA-A0201. Subsequently, the levels of cytokines and chemokines in the culture media were evaluated using the Bio-Plex system, which enables simultaneous determination of the levels of 48 different cytokines and chemokines, excluding CXCL11. We found that the protein levels of nine molecules, including CXCL9, CXCL10, and IFNγ substantially increased in an HLA-A0201 dependent manner ().
Figure 1. CXCR3 ligand mRNAs are sensitive indicators of specific immune responses. (A) Peripheral blood T cells from a healthy donor were expanded in vitro and retrovirally transduced with the NY-ESO-1p157–165/HLA-A0201-specific TCR. The CD8+ T cells positively stained by the NY-ESO-1 p157–165/A0201 tetramer were isolated by sorting. (B) Following overnight incubation of NY-ESO-1p157–165 specific CD8+ T cells with 397mel or 397melA0201, the levels of 48 cytokines/chemokines in the culture supernatants were evaluated using the Bio-Plex system. Data for nine selected cytokines/chemokines that increased in an HLA-A2 dependent manner are shown. (C) PBMC from A24+ donors latently infected with EB virus were stimulated with EBNA3A246–254 (RYSIFFDYM) peptide or DMSO as a control, and total RNA was extracted at the indicated time points. The fold increase of mRNA levels of the nine selected cytokines/chemokines and of CXCL11 compared with the DMSO control was evaluated by RT-qPCR. Expression of each gene was normalized to that of GAPDH. One representative data set out of three independent experiments is shown. Data represent relative quantity means ± SD.
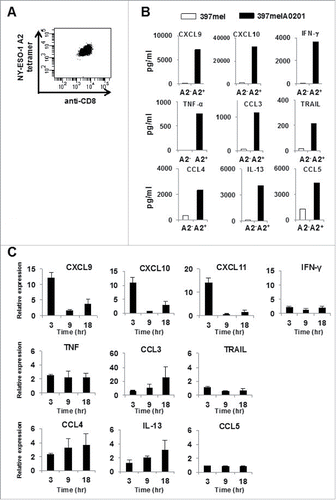
We verified that the same phenomenon also occurred in a human immune response model against Epstein–Barr (EB) virus, which elicits CD8+ T-cell immune responses in infected individuals. As previously reported, CD8+ T cells recognizing the immunogenic EB nuclear antigen 3A (EBNA3A)-derived 9-mer peptide (RYSIFFDYM) in an HLA-A24-restricted fashion are found in the peripheral blood of latently infected with EB virus HLA-A24+ donors.Citation17 Following an overnight incubation of peripheral blood mononuclear cells (PBMC) of latently infected with EB virus HLA-A24+ donors in the presence of either this peptide or the control, DMSO, the levels of the same 9 proteins in the culture media increased in an antigen-dependent manner (Fig. S1).
We next tested whether synthesis of mRNA encoding these proteins was rapidly triggered by antigenic stimulation, which could be used for the sensitive detection of a specific immune response. Following the addition of the antigenic peptide, we periodically extracted whole cell RNA and quantified the fold increase in the RNA levels of the nine selected cytokines/chemokines, in addition to CXCL11, compared with the DMSO control. As expected, a rapid increase in the mRNA expression of CXCR3 ligands was detected as early as 3 h following the peptide addition, whereas only a minor increase of IFNγ mRNA expression was detected during the time periods examined (). This marked increase in mRNA expression was dependent not only on the peptide epitope, but also on HLA-A24 expression, as indicated by the lack of such an increase in PBMC from a HLA-A24− donor latently infected with EB virus (Fig. S2).
The kinetics of mRNA synthesis of the CXCR3 ligands differs with the long peptide
We hypothesized that the kinetics of mRNA synthesis might be different for the long (> 20-mer) peptide that is often used in immunogenic neo-epitope searching. To test this possibility using the same experimental setting as described above, we incubated PBMC derived from HLA-A24+ donors latently infected with EB virus with the long peptide (20-mer), which included the EBNA3A 9-mer short peptide in its center. As expected, it took as long as 8 h following the peptide addition for CXCL9 mRNA expression to reach maximum levels. Again, we observed a minor increase in IFNγ mRNA expression during the time periods examined ().
Figure 2. Kinetics of CXCR3 ligand mRNA synthesis following treatment with long peptides. (A) PBMC from A24+ donors latently infected with EB virus were stimulated with EBNA3A240–259 (20-mer; VQSCNPRYSIFFDYMAIHRS) peptide or DMSO as a control. Relative fold increase of mRNA levels of CXCL9 and IFNγ compared with the DMSO control was evaluated by RT-qPCR. One representative data out of three independent experiments is shown. Data represent relative quantity means ± SD. (B) In this experiment, we used the same PBMC aliquot from which we had previously established an HLA-B35 restricted NY-ESO-1p94–104 peptide specific CTL clone. After addition of NY-ESO-1p91–110 (20-mer) peptide or DMSO as a control, total RNA was extracted from PBMC at the indicated time points. The relative fold increase of mRNA levels of CXCL9 and IFNγ compared with DMSO control was quantified.
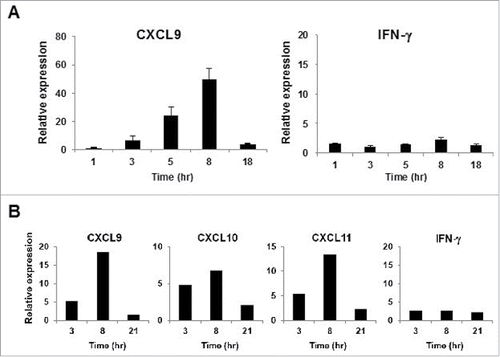
To determine whether this phenomenon is similarly observed in a different model of human immune response, we used PBMC from one patient, from which we had previously established an HLA-B35-restricted NY-ESO-1p94–104 specific CTL clone. After the addition of the NY-ESO-1p91–110 20-mer peptide containing the NY-ESO-1p94–104 epitope to the cells, we periodically quantified the fold increase in mRNA levels of the CXCR3 ligands compared with the DMSO control. As expected, it also took as long as 8 h for mRNA expression of these chemokines to reach maximum levels (), while we observed no increase in the synthesis of CXCR3 ligand mRNAs when using HLA-35+ PBMC from a healthy donor (data not shown). These observations might be explained by the possibility that it takes more time for the long peptide to be processed and presented on the cell surface of antigen presenting cells.
This method is applicable to murine immune systems as well
We tested the applicability of our method to the murine immune system as well, by using DUC18 TCR transgenic mice, expressing an α/β TCR gene derived from a CD8+ T-cell clone, which recognizes a mutated mitogen-activated protein kinase 2 (ERK2)-derived neo-epitope (mERK2–9m) presented by H-2Kd on the cell surface of murine fibro-sarcoma cell line CMS5.Citation18,19 We mixed 1 × 106 splenic cells from wild-type BALB/c mice with 2×102 splenic cells from DUC18 mice, and following the addition of the mERK2–9m peptide or the wERK2–9m peptide, we periodically quantified the fold increase in mRNA levels of the murine CXCR3 ligands compared with the DMSO control. As shown in , we observed significant increases in the mRNA expression of murine CXCR3 ligands as early as 5 h following the mERK2–9m peptide addition, similarly to human CXCR3 ligands ().
Figure 3. Kinetics of CXCR3 ligand mRNA synthesis are similar in murine and human immune system. (A) Splenocytes from naive BALB/c mice were mixed with splenocytes isolated from DUC18 mice. Mixed splenocytes were incubated with mERK2–9m peptide, wERK2–9m peptide or DMSO as a control. The relative fold increase of mRNA levels of murine CXCL9 compared with the DMSO control was quantified by RT-qPCR at the indicated time points. (B) CHP-NY-ESO-1 was subcutaneously injected into the back of BALB/c mice, twice at a 1-week interval. Splenic cells from pooled spleens (n = 3 per experiment) were prepared for analysis 7 d after the second immunization. Pooled splenic cells from naive BALB/c (left) or immunized BALB/c (right) mice were incubated with one of 17 overlapping peptides spanning the whole amino acid sequence of NY-ESO-1 (Table S1) or DMSO as a control for 8 h. Total RNA was extracted, and the relative fold increase of mRNA levels of murine CXCL9 compared with DMSO was quantified.
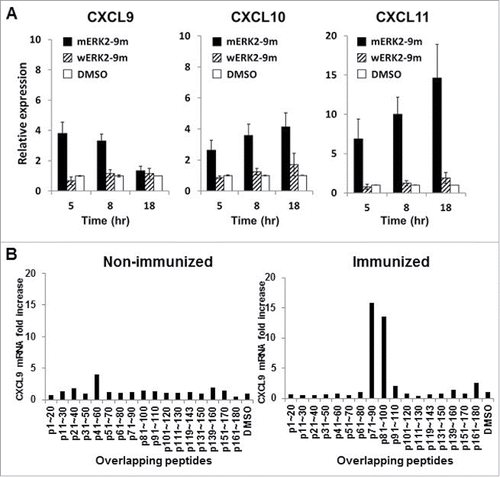
We then tested whether the long (> 20-mer) peptide is also applicable to the murine immune system. We immunized BALB/c mice with the protein-based vaccine CHP-NY-ESO-1, twice at a 1-week interval,Citation20 and prepared splenic cell suspensions from pooled spleens one week following the last vaccination. Each peptide (20–25-mer) from 17 overlapping peptides spanning the entire amino acid sequence of NY-ESO-1 (Table S1) or DMSO as a control were added to splenic cells from immunized mice, and the fold increase in murine CXCL9 mRNA levels was quantified following an 8-h incubation. We observed that murine CXCL9 mRNA expression significantly increased after the addition of 2 distinct peptides, NY-ESO-1p71–90 and NY-ESO-1p81–100 (). Interestingly, the data show that both peptides contained the identical 9-mer epitope, NY-ESO-1p81–88 (RGPESRLL), which is the immunogenic and unique epitope recognized by murine CD8+ T cells in an H-2Dd-restricted manner, as our group has previously reported.Citation21
Collectively, we concluded that our method is applicable for the detection of specific immune responses in the murine system as well.
Rapid synthesis of CXCR3 ligand mRNAs after low IFNγ secretion by CD8+ T cells
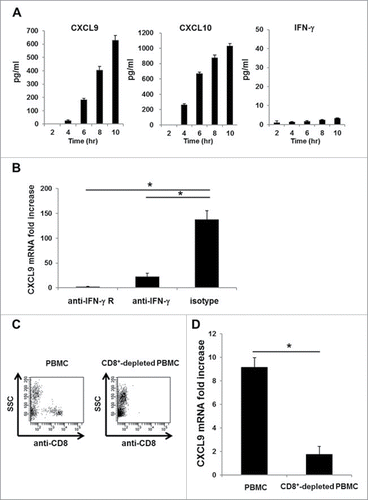
Although mRNA synthesis of the CXCR3 ligand appears to respond rapidly to specific immune responses than synthesis of IFNγ mRNA, CXCR3 ligands are well-known IFNγ inducible proteins. To determine the effect of IFNγ, we periodically quantified the protein levels of CXCL9, CXCL10, and IFNγ in the same culture medium after the addition of the EBNA3A 9-mer peptide to PBMC from HLA-A24+ donors latently infected with EB virus. We observed that CXCL9 and CXCL10 in the culture medium became detectable Shortly after the peptide addition, whereas IFNγ was not detectable at the indicated time points (). However, the robust increase in CXCL9 mRNA expression was completely diminished when we blocked IFNγ signaling by the addition of anti-IFNγ or anti-IFNγ receptor neutralizing antibodies into the culture medium (). Moreover, IFNγ secreted by CD8+ T cells was essential for the robust synthesis of CXCL9 ligand mRNA, since we could not detect an increase in CXCL9 mRNA expression when CD8+ T cells from IFNγ knockout DUC18 mice were co-cultured with wild-type BALB/c splenic cells that were pulsed with the mERK2–9m peptide (data not shown). An increase in CXCL9 mRNA expression was detected when using the control IFNγ competent DUC18 mice. Similar to the murine immune system, we found that CXCL9 mRNA synthesis was also diminished in the immune response model against EB virus when we used CD8+-depleted PBMC instead of whole PBMC (). These data suggest that CD8+ T cells capable of producing IFNγ exist in peripheral blood, although we cannot detect such IFNγ producing CD8+ T cells in PBMCs by IFNg ICS assay before incubation with EBNA3A246–254 peptide for a certain period of time (Fig. S3). Furthermore, we also observed the same phenomenon when we used CD14+-depleted PBMC (Fig. S4), as has been previously reported.Citation14,22 Taken together, these results clearly indicate that the minute amount of IFNγ secreted by CD8+ T cells is sufficient and essential for CD14+ cells to rapidly and robustly produce CXCR3 ligands at both the protein and mRNA levels.
Figure 4. A minute amount of IFNγ secreted by CD8+ T cells is sufficient to induce rapid and robust CXCR3 ligand mRNA synthesis. (A) PBMC from A24+ donors latently infected with EB virus were incubated with EBNA3A246–254 peptide. At the indicated time points, the levels of CXCL9, CXCL10 and IFNγ in the culture supernatant were evaluated by ELISA. One representative data out of three independent experiments using different PBMC donors is shown. Data represent means ± SD. (B) Blocking assay, whereby PBMC from A24+ donors latently infected with EB virus were cultured for 1 h in the presence of an anti-IFNγ receptor mAb, an anti-IFNγ mAb or an isotype control mAb before incubation with EBNA3A246–254 peptide or DMSO as a control. After 5-h incubation with EBNA3A246–254 peptide, the relative fold increase of mRNA levels of CXCL9 compared with DMSO was quantified. Data represent relative quantity means ± SD. Asterisks indicate statistically significant differences (p < 0.01). (C) Flow cytometry analysis before (left) and after (right) cell sorting. The CD8+ population was depleted from PBMC by cell sorting. (D) Each population was incubated with EBNA3A246–254 peptide. The relative fold increase in mRNA levels of CXCL9 over the DMSO control was quantified. Data represent relative quantity means ± SD. Asterisks indicate statistically significant difference (p < 0.01).
Detection of CD8+ T-cell immune responses against tumors in tumor-bearing mice
To determine whether our method was sufficiently sensitive to detect CD8+ T-cell immune responses against tumors in tumor-bearing mice, we used at first the murine colorectal tumor cell line CT26 which expresses the murine leukemia virus (MuLV) gp70-derived dominant CD8+ T-cell epitope AH-1.Citation23 In pooled spleen cells, isolated 2 weeks after the subcutaneous administration of CT26 tumor cells into the flanks of BALB/c mice, we added the AH1 or mERK2–9m peptides, or DMSO as a control, and quantified the fold increase in CXCL9 mRNA levels following a 5-h incubation. As shown in , we successfully detected a specific immune response to AH-1, as indicated by the significant increase in CXCL9 mRNA expression observed only following the addition of the AH-1 peptide.
Figure 5. Detection of cellular immune responses against tumors in tumor-bearing mice. (A) Splenocytes from CT26 tumor-bearing BALB/c mice were incubated with mERK2–9m peptide, AH1 peptide, or DMSO as a control. The relative fold increase in mRNA levels of murine CXCL9 compared with DMSO was quantified 5 h later. One representative data out of three independent experiments is shown. Data represent relative quantity means ± SD. Asterisks indicate statistically significant differences (p < 0.01). (B) After subcutaneous inoculation with 1 × 106 CMS5 cells, mice were randomly divided into 2 groups. One group was left untreated, and the other was intraperitoneally administered with 100 μg of anti-CTLA-4, 200 μg of anti-PD-1 and 150 μg of anti-GITR mAbs at days 7, 9, and 11. Both mouse groups were sacrificed at day 21 and splenic cell suspensions were prepared from pooled spleens (n = 3 per experiment). Splenic cells (1 × 106) from the antibody-treated group were incubated with mERK2–9m peptide, AH1 peptide, or DMSO as a control. The relative fold increase in mRNA levels of murine CXCL9 compared with DMSO was quantified 5 h later. One representative data set out of three independent experiments is shown. Data represent relative quantity means ± SD. Asterisks indicate statistically significant differences (p < 0.01). (C) For IFNγ intracellular staining, splenic cells from both mouse groups were incubated with mERK2–9m peptide, AH1 peptide or DMSO as a control for 15 min at room temperature, and subsequently with GolgiPlug for 4 h. Following stimulation, cells were stained for cell surface CD8+ and intracellular IFNγ. Representative dot plots gated on CD8+ splenocytes were shown. These experiments were repeated three times with similar results. (D) Pooled splenic cells from antibody treated or untreated CMS5 tumor-bearing mice were subjected to IFNγ ELISPOT assays. Pooled splenic cells were incubated in vitro with mERK2–9m, AH-1 or DMSO for 18 h. One representative data out of three independent experiments is shown.
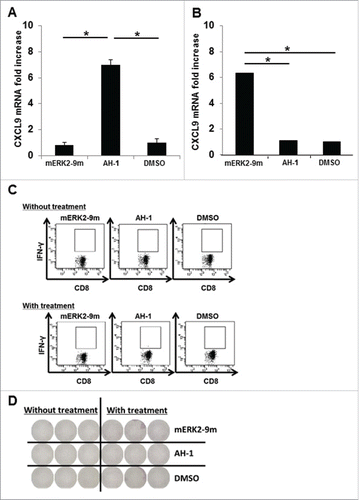
Subsequently, we investigated whether we could detect neo-epitope-specific CD8+ T-cell immune responses in tumor-bearing mice. To this end, we used the murine fibro-sarcoma cell line CMS5, which expresses the immunogenic neo-epitope, mERK2–9m, following the same procedure as described for the CT26 cell line. In pooled spleen cells isolated from CMS5 tumor-bearing mice, we added the mERK2–9m or AH-1 peptides, or DMSO as a control, and quantified the fold increase in CXCL9 mRNA levels. However, no significant increase in CXCL9 mRNA expression was observed in any of the conditions tested (data not shown). Recent advances in immunology indicate that the administration of immune-stimulating agonistic antibodies, such as anti-GITR antibody and anti-CD40 antibody, can also augment cellular immune responses against tumors, as well as checkpoint blockade antibodies. Based on this information, we tested whether immune responses to the mERK2–9m peptide became detectable following intra-peritoneal administration of anti-CTLA-4, PD-1 and GITR antibodies. Indeed, under these conditions, we successfully detected a specific immune response to mERK2–9m, as indicated by the significant increase of CXCL9 mRNA synthesis observed only following mERK2–9m addition (). However, a specific immune response to this neo-epitope was not detected by the IFNγ intracellular staining (ICS) and IFNγ ELISPOT assay regardless of the antibody treatment (). These data suggest that our method can detect immune responses only of a certain magnitude, although it seems to be more sensitive than IFNγ ICS or IFNγ ELISPOT assay.
Successful identification of an immunogenic neo-epitope encoded by mouse sarcoma CMS7
By combining whole-exome and transcriptome sequencing analyses, we aimed at identifying tumor immunogenic neo-epitopes using the murine fibro-sarcoma cell line CMS7. To this end, we identified nonsynonymous somatic point mutations specifically expressed in the tumor by comparing exome sequencing data of CMS7 tumors with that of BALB/c mice tails. Then, transcriptome sequencing data were applied to deduce the expression potential of the mutated antigens at the protein level. Complete lists of mutation-containing peptides, each ranging from 8 to 10 amino acids, were prepared and filtered based on their predicted binding affinities to each of the murine H-2Kd, Dd, or Ld alleles using the IEDB analysis resource (http://tools.immuneepitope.org/analyze/html/mhc_binding.html). Finally, we prepared a total of 57 candidate neo-epitope peptides encoded by CMS7 (), and evaluated their immunogenicity using splenic cells derived from CMS7 tumor-bearing mice as described below.
Table 1. The list of candidate neo-epitopes encoded by CMS7 tumor.
BALB/c mice were subcutaneously inoculated with CMS7 tumor cells in their flanks at day 0 and were divided into two groups. One group was left untreated, while in the other anti-CTLA-4, anti-PD-1 and anti-GITR antibodies were intraperitoneally administered at days 7, 9, and 11. At day 21, the two groups of mice were sacrificed and splenic cell suspensions were prepared from pooled spleens. Following the addition of a panel of neo-epitope peptides or DMSO as a control to splenic cells, the specific immune response to each peptide was measured by our method. In both mouse groups, we observed a significant increase in CXCL9 mRNA levels following the addition of only a single neo-epitope derived from mutated Snd1 (staphylococcal nuclease domain-containing protein 1) (). In addition, we confirmed that a significant increase in CXCL9 mRNA levels was observed only following the addition of mSnd1 (). The data obtained from IFNγ ICS and ELISPOT assays also strongly supported our assertion that the immune responses to mSnd1, not to wSnd1, is elicited in CMS7 tumor-bearing mice and augmented by antibody treatment (). It should also be noted that in the case of untreated mouse group, the specific immune response to this neo-epitope could be detected by our method, but not by the IFNγ ICS method ().
Figure 6. Successful identification of an immunogenic neo-epitope encoded by mouse sarcoma CMS7. (A) After subcutaneous inoculation with CMS7 cells, mice were randomly divided into two groups. One group was left untreated, and the other was administered intraperitoneally with anti-CTLA-4, anti-PD-1 and anti-GITR mAbs on days 7, 9, and 11. Both mouse groups were sacrificed at day 21 and splenic cell suspensions were prepared from pooled spleens (n = 3 per experiment). Splenic cells from untreated (upper) or treated mice (lower) were incubated with each panel of neo-epitope peptides for 5 h and the fold increase in CXCL9 mRNA levels compared with DMSO was quantified. One representative data set out of three independent experiments is shown. (B) Splenic cells from untreated (upper) or treated mice (lower) were incubated with mutated Snd1 peptide, its wild-counterpart or DMSO as a control for 5 h and the fold increase in CXCL9 mRNA levels compared with DMSO was quantified. (C) Splenic cells from untreated (upper) or treated mice (lower) were incubated with mutated Snd1 peptide, its wild-counterpart or DMSO as a control for 15 min at room temperature and subsequently with GolgiPlug for 4 h. Following stimulation, cells were stained for cell surface CD8+ and intracellular IFNγ. Representative dot plots gated on CD8+ splenic T cells are shown. The number indicates the percentage of CD8+ IFNγ+ T cells. These experiments were repeated three times with similar results. (D) CD8+ splenic T cells were obtained from antibody-treated mice pooled spleens by positive enrichment using the MACS system, and were stimulated in vitro with mutated Snd1-pulsed CD8− splenic cells. Cultured CD8+ splenic T cells were subjected to ELISPOT assays 10 d later. The target cells were CD8− splenic cells pulsed with mutated Snd1 peptide, or its wild counterpart. Splenic cells pulsed with DMSO were used as control targets. (E) Age-matched female BALB/c mice were inoculated with CMS7 at day 0 following two injections (at days −14 and −7) with mSnd1 peptide and poly (I:C) formulated in PBS or poly (I:C) alone using prophylactic schedules. The tumor size was monitored three times a week. Each group consisted of five mice. Mice without any immunization and mice treated by the co-administration of antibodies (anti-CTLA-4/PD-1/GITR Abs) at days 7, 9, 11 served as a negative control group and a positive control group, respectively.
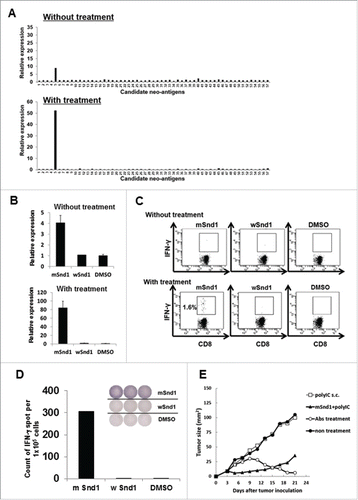
To assess the immunogenicity of the mSnd1 neo-epitope, we immunized mice with this peptide twice, 14 and 7 d before the CMS7 tumor challenge. At day 0, CMS7 cells were subcutaneously injected to the mice. As shown in , mice immunized with the mSnd1 peptide developed resistance to the CMS7 challenge, whereas no tumor immunity was observed in any of the other groups, except for the antibody-treated group that showed complete reduction of tumor growth.
Discussion
The chemokines CXCL9, 10, and 11 play crucial roles in a wide variety of inflammatory immune responses,Citation24,25 including tumor immunity. Indeed, in many types of malignancies, there is close correlation between higher expression of CXCR3 ligands at both the mRNA and protein levels and a higher number of tumor-infiltrating T lymphocytes.Citation26-29 In addition, recent clinical studies have shown that the expression of these chemokines in tumor tissue before treatment was positively associated with a favorable clinical response to immunotherapies, such as a cancer vaccine targeting MAGE-A3,Citation30 an immunogenic cancer/testis antigen, and an immunotherapy using an anti-CTLA-4 antibody for melanoma patients.Citation31 These results are not unexpected, considering that IFNγ, the pivotal player in anti-tumor immune response, triggers the production of these chemokines and thereby recruits CXCR3+ immune effector cells, such as Th1 cells, NK cells, and activated CD8+ T cells, to tumor sites.
The in vitro data of our present study indicate that CXCR3 ligands appeared to be sensitive indicators of specific immune responses, which are consistent with the results reported by Chakera et al.Citation14 In particular, we often observed a significant increase in CXCR3 ligand mRNA levels, which was however, not accompanied by an increase in IFNγ mRNA levels. Likewise, we observed a rapid increase of CXCR3 ligands at the protein level, which was substantially diminished by blocking IFNγ signaling, although no measurable IFNγ was detected in the same culture medium. These data exemplify the amplification of IFNγ signals as a prominent property of CXCR3 ligands. In conclusion, we have shown that the utilization of CXCR3 ligand mRNA as a sensitive and specific immune response indicator is a promising strategy.
To examine the feasibility of this approach for identifying immunogenic neo-epitopes, we used a murine CMS5 tumor model since our group has previously identified a mutated ERK2-derived neo-epitope (mERK2–9m) as a tumor rejection antigen of CMS5 tumors, by cDNA cloning from a CD8+ T-cell clone (C18), established from draining lymph node cells of CMS5 tumor-bearing mice.Citation18 Moreover, adoptive transfer of CD8+ T cells from DUC18 mice, expressing a TCR α/β gene derived from clone C18, has been shown to potently eradicate established CMS5 tumors.Citation19 However, an immune response to this epitope could not be detected by our approach in untreated mice. The cause of this unexpected result could be the low antigenicity of this peptide. Consistently with this speculation, another group recently reported that specific immune responses to this epitope were hardly detectable even after immunization with this peptide.Citation32 However, the fact that our approach successfully detected this specific immune response following the checkpoint antibody treatment encouraged us to validate our approach for identifying other neo-epitopes with higher antigenicity in another mouse model.
As shown in , we successfully detected CD8+ T-cell immune response to a single neo-epitope (YAPCRGEF) derived from mutated Snd1 in the CMS7 tumor model even without the checkpoint antibody treatment. Furthermore, we observed that the antibody treatment augmented the immune response and resulted in successful detection of the immune response to the neo-epitope (YAPCRGEF) but not its wild-type counterpart (YAPRRGEF) by the IFNγ ICS method (). These data clearly demonstrate the higher sensitivity of our method than that of IFNγ ICS. In addition, as shown in , the sensitivity of this method seems to be almost equal to that of IFNγ ELISPOT assay, since we were able to detect the existence of approximately 20 antigen-specific CD8+ T cells among 1 × 106 splenic cells (0.002% antigen-specific CD8+T cells/whole cells). However, under different conditions, our approach might be more sensitive compared with the IFNγ ELISPOT assay, since we could not detect specific immune responses to mERK2–9m or the mutated Snd1-derived neo-epitope by the IFNγ ELISPOT assay, in antibody-treated CMS5 tumor-bearing mice () or in antibody-untreated CMS7 tumor-bearing mice (data not shown), respectively. Further investigation is required to clarify this point.
Contrary to our expectations, our data show that we could detect an immune response to only one neo-epitope in the CMS7 tumor model. One major reason might stem from the limitations of in silico technics for the prediction of immunogenic neo-epitopes. Although the list of our candidate neo-epitope peptides was based on the binding strength to H-2 alleles, selection of candidate neo-epitopes according to different criteria may improve the outcome, as proposed by another group.Citation32
Personalized cancer vaccines using neo-epitopes encoded by an individual's tumor could be a promising strategy for the treatment of cancer patients. In the clinical setting, the quick and accurate identification of immunogenic neo-epitopes is necessary before the immunotherapy treatment and therefore, our simple and quick approach is promising. We are currently investigating whether our approach is applicable for the identification of immunogenic neo-epitopes using PBMC from cancer patients.
In conclusion, we hope that our novel method would prove useful for the identification of neo-antigens, which show therapeutic potential.
Materials and methods
Ethics statement (human samples)
Written informed consents were obtained from the one patient and healthy volunteers according to the guidelines of the Declaration of Helsinki. The experimental protocol was approved by the Institutional Review Board at the Mie University School of Medicine.
Ethics statement (mouse experiments)
All mice were housed under specific pathogen free conditions and used at 8–10 weeks of age. All animal experiments were conducted using protocols approved by the Animal Care and Use Committee at the Mie University Life Science Center.
Animal studies
Female BALB/c mice were purchased from the Shizuoka Animal Laboratory Center (Shizuoka, Japan). DUC18 mice, transgenic for α/β-TCR reactive with the H-2Kd-restricted mERK2136–144, were established as described previously.Citation18
Antibodies
All fluorescence-conjugated mAbs, including anti-human CD8+ (HIT8a), were purchased from BioLegend (San Diego, CA, USA). Anti-human CD119 (IFNγ receptor α chain, GIR-208) mAb, anti-human IFNγ (NIB42) mAb, and mouse IgG1 isotype control were purchased from BioLegend. Anti-human CD3 (OKT3) mAb was purchased from BD Biosciences (San Jose, CA, USA). Anti-mouse PD-1 (RMP1–14), anti-mouse CTLA-4 (9D9) and anti-mouse GITR (DTA-1) mAbs were produced from hybridomas and were purified by protein G columns.
Cell lines
CT26 is a colon epithelial tumor cell line derived from intra-rectal injections of N-nitroso-N-methylurethane into BALB/c mice.Citation33 CMS5 and CMS7 are 3-methylcholanthrene-induced sarcoma cell lines of BALB/c origin.Citation34 NY-ESO-1 expressing human melanoma cell line 397mel was a kind gift from Dr. Y. Kawakami (Keio University, Tokyo, Japan). 397melA0201 was established by transfecting plasmid DNA encoding HLA-A0201 into 397mel cells. All tumor lines were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated Fetal Calf Serum (FCS) (Biowest, Kansas, USA), 50 µM 2-mercaptoethanol (2-ME), and 0.2 mg/mL glutamine.
Cytokine/chemokine measurements
The levels of 48 kinds of human cytokines and chemokines in culture supernatants were determined using Bio-Plex kits (BioRad, Hercules, CA, USA) according to the manufacturer's instructions. The analytes included IL-1α, IL-1β, IL-1Ra, IL-2, IL-2Rα, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12(p40), IL-12(p70), IL-13, IL-15, IL-16, IL-17, IL-18, eotaxin, FGF basic, G-CSF, GM-CSF, IFNγ, MCP-1, CCL3, CCL4, CCL5, PDGF-BB, CTACK, GRO-α, HGF, IFNα2, LIF, MCP3, M-CSF, MIF, CXCL9, CXCL10, β-NGF, SCF, SCGF-β, SDF-1α, TNF-α, TNF-β, TRAIL, and VEGF. In some experiments, the levels of human CXCL9, CXCL10, and IFNγ were analyzed using quantitative human DuoSet ELISA kits (R&D Systems, Minneapolis, MN).
Cell staining
For intracellular cytokine staining, splenocytes (1 × 106) were incubated with 50 μM synthetic peptide or DMSO for 15 min at room temperature, and subsequently with GolgiPlug (BD Bioscience) for 4 h. The cells were stained with a CD8α-specific mAb for 15 min at 4 °C. After permeabilization and fixation using a Cytofix/Cytoperm kit (BD Biosciences) according to manufacturer's instructions, the cells were stained intracellularly using an allophycocyanin-conjugated anti-IFNγ mAb.
Enzyme-linked immunospot (ELISPOT) assay
Murine IFNγ ELISPOT assays were performed as described previously.Citation35 Briefly, 96-well nitrocellulose ELISPOT plates (MAHA S4510; Millipore, Bedford, MA) were coated with 2 μg/mL anti-mouse IFNγ mAb (mAb, clone R4–6A2; PharMingen, San Diego, CA) overnight at 4 °C, washed with 0.05% Tween 20 in PBS (PBS/Tween) and blocked with FCS-containing culture medium for 2 h at 37 °C. Cultured CD8+ T cells (1 × 105 per well) and mutated Snd1 peptide-pulsed CD8− splenic cells (1 × 106 per well) were plated at a final volume of 200 μL per well, and were incubated for 18 h at 37 °C in a CO2 incubator. Following a thorough wash with PBS/Tween, 1.25 μg/mL biotinylated anti-mouse IFNγ mAb (PharMingen) was added, and the plate was incubated overnight at 4 °C, washed with PBS/Tween, and further incubated with 1 μg/mL streptavidin-alkaline phosphatase conjugate (Mabtech, Nacka, Sweden) in 100 μL PBS per well for 90 min at room temperature. Following three washes with PBS/Tween, the staining was performed with an alkaline phosphatase conjugate substrate kit (BioRad), the reaction was stopped by adding distilled water, and the stained spots were counted using an ELISPOT Plate Reader (ImmunoSpot, CTL-Europe GmbH) after drying.
Peptides
The EB virus-derived peptides EBNA3A246–254 (RYSIFFDYM), EBNA3A240–259 (VQSCNPRYSIFFDYMAIHRS), AH-1 (SPSYVYHQF), the mutated ERK2-derived peptide (mERK2–9m; QYIHSANVL), the non-mutated ERK2-derived peptide (wERK2–9m; KYIHSANVL),Citation18 the mutated Snd1-derived peptide (mSnd1; YAPCRGEF), the non-mutated Snd1-derived peptide (wSnd1; YAPRRGEF), and 17 overlapping peptides spanning the NY-ESO-1 protein (Table S1) were synthesized at a purity higher than 80% from Invitrogen (Carlsbad, CA, USA). All peptides were dissolved in DMSO at a concentration of 10 mM and stored in aliquots at −80 °C before use.
Immunization
The complex of cholesterol-bearing hydrophobized pullulan (CHP) and NY-ESO-1 protein (CHP-NY-ESO-1) was provided by ImmunoFrontier, Inc. (Tokyo, Japan).Citation20 CHP-NY-ESO-1 complex vaccines were subcutaneously injected into the back of BALB/c mice at a dose of 50 μg, twice, at a 1-week interval. Following 7 d from the second vaccination, spleen cells were harvested for analysis. In another experiment, before CMS7 tumor challenge, 200 μg mSnd1 peptide and 50 μg poly(I:C) formulated in PBS (200 μL total volume) were subcutaneously injected into the back of BALB/c mice, twice, at a 1-week interval.
Preparation of murine splenic cells
Mice spleens were mashed with a microscope slide, treated with ammonium chloride-potassium (ACK) and filtrated with Cell Trics® 30 µm (Sysmex Partec, Kobe, Japan). Single cell suspensions from pooled spleen were cultured in 96-well V-bottom plates at a density of 5 × 106 cells/mL, with the appropriate peptide. In some experiments, CD8+ splenic T cells were obtained by positive enrichment using the MACS system (Miltenyi Biotec, Bergisch Gladbach, Germany). Flow cytometry confirmed that the T-cell fractions contained more than 95% CD8+ T cells.
Preparation of human cell populations
Peripheral blood mononuclear cells (PBMC) were isolated from fresh heparinized venous blood samples with density-gradient centrifugation in Ficoll-Paque PLUS (GE Healthcare, Buckinghamshire, UK). CD8+ cell-depleted or CD14+ cell-depleted subsets were obtained from PBMC using FACSAria (BD Biosciences).
T-cell transduction
PBMC obtained from healthy volunteers were stimulated using a plate-coated anti-CD3 antibody and RetroNectin (Takara Bio, Inc., Otsu, Japan) in the presence 600 IU/mL of recombinant IL-2. Proliferating lymphocytes were transduced with a retrovirus encoding NY-ESO-1p157–165/HLA-A0201-specific TCR-α and -β chains, and were expanded further in vitro. Ten days after transduction, CD8+ T cells expressing NY-ESO-1-specific TCR, determined by the specific tetramer, were sorted using FACSAria (BD Biosciences).
Blocking assay
Human lymphocytes (1 × 106) were cultured in 200 μL X-VIVO™15 (Lonza, Walkersville, MD, USA) in 96-well V-bottom plates in the presence of anti-human CD119, anti-human IFNγ, or mouse IgG1 isotype control (final 10 µg/mL) for 1 h before the addition of the EBNA3A246–254 peptide.
Next generation sequencing and data analysis
DNA extracted from CMS7 tumor cells or BALB/c tails by conventional methods was subjected to exome capturing using the SureSelectXT mouse exon kit (Agilent Technologies, Santa Clara, CA, USA). Exome capture libraries were subsequently sequenced with a HiSeq2000 (Illumina, CA) sequencing system, using the Illumina V3 kit (Riken Genesis Co., Ltd.). From each library, 20M (2 × 150 bp) exome reads were sequenced. RNA samples extracted from CMS7 tumor cells were used to generate RNA-seq libraries, which were sequenced by Hokkaido System Science Co., Ltd., with an Illumina HiSeq 2000 (Illumina) sequencing system. From each library, 30M (2 × 100 bp) RNA fragments were sequenced. Whole exome sequence reads were aligned to the mm9 reference sequence using the Burrows-Wheeler Aligner (BWA) software.Citation36 Somatic single nucleotide variants (SNVs) were detected from read count comparisons between normal and tumor samples using the Fisher exact test, and variants whose p-value was less than 10−10 were considered as statistically significant. From the identified SNVs, we extracted only non-synonymous SNVs. We also aligned sequence reads from RNA-seq libraries to mm9 using the Bowtie algorithm of the TopHat softwareCitation37 to confirm whether the variants detected in whole exome sequencing were indeed transcribed to RNA. Based on the aligned RNA sequence reads, the candidate variants from whole exome sequencing were tested by the Fisher exact test, and variants with a p-value less than 10−10 were considered as transcribed SNVs.
Selection of candidate neo-epitopes
To identify potential neo-epitopes that could bind murine MHC class I, we used the IEDB Analysis Resource (http://www.iedb.org/). Briefly, all candidate neo-epitopes (8–10-mer) containing missense mutations were analyzed in silico for their binding affinities to either H-2Kd, H-2Dd or H-2Ld molecules based on the IEDB recommended method (Consensus) consisting of the Artificial Neural Network (ANN), the Stabilized Matrix Method (SMM), and the Scoring Matrices derived from Combinatorial Peptide Libraries (Comblib_Sidney2008) algorithms.Citation38 Based on percentile rank (≤ 0.6) and IC50 (< 4,000 nM) values (), we selected 57 different candidate neo-epitopes for CMS7 cells.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
We performed the experiment protocol described below, referring to a previous report.Citation14 Total RNA from cultured cells was extracted using an RNA Blood Mini Kit (Qiagen, Hilden, Germany) and was reverse-transcribed into first-strand cDNA (cDNA) using a QuantiTect Reverse Transcription Kit (Qiagen). Quantitative PCR was performed using a Step One Plus (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturer's protocol. Primers and probes were selected from the ABI TaqMan Gene Expression Assay catalog (for human targets: CXCL9, Hs00171065_m1; CXCL10, Hs01124251_g1; CXCL11, Hs04187682_g1; IFNγ, Hs00989291_m1; TNFα, Hs01113624_g1; CCL3, Hs00234142_m1; CCL4, Hs00237011_m1; CCL5, Hs00174575_m1; TRAIL, Hs00921974_m1; and IL-13, Hs00174379_m1; for mouse targets: CXCL9, Mm00434946_m1; CXCL10, Mm00445235_m1; CXCL11, and Mm00444662_m1). Expression of each gene was normalized to that of GAPDH. Fold change was determined by the ΔΔCt method. Each experiment was performed in triplicate.
Statistical analysis
Statistical significance was evaluated using the unpaired Student's t-test and p values less than 0.05 were considered as statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1306617_supplementary_data.zip
Download Zip (323.4 KB)Acknowledgment
The authors thank Dr. Toshitada Takahashi for helpful discussions.
Funding
This work was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (16K07168).
References
- Matthew MG, Xiuli Z, Heiko S, Etienne C, Jeffrey PW, Takuro N, Yulia I, Jasreet H, Cora DA, Willem-Jan K et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014; 515:577-81; PMID:25428507; https://doi.org/10.1038/nature13988
- John CC, Sebastian K, Jan D, Martin L, Niels van de R, Jos de G, Abderraouf S, Mustafa D, Sebastian B, Claudia P et al. Exploiting the mutanome for tumor vaccination. Cancer Res 2012; 72:1081-91; PMID:22237626; https://doi.org/10.1158/0008-5472.CAN-11-3722
- Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res 2014; 24:743-50; PMID:24782321; https://doi.org/10.1101/gr.165985.113
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014; 371:2189-99; PMID:25409260; https://doi.org/10.1056/NEJMoa1406498
- van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJ, Behjati S, Hilkmann H, El Atmioui D et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 2013; 31:e439-42; PMID:24043743; https://doi.org/10.1200/JCO.2012.47.7521
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711-23; PMID:20525992; https://doi.org/10.1056/NEJMoa1003466
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; 348:124-8; PMID:25765070; https://doi.org/10.1126/science.aaa1348
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015; 372:2509-20; PMID:26028255; https://doi.org/10.1056/NEJMoa1500596
- Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 2013; 19:747-52; PMID:23644516; https://doi.org/DOI:10.1038/nm.3161
- Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014; 344:641-5; PMID:24812403; https://doi.org/10.1126/science
- Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, Prickett TD, Gartner JJ, Crystal JS, Roberts IM et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med 2016; 22:433-8; PMID:26901407; https://doi.org/10.1038/nm.4051
- Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014; 515:572-6; PMID:25428506; https://doi.org/10.1038/nature14001
- Bakker AH, Hoppes R, Linnemann C, Toebes M, Rodenko B, Berkers CR, Hadrup SR, van Esch WJ, Heemskerk MH, Ovaa H et al. Conditional MHC class I ligands and peptide exchange technology for the human MHC gene products HLA-A1, -A3, -A11, and -B7. Proc Natl Acad Sci USA 2008; 105:3825-30; PMID:18308940; https://doi.org/10.1073/pnas.0709717105
- Chakera A, Bennett SC, Cornall RJ. A whole blood monokine-based reporter assay provides a sensitive and robust measurement of the antigen-specific T cell response. J Transl Med 2011; 9:143; PMID:21871084; https://doi.org/10.1186/1479-5876-9-143
- Chen YT, Scanlan MJ, Sahin U, Türeci O, Gure AO, Tsang S, Williamson B, Stockert E, Pfreundschuh M, Old LJ. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci USA 1997; 94:1914-8; PMID:9050879
- Chen JL, Dunbar PR, Gileadi U, Jäger E, Gnjatic S, Nagata Y, Stockert E, Panicali DL, Chen YT, Knuth A et al. Identification of NY-ESO-1 peptide analogues capable of improved stimulation of tumor-reactive CTL. J Immunol 2000; 165:948-55; PMID:10878370; https://doi.org/10.4049/jimmunol.165.2.948
- Burrows SR, Gardner J, Khanna R, Steward T, Moss DJ, Rodda S, Suhrbier A. Five new cytotoxic T cell epitopes identified within Epstein-Barr virus nuclear antigen 3. J Gen Virol 1994; 75:2489-93; PMID:7521394; https://doi.org/10.1099/0022-1317-75-9-2489
- Ikeda H, Ohta N, Furukawa K, Miyazaki H, Wang L, Kuribayashi K, Old LJ, Shiku H. Mutated mitogen-activated protein kinase: a tumor rejection antigen of mouse sarcoma. Proc Natl Acad Sci USA 1997; 941:6375-9; PMID:9177225; https://doi.org/10.1073/pnas.94.12.6375
- Hanson HL, Donermeyer DL, Ikeda H, White JM, Shankaran V, Old LJ, Shiku H, Schreiber RD, Allen PM. Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity 2000; 13:265-76; PMID:10981969; https://doi.org/10.1016/S1074-7613(00)00026-1
- Kageyama S, Wada H, Muro K, Niwa Y, Ueda S, Miyata H, Takiguchi S, Sugino SH, Miyahara Y, Ikeda H et al. Dose-dependent effects of NY-ESO-1 protein vaccine complexed with cholesteryl pullulan (CHP-NY-ESO-1) on immune responses and survival benefits of esophageal cancer patients. J Transl Med 2013; 11:246; PMID:24093426; https://doi.org/10.1186/1479-5876-11-246
- Muraoka D, Nishikawa H, Noguchi T, Wang L, Harada N, Sato E, Luescher I, Nakayama E, Kato T, Shiku H. Establishment of animal models to analyze the kinetics and distribution of human tumor antigen-specific CD8+ T cells. Vaccine 2013; 31:2110-8; PMID:23499606; https://doi.org/10.1016/j.vaccine.2013.02.056
- Dengel LT, Norrod AG, Gregory BL, Clancy-TE, Burdick MD, Strieter RM, Slingluff CL Jr, Mullins DW. Interferons induce CXCR3-cognate chemokine production by human metastatic melanoma. J Immunother 2010; 33:965-74; PMID:20948440; https://doi.org/10.1097/CJI.0b013e3181fb045d
- Huang AY, Gulden PH, Woods AS, Thomas MC, Tong CD, Wang W, Engelhard VH, Pasternack G, Cotter R, Hunt D, Pardoll DM, Jaffee EM. The immunodominant major histocompatibility complex class I-restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product. Proc Natl Acad Sci USA 1996; 93:9730-5; PMID:8790399; https://doi.org/10.1073/pnas.93.18.9730
- Lacotte S, Brun S, Muller S, Dumortier H. CXCR3, inflammation, and autoimmune diseases. Ann NY Acad Sci 2009; 1173:310-7; PMID:19758167; https://doi.org/10.1111/j.1749-6632.2009.04813.x
- Wendel M, Galani IE, Suri-PE, Cerwenka A. Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res 2008; 68:8437-45; PMID:18922917; https://doi.org/10.1158/0008-5472.CAN-08-1440
- Ohtani H, Jin Z, Takegawa S, Nakayama T, Yoshie O. Abundant expression of CXCL9 (MIG) by stromal cells that include dendritic cells and accumulation of CXCR3+ T cells in lymphocyte-rich gastric carcinoma. J Pathol 2009; 217:21-31; PMID:18980207; https://doi.org/10.1002/path.2448
- Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 2009; 69:3077-85; PMID:19293190; https://doi.org/10.1158/0008-5472.CAN-08-2281
- Kondo T, Nakazawa H, Ito F, Hashimoto Y, Osaka Y, Futatsuyama K, Toma H, Tanabe K. Favorable prognosis of renal cell carcinoma with increased expression of chemokines associated with a Th1-type immune response. Cancer Sci 2006; 97:780-6; PMID:16863511; https://doi.org/10.1111/j.1349-7006.2006.00231.x
- Kunz M, Toksoy A, Goebeler M, Engelhardt E, Bröcker E, Gillitzer R. Strong expression of the lymphoattractant C-X-C chemokine Mig is associated with heavy infiltration of T cells in human malignant melanoma. J Pathol 1999; 189:552-8; PMID:10629557; https://doi.org/10.1002/(SICI)1096-9896(199912)189:4%3c552::AID-PATH469%3e3.0.CO;2-I
- Ulloa-Montoya F, Louahed J, Dizier B, Gruselle O, Spiessens B, Lehmann FF, Suciu S, Kruit WH, Eggermont AM, Vansteenkiste J et al. Predictive gene signature in MAGE-A3 antigen-specific cancer immunotherapy. J Clin Oncol 2013; 31:2388-95; PMID:23715562; https://doi.org/10.1200/JCO.2012.44.3762
- Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, Alaparthy S, Berman D, Jure-Kunkel M, Siemers NO, Jackson JR, Shahabi V. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother 2012; 61:1019-31; PMID:22146893; https://doi.org/10.1007/s00262-011-1172-6
- Duan F, Duitama J, Al Seesi S, Ayres CM, Corcelli SA, Pawashe AP, Blanchard T, McMahon D, Sidney J, Sette A et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med 2014; 11:2231-48; https://doi.org/10.1084/jem.20141308
- Griswold DP, Corbett TH. A colon tumor model for anticancer agent evaluation. Cancer 1975; 36:2441-4; PMID:1212662; https://doi.org/10.1002/1097-0142(197512)36:6%3c2441::AID-CNCR2820360627%3e3.0.CO;2-P
- DeLeo AB, Shiku H, Takahashi T, John M, Old LJ. Cell surface antigens of chemically induced sarcomas of the mouse. I. Murine leukemia virus-related antigens and alloantigens on cultured fibroblasts and sarcoma cells: description of a unique antigen on BALB/c Meth A sarcoma. J Exp Med 1977; 146:720-34; PMID:197192
- Power CA, Grand CL, Ismail N, Peters NC, Yurkowski DP, Bretscher PA. A valid ELISPOT assay for enumeration of ex vivo, antigen-specific, IFNgamma-producing T cells. J Immunol Methods 1999; 227:99-107; PMID:10485258; https://doi.org/10.1016/S0022-1759(99)00074-5
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009; 25:1754-60; PMID:19451168; https://doi.org/10.1093/bioinformatics/btp324
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009; 25:1105-11; PMID:19289445; https://doi.org/10.1093/bioinformatics/btp120
- Moutaftsi M, Peters B, Pasquetto V, Tscharke DC, Sidney J, Bui HH, Grey H, Sette A. A consensus epitope prediction approach identifies the breadth of murine T (CD8+)-cell responses to vaccinia virus. Nat Biotechnol 2006; 24:817-9; PMID:16767078; https://doi.org/10.1038/nbt1215