
ABSTRACT
The mechanisms of immunogenicity underlying mild heat-shock (mHS) treatment < 42°C of tumor cells are largely attributed to the action of heat-shock proteins; however, little is known about the immunogenicity of tumor cells undergoing severe cytotoxic heat-shock treatment (sHS > 43°C). Here, we found that sHS, but not mHS (42°C), induces immunogenic cell death in human cancer cell lines as defined by the induction of ER stress response and ROS generation, cell surface exposure of calreticulin, HSP70 and HSP90, decrease of cell surface CD47, release of ATP and HMGB1. Only sHS-treated tumor cells were efficiently killed and phagocytosed by dendritic cells (DCs), which was partially dependent on cell surface calreticulin. DCs loaded with mHS or sHS-treated tumor cells displayed similar level of maturation and stimulated IFNγ-producing CD8+ T cells without any additional adjuvants in vitro. However, only DCs loaded with sHS-treated tumor cells stimulated antigen-specific CD4+ T cells and induced higher CD8+ T-cell activation and proliferation. sHS-treated murine cells also exposed calreticulin, HSP70 and HSP90 and activated higher DC maturation than mHS treated cells. Vaccination with sHS-treated tumor cells elicited protective immunity in mice. In this study, we defined specific conditions for the sHS treatment of human lung and ovarian tumor cells to arrive at optimal ratio between effective cell death, immunogenicity and content of tumor antigens for immunotherapeutic vaccine generation.
Abbreviations
| ATP | = | adenosine triphosphate |
| CRT | = | calreticulin |
| DAMPs | = | danger-associated molecular patterns |
| DC | = | dendritic cells |
| ER | = | endoplasmic reticulum |
| HHP | = | high hydrostatic pressure, HMGB1, high-mobility group box 1 |
| HS | = | heat-shock treatment |
| HSP | = | heat-shock protein |
| ICD | = | immunogenic cell death |
| IFNγ | = | interferon-gamma |
| ROS | = | reactive oxygen species |
Introduction
Hyperthermia (HT) has been studied for the treatment of malignant diseases since 1970s.Citation1 In 2011, HT treatment prior to radiotherapy was approved by FDA to treat cervical cancer patients. The immune system mediates the major therapeutic effect of HT;Citation2 therefore, a major unanswered question is whether the combination of HT with various immunotherapeutic approaches would translate into a better clinical outcome in cancer patients. However, there are no clinical data on the use of heat-shock (HS)-killed tumor cells alone or as a source for antigens for active cellular immunotherapy in clinical protocols.
Several cytotoxic drugs, oncolytic viruses and some physical modalities were shown to induce immunogenic cell death (ICD) of tumor cells, subsequently leading to the induction of effective antitumor immune responses in vitro and in vivo.Citation3-6 On the molecular level, ICD is characterized by elevated levels of reactive oxygen species (ROS) and endoplasmic reticulum (ER)-stress unfolded protein response (UPR) during which several immunogenic proteins such as an “eat-me” signal calreticulinCitation7 or danger-associated molecular patterns (DAMPs)Citation8,9 such as HSP70 and 90,Citation10 ATPCitation11 or HMGB112 are exposed at the cell surface or released to the cell vicinity.Citation5,13,14 These molecules facilitate the engulfment of dying tumor cells by immune cells and stimulate antigen presentation. Activated dendritic cells, in particular, prime antitumor adaptive T-cell responses.Citation4,5 We have recently described ICD induced by high hydrostatic pressure (HHP), which bears the advantage of leaving behind no residual chemical;Citation15 therefore, being suitable for generation of immunogenic tumor cells for active cancer immunotherapy approaches.Citation6
HS-treated tumor cells were documented to die by various modes of cell death depending on the thermal dose.Citation1,16,17 Thermal dose encompasses the temperature (ranging in most studies from 41°C to 44°C), the duration of heat treatment (30 min or 1 h), tumor cell type and the cell cycle phase.Citation1,16 Mouse and human tumor cells subjected to mild HS (mHS) ≤ 42°C showed increased immunogenicity as well as the induction of tumor-antigen specific T-cell responses in vitro and in vivo, which has been attributed mainly to the action of HS proteins such as HSP70.Citation18-26 Furthermore, HS treatment at 42°C was shown to increase the mRNA for tumor-specific antigens in several cancer cell lines.Citation21,27,28 However, despite increased immunogenicity, mHS treatment or repeated mHS application on cancer cells often leads to the induction of thermotolerance and cancer cell survival.Citation1
Little is known about the mechanisms of immunogenicity of tumor cells that undergo treatment with severe cytotoxic temperatures (≥ 43°C). The induction of tumor-specific T cells in patients with hepatocellular carcinoma after in situ radiofrequency ablation of tumors (> 45–50°C) was documented.Citation29 The recent development of new techniques based on magnetic nanoparticlesCitation30 shows that heat treatment at 46.8°C or 50–55°C can significantly reduce the tumor growth or induce abscopal antitumor immune effects in vivo.Citation31,32 Recently, it has been shown that microwave thermal ablation enhanced immunogenicity of osteosarcoma in vitro and in vivo accompanied by an increased abundance of calreticulin and release of ATP and HMGB1.Citation33 Similarly, prostate cancer cells or NHL cell lines treated with cytotoxic HS in vitro were shown to expose calreticulin, HSP90, HSP70 and release HMGB1 and ATP and stimulate DC maturation.Citation34,35 This suggests that severe heat shock (sHS) treatment might be considered as another physical modality inducing ICD.Citation6
In this study, we examined the immunogenic features of human cancer cell lines treated with mHS (42°C) and sHS (47°C). We found that only sHS induced ICD as defined by activation of molecular pathways underlaying the immunogenic DAMPs exposure/release in human cancer cells and by the induction of a prophylactic immunity in in vivo. Moreover, we defined specific conditions of the sHS treatment to obtain an optimal ratio between cell death, immunogenicity and the presence of tumor antigens to generate an immunotherapeutic vaccine.
Materials and methods
Cell lines
A549, OV90 and CT26 were obtained from ATCC. OV90 and CT26 were cultured in RPMI 1640 (Gibco) containing 10% heat-inactivated FCS, 100 U/mL of penicillin/streptomycin (Gibco) and 2 mmol/L L-glutamine (Gibco). A549 cells were cultured in F12 medium (Gibco) containing 10% heat-inactivated FCS and 100 U/mL of penicillin/streptomycin. Mouse embryonic fibroblasts (MEFs) wt and MEFs Bax/Bak −/− were kindly provided by Prof. Guido Kroemer MD, INSERM U848, Institut Gustave Roussy, France. Wild-type (wt) and calreticulin-deficient MEFs (Calr−/−) were kindly provided by Prof. Marek Michalak, University of Alberta, Edmonton, Canada. MEFs were cultivated in DMEM (Sigma) containing glucose (1 g/L), 10% heat-inactivated FCS, 100 U/mL of penicillin/streptomycin, 2 mmol/L L-glutamine, non-essential amino acids (1%; Gibco) and β-mercaptoethanol (0.1%; Gibco).
Antibodies
The following antibodies were used for human studies: anti-human Calreticulin, HSP90 (both from Enzo Life Sciences), HSP70 (R&D Systems), CD47 (B6H12; Santa Cruz Biotechnology), anti-CD80-FITC, CD86-PE, CD83-PerCP-Cy5.5 (Beckman Coulter, Inc.), HLA-DR-PE-Cy7, IFNγ-FITC (BD Biosciences), CD11c-APC, CD8-PE-Dylight594, CD3-Alexa 700, CD8-Alexa Fluor 700, CD127-Alexa647, CD45RO-APC (ExBio), CD4-PE-Cy7, Foxp3-Alexa488, CD3-PerCP-Cy5.5, (eBioscience), CD137-PE (4–1BB), CD25-PerCP-Cy5.5, Ki-67-PE (BioLegend). The following antibodies were used for mouse studies: anti-mouse Calreticulin (Abcam), CD80-FITC, CD86-PE, CD40-PerCP-eFluor710, I-A/I-E-PE-Cy7, CD11c-APC (all from eBioscience), Following antibodies were used for immunoblot analyses: anti-human Calreticulin, α-fodrin (both from Enzo Life Sciences), actin (Sigma), BiP, caspase-8, caspase-9, caspase-3, caspase-7, caspase-6, caspase-4, CHOP, lamin, phospho-eIF2α (Ser51), eIF2α, VDAC, cytochrome c, Her2/neu, PERK, IRE1α, XBP1 (all from Cell Signaling Technology), CEA (3G12; Pierce), MAGE-A1, MAGE-A3, MAGE-A4 (Santa Cruz Biotechnology). Secondary anti-rabbit and anti-mouse antibodies conjugated to horseradish peroxidase (Jackson ImmunoResearch Laboratories) were used.
Cell death inducing treatment of tumor cells
For HS treatment, cells at 1–4 × 106/mL in 1.5-mL tube were placed in heating blocks (Eppendorf) and incubated at 42°C or 47°C for 1 h. Control non-treated cells were incubated at 37°C. Subsequently, cells at 1 × 106/mL were transferred into tissue culture plates and incubated at 37°C for additional 6 h or 24 h. For UV treatment, cells were irradiated by UV-B (312 nm, 7.6 J/cm2) for 10 min. For treatment with chemotherapeutics, cells were incubated with idarubicin hydrochloride (37 μM; Sigma Aldrich) or cisplatin (A549 1000 μM or OV90 300 μM; Ebewe Pharma).
Flow cytometry analysis of cell death and immunogenic molecules
To detect Calreticulin, HSP70, HSP90 and CD47 cells at 6 h or 24 h were incubated with primary antibodies for 30 min, at 4°C and then stained with APC-conjugated secondary anti-rabbit antibody (Jackson ImmunoResearch Laboratories) for additional 30 min, at 4°C. Cell viability was determined by staining with AnnexinV-PE (BioLegend) or AnnexinV-PerCP-eFluor710 (eBioscience) for 15 min in Annexin V-staining buffer (eBioscience) and DAPI by flow cytometry (LSRFortessa, BD Biosciences). Data were evaluated with FlowJo software (Tree Star). The MFI of markers was determined in AnnexinV+/Dapi negative population. A549 cells were pretreated with caspase-9 inhibitor (Z-LEHD-FMK, 50 μM) (Enzo Life Sciences, Inc.) overnight before HS treatment of 1 h. Cells death was detected by DAPI and AnnexinV-PE (BioLegend) staining by flow cytometry.
Colony-forming assay
Human tumor cells A549 and OV90 were heat-treated as described above and incubated at 37°C for additional 24 h. Mouse CT26 were heat-treated for 1 h and incubated at 37°C for 1h. Cells at concentration of 1.66 × 105/well/3 mL were seeded in six-well plate in fresh medium mixed with a conditioned media at ratio 1:1 and incubated at 37°C for 10 d (human cells) or 14 d (mouse cells), respectively. After that, medium was discarded, cells stained with crystal violet for 5 min, washed with distilled water and colony-forming units (CFU) were counted where applicable. A low CFU growth (30 CFU/well) was set as a positive control of growth-favoring conditions.
Detection of changes in mitochondrial membrane potential, reactive oxygen species and the intracellular Ca2+ release
To detect changes in mitochondrial membrane potential (ΔΨm), 1 × 106/mL of tumor cells were stained with tetramethylrhodamine, ethyl ester (TMRE, 100 nM, Invitrogen) for 15 min at 37°C directly after HS treatment (0 h = 1 h at 47°C). To detect ROS, 1 × 106/mL of tumor cells were allowed to rest at 37°C for 1 h before staining with cell permeable 2′,7′-dichlorofluorescein diacetate (DCFDA) fluorogenic dye (20 μM) (Abcam) for 30 min at 37°C. Stained cells were treated with HS as indicated above and placed to 37°C for 3 h. For measurement of intracellular Ca2+ release 1 × 106/mL tumor cells after HS treatment were stained with Fluo-4-AM (4 μM, Molecular Probes Invitrogen) for 20 min at 37°C. ΔΨm, ROS and Ca2+ release were detected by flow cytometry. Cells were incubated with L-glutathione (GSH; 5 mM) and N-acetyl-L-cysteine (NAC; 5 mM) (Sigma Aldrich) were used to inhibit ROS induction in HS-treated cells. NAC and GSH were prepared in complete media with adjusted pH to 7.3–7.4.
Confocal microscopy
HS treated tumor cells (1 × 106/mL) were incubated for 6 h at 37°C. Cells were stained with primary antibodies to Calreticulin, CD47, HSP70 and HSP90, and Alexa488-conjugated secondary anti-rabbit antibody (Life Technologies) to detect the immunogenic molecules and CD47 as described above and. After that cells were fixed with 4% paraformaldehyde for 20 min and mounted on slides using cytocentrifuge (StatSpin). Cells were analyzed Leica TCS A0BS SP5 tandem scanning confocal microscope.
HMGB1 and ATP release
Tumor cells at 1 × 106/mL heated as described above were incubated for additional 1 h at 37°C. For measurement of extracellular ATP release cell culture supernatant was used and for intracellular ATP detection, cells were centrifuged 2,200 rpm, 2 min and pellet resuspended in cell lysis buffer (eBioscience). ATP content was determined according to manufacturer's instructions (ATP assay kit, Sigma-Aldrich). HMGB1 release was determined by HMGB1 ELISA kit (IBL International) from cell culture supernatant according to manufacturer's instructions after 6 h and 24 h.
DC maturation and cytokine production
Immature monocyte-derived dendritic cells (MDDCs) were generated using EasySep human CD14-positive selection kit (StemCell Technologies, Inc.) from human PBMCs isolated from buffy coats (provided by the Institute of Hematology and Blood Transfusion, Prague, Czech Republic). Monocytes were cultured for 5 d with human GM-CSF (500 U/mL; Gentaur) and IL-4 (20 ng/mL; PeproTech) in RPMI 1640 (Gibco) supplemented with L-glutamine (2 mM, Sigma), 100 U penicillin/mL and 10% heat-inactivated FCS at 37°C. DCs were pulsed with HS-treated tumor cells (1 h HS + 23 h at 37°C) at ratio 5:1 for 24 h or incubated separately in transwell system (Corning). Bone marrow cells were flushed from femurs and tibias of BALB/c mice, and cultured at 2 × 106/mL in 100-mm dishes in 10 mL of in the same medium as human DCs, but supplemented with murine granulocyte-macrophage colony-stimulating factor (GM-CSF) (20 ng/mL, PeproTech) and IL-4 (5 ng/mL, Peprotech). Fresh medium was added on day 3 or changed on day 6. Loosely attached cells were used for experiments at day 7. DCs were pulsed with HS-treated CT26 (1 h sHS + 5 h at 37°C) at ratio 5:1 for 24h. DCs incubated with poly(I:C) VacciGrade™ (InvivoGen) 25 μg/mL for 24 h were used as a positive control of DC maturation. The expression of CD80, CD86, CD83 and HLA-DR (human DCs) or CD80, CD86, I-A/I-E and CD40 (in mouse DC) in CD11c+DAPI negative cells were analyzed by flow cytometry. MFI of control unpulsed immature DCs were set to 1. The production of cytokines IL-10, IL-12p70 and TNF-α by human DCs was detected by ELISA (R&D Systems) according to manufacturer's instruction from cell culture supernatants after 24 h.
Phagocytosis of HS-treated cells by DCs
Tumor cells A549, OV90 or wt MEFs, Calr−/− MEFs (2 × 106/mL) were stained with VybrantVR DiD (2.5 μL/mL, Invitrogen), in serum free RPMI medium for 20 min, at 37°C prior the HS treatment. Immature monocyte-derived DCs (day 5) or mouse bone marrow-derived dendritic cells (BMDCs)Citation36 were stained with VybrantVR DiO (2.5 μL/mL, Invitrogen) for 20 min, at 37°C. Cells were washed twice with serum-containing medium. DCs were pulsed with tumor cells or MEFs at a DC/cell ratio 5:1 for 24 h and the amount of phagocytosed tumor cells was analyzed by flow cytometry and plotted as a percentage of DiO+DiD+ DCs. DCs pulsed with tumor cells incubated at 4°C for 24 h were used as a negative control for phagocytosis.
Detection of antigen-specific IFNγ-producing T cells and CD4+CD25+Foxp3+CD127low T regulatory cells
DCs pulsed with HS-treated tumor cells at ratio 5:1 for 24 h were added to autologous lymphocytes at T cell:DC ratio of 10:1 for 7 d. IL-2 (50 U/mL; PeproTech) was added on days 3 and 5. On day 7, lymphocytes were re-stimulated with fresh DCs treated accordingly to original samples. After 1 h, Brefeldin A (eBioscience) was added to block the release of IFNγ for 5 h. After staining with extracellular antibodies for 20 min, 4°C cells were fixed and permeabilized (Fix-PermBuffer Bioscience), and stained to detect intracellular IFNγ or Ki-67 for 30 min at 4°C. To detect MP158–66-specific CD8+ T cells, DCs were pulsed with HS-treated A549 stably expressing influenza matrix protein 1(MP1)Citation37 and incubated with autologous HLA-A2+ T cells as described above. After 8 d, cells were stained with MP158–66-HLA-A*201 Tetramer-PE (2 μL/sample; MBL International) for 30 min together with CD3-PerCP-Cy5.5, CD4-PE-Cy7, CD8-Alexa Fluor 700 and CD45RO-APC antibodies and analyzed by flow cytometry. DCs incubated with MP158–66 (GILGFVFTL) (10 μg/mL; MBL International) and CEF-E (peptides from CMV, EBV, influenza, JPT Technologies) were used as a positive control for CD8+ T-cell stimulation. To detect T regulatory cells, DC–T cell culture was analyzed without restimulation after 7 d of incubation. T regulatory cells were determined as a percentage of Foxp3+CD127low from CD4+CD25+ T cells by flow cytometry.
Immunoblot analysis
HS-treated tumor cells at 4 × 106/mL incubated at 37°C were collected at various time points for cell extract preparation. In some experiments cells treated with thapsigardin (TPG, 5 μM) and staurosporin (1 μM) (all from Sigma-Aldrich) incubated for 24 h were used as positive controls. A549 cells were pretreated with caspase-9 inhibitor (Z-LEHD-FMK, 50 μM) (Enzo Life Sciences, Inc.) overnight before HS treatment. Cells were lysed in ice-cold RIPA buffer (10 mM Tris pH 7.5, 150 mM NaCl, 5 mM EDTA and 1% Triton X-100) containing a protease inhibitor cocktail (Roche Diagnostics) and 1 mM phenylmethylsulfonylfluoride (PMSF, Roche Diagnostics) 10 min on ice. Proteins were separated on a 10–12% SDS-PAGE, electrophoretically transferred to nitrocellulose membranes (Bio-Rad) and blocked with 5% skim milk in TBST buffer (50 mM Tris, 150 mM NaCl and 0.05% Tween 20) for 1 h at room temperature. Membranes were incubated with primary antibodies overnight at 4°C, washed with TBST buffer and immunoreactive bands were visualized with horseradish peroxidase conjugated secondary antibodies using enhanced chemiluminescence reagents (ECL, Amersham Biosciences).
Isolation of cell surface proteins and separation of cellular fractions
Tumor cells grown in flasks were incubated in water bath for 1 h at 37°C, 42°C or 47°C, respectively. Cell were further incubated for additional 2.5 h at 37°C, than washed with PBS and stained with cell non-permeable sulfosuccinimidobiotin (EZ-link™ Sulfo-NHS-Biotin; Thermo Scientific) in PBS on rocking shaker at room temperature for 30 min. Labeled plasma membrane proteins were then affinity-purified using NeutrAvidin Agarose according to the manufacturer's instructions. The presence of calreticulin among plasma membrane proteins was detected by immunoblot analysis. Mitochondria and cytosol were separated using Mitochondria/Cytosol Fractionation Kit (Abcam) according to manufacturer's instructions.
In vivo experiments
BALB/ mice were obtained from a breeding colony at the Institute of Physiology, Academy of Sciences of the Czech Republic (ASCR), v.v.i. Mice were used at 9–15 weeks of age and kept in the conventional animal facility of Institute of Microbiology of ASCR, v.v.i. Mice were regularly screened for MHV and other pathogens according to FELASA. All experiments were approved by the Welfare Animal Committee at the Institute of Microbiology, ASCRv.v.i. CT26 cells were treated with mitoxantrone (1 µM, Sigma Aldrich) for 24 h or heated at 47°C for 1 h followed by 1 h incubation at 37°C. 5 × 106 cells in 200 μL PBS were injected s.c. into lower left flank on days 0 and 21. 1 × 105 live CT26 cells in 100 μL PBS were injected s.c. into lower right flank on day 31. Mice were monitored for tumor growth and survival. Mice surviving day 100 without tumor were considered long-term survivors. Tumor size was determined every 2–4 d by a caliper. The experiments were repeated thrice using 10 mice per group.
Statistical analysis
Statistical analysis was performed using software GraphPad (PRISM 6.0). The significance of the differences between groups was determined by using paired Student's t-test. Differences were considered statistically significant if p < 0.05 (*). Data were expressed as mean ± SEM.
Results
Severe, but not mild heat shock treatment of human cancer cells induces cell death
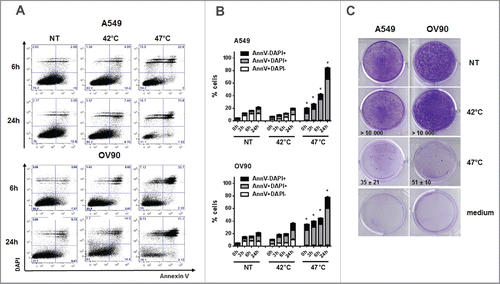
A549 and OV90 cells were treated with mHS (42°C) or sHS (47°C) for 1 h and subsequently incubated at 37°C for 0 h, 3 h, 6 h and 24 h. Cell viability was determined with Annexin V and DAPI (). sHS treatment decreased viability of both cell lines by 40–50% within 6 h (). Contrary to untreated or mHS-treated tumor cells, 80–90% of sHS-treated tumor cells were Annexin V/DAPI double positive by 24 h. High mortality rate of sHS-treated tumor cells was also confirmed in a 10 d colony-forming assay (). These results show that sHS, but not mHS, induces cell death in human tumor cells.
Figure 1. Severe, but not mild heat shock treatment of human cancer cells induces cell death. (A, B) The viability of untreated A549 and OV90 cells and cells treated with mHS (42°C, 1 h) or sHS (47°C, 1 h) followed by an incubation at 37°C for 0 h, 3 h, 6 h and 24 h. (C) Colony-forming assays were assessed after 10 d.
sHS-treated tumor cells expose calreticulin, HSP70 and HSP90, decrease CD47 on the cell surface and release HMGB1 and ATP
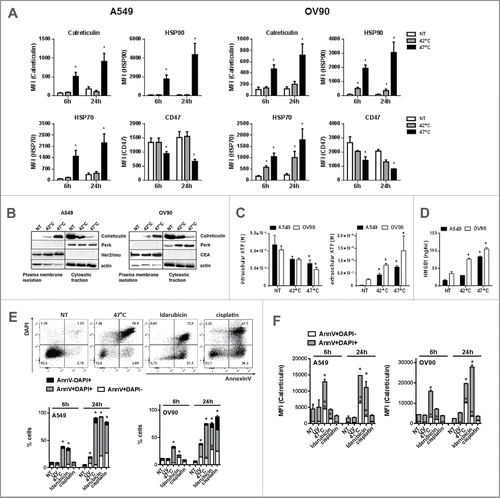
It has been shown that physical modalities such as HHP or hypericin-based photodynamic therapy (Hyp-PDT) induce ICD in cancer cells.Citation15 Therefore, we analyzed if sHS-treatment of human tumor cells inducer might induce, similarly to HHP or Hyp-PDT, cell surface translocation of calreticulin, HSP70 and HSP90 and release of ATP and HMGB1. Cells were treated with mHS (42°C) or sHS (47°C) for 1 h and subsequently incubated at 37°C for 6 h and 24 h. We found that sHS, but not mHS, induces cell surface exposure of calreticulin (CRT) and HSP90 and decrease of cell surface CD47 in both cancer cell lines after 6 h in AnnexinV+DAPI− cells gated from flow cytometry dotplots (). HSP70 was detected also on the cell surface of mHS-treated cells, albeit at lower amounts than on cells treated with sHS. To further confirm the exposure of calreticulin, the proteins of plasma membrane of HS-treated cells were analyzed by a western blot. Calreticulin was enriched on the cell surface 3 h after sHS treatment, whereas it was retained in cytosolic/intracellular membranes fraction in control and mHS-treated cells (). Intracellularly located ER-sessile protein PERK or plasma membrane-located proteins Her2/neu (A549) and CEA (OV90) were used as membrane isolation controls. As shown in , we observed that both mHS and sHS induce the release of ATP from tumor cells 1 h after HS treatment; however, only sHS displayed significant loss of intracellular ATP. Similarly, we detected HMGB1 release in mHS-treated OV90, but profound release of HMGB1 in both A549 and OV90 cancer cell lines 24 h after HS treatment (). These data suggest that only sHS treatment of cancer cells induces typical molecular characteristics of ICD, described for other ICD inducers.Citation4,6,38 We compared sHS-mediated exposure of calreticulin to an ICD inducer idarubicinCitation39 and two non-ICD inducers UV-B and cisplatin in both cell lines (). We show that HS induces faster and higher calreticulin exposure in early apoptotic cells (AnnexinV+DAPI-) over the course of 24 h when compared with idarubicin. The percentage of dead cells was comparable between treatments (). Cisplatin, on the other hand, while inducing a similar percentage of cell death after 24 h, did not induce calreticulin exposure in cells similarly to UV-B treatment ().
Figure 2. sHS-treated tumor cells expose calreticulin, HSP70 and HSP90, decrease CD47 on the cell surface and release HMGB1 and ATP. A549 or OV90 cells were left untreated or were treated with mHS (42°C, 1 h) or sHS (47°C, 1 h) followed by an incubation at 37°C for 6 h and 24 h. The exposure of HSP70, HSP90, calreticulin and the decrease of CD47 on the cell surface was determined by flow cytometry from AnnV+DAPI− cells (A). (B) Immunoblot of calreticulin cell surface exposure 2.5 h after HT treatment. The expression of PERK, CEA and Her2/neu are shown as controls of cellular and membrane fraction purity. (C) Intra- and extra-cellular levels of ATP 1 h after HS treatment (D) HMGB1 release 24 h after HS treatment. Graphs represent means ± SEM of n = 3–5 (* p < 0.05). Pictures and immunoblots are representative of n = 3–4. Comparison of cell death (E) and calreticulin exposure (F) in A549 and OV90 cells induced by treatment with sHS, UV, idarubicin and cisplatin after 6 h and 24 h. Calreticulin exposure was analyzed by flow cytometry from early (AnnV+DAPI−) and late (AnnV+DAPI+) apoptotic cells (F). Graphs represent means ± SEM of n = 3 (*p < 0.05).
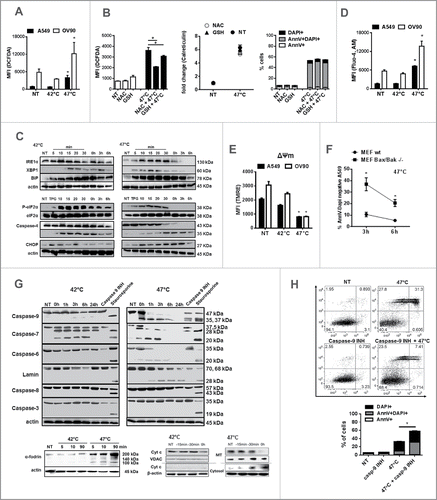
sHS treatment of human cancer cells induces intracellular Ca2+ release, ROS generation and ER stress response leading to a cell death with apoptotic features
Intracellular Ca2+ release, ROS generation and ER stress are required for DAMPs release during ICD in tumor cells.Citation40-43 Therefore, we further analyzed whether sHS treatment of tumor cells induces intracellular Ca2+ release, ER stress and ROS production (). sHS but not mHS treatment of both cancer cell lines triggered ROS production 3 h after treatment (). The sHS-mediated induction of ROS could be only partly inhibited by L-glutathione (GSH) and N-acetyl-L-cysteine (NAC), two well-established ROS scavengers, 3 h post-treatment (). However, the reduction of oxidative stress was not sufficient to diminish calreticulin exposure or cell death of tumor cells induced by sHS. Both heat treatments induced ER stress in tumor cells as documented by generation of a spliced XBP1, overexpression of BiP and eIF2α phosphorylation (). sHS induced higher BiP expression than mHS treatment. Both HS treatments also led to a cleavage of caspase-4; however, only sHS treatment induced expression of proapoptotic CHOP protein in A549 cells (), a protein involved in ER-mediated apoptotis. Only sHS treatment induced an increase in intracellular Ca2+ () and led to a decrease in mitochondrial membrane potential (ΔΨm) () in both cell lines by the end of the sHS incubation period (1 h HS = 0 h). The loss of mitochondrial functions might be related to the action of Bax and Bak proteins as there was a significant survival benefit in sHS-treated mouse embryonic fibroblast (MEFs) lacking Bax/Bak proteins 3 h after treatment (). We further observed that sHS treatment did not lead to caspase-8 and caspase-3 activation in A549 cells (), but to a rapid activation of caspase-9, followed by a cleavage of caspase-6 and −7 and lamins. The initial cleavage of caspase-7, but not caspase-6, was also observed in mHS treated tumor cells. Pre-incubation of A549 cells with caspase-9 inhibitor prevented the activation of executioner caspases-6 and −7 immediately after 1 h of sHS-treatment, which led to a survival benefit of sHS-treated cells by 20% 6 h post-treatment (). These data point at the activation of intrinsic mitochondrial apoptotic pathway as we also observed a rapid release of cytochrome c from mitochondria 15 min after sHS treatment (). We also detected cleavage of α-fodrin, which suggests the involvement of Ca2+-activated calpains in sHS-induced cell death (). In summary, these results show that sHS, but not mHS induces cell death with apoptotic features in human cancer cells.
Figure 3. sHS treatment of human cancer cells induces intracellular Ca2+ release, ROS generation and ER stress response leading to a cell death with apoptotic features. HS-induced (A, B) ROS generation after 3 h, (C) time course analyses of ER stress response, (D) intracellular Ca2+ release and (E) the loss of mitochondrial potential after HS treatment (1 h = 0 h) were analyzed. (F) Survival analyses of sHS-treated MEF wt and MEF Bax/Bak −/− cells. (G) Time course analyses of cleavage of caspases, lamin, fodrin and cytochrome c release. (H) Flow cytometry analyses of sHS-treated A549 incubated with capase-9 inhibitor. Dotplots and immunoblots are representative of n = 3–6. Graphs represent means ± SEM of n = 3–5 (*p < 0.05).
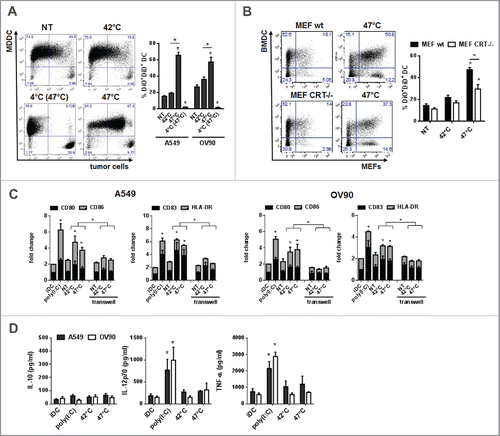
Phagocytosis of sHS-treated tumor cells partially depends on calreticulin and leads to a phenotypic maturation of DC in vitro
We analyzed whether sHS-induced ICD of tumor cells would impact on phagocytosis and functional characteristics of DCs in vitro. mHS or sHS-treated tumor cells (1 h + 24 h, 37°C) were added to human MDDCs at ratio 1:5 and incubated for additional 24 h. shows that 70% (A549) and 60% (OV90) of DCs phagocytosed sHS-treated tumor cells, whereas only 20% of DCs phagocytosed mHS-treated or control non-treated tumor cells. The phagocytosis of sHS-treated cells was partially dependent on calreticulin as sHS-treated MEFs (Calr−/−) were phagocytosed less than sHS-treated MEFs wt (). Interestingly, mHS-and sHS-treated tumor cells induced a similar degree of expression of maturation markers CD80, CD86, CD83 and HLA-DR on DCs as poly(I:C) (). However, only poly(I:C) stimulated TNF-α and IL-12p70 production from DCs. Neither poly(I:C) nor HS-treated tumor cells induced IL-10 (). The capacity of HS-treated tumor cells to activate DC maturation was cell contact dependent as separation of HS-treated tumor cells and DC in a transwell system completely prevented DC maturation ().
Figure 4. Phagocytosis of sHS-treated tumor cells partially depends on calreticulin and leads to a phenotypic maturation of DC in vitro. (A, B) Untreated cells or cells treated with mHS (42°C, 1 h) or sHS (47°C, 1 h) followed by incubation at 37°C for 24 h were incubated with DC for additional 24 h. Graphs show percentage of DAPI negative DiO+DiD+ DCs and represent means ± SEM of five donors or four mice (*p < 0.05). (C) The expression of maturation markers on DCs after 24 h in cultures where DC + HS-treated tumor cells were co-incubated or where HS-treated cells were separated from DCs by a transwell. (D) IL-10, IL-12p70 and TNF-α production was determined from cell culture supernatants by ELISA after 24 h. The graphs are means ± SEM of 6–8 donors (*p < 0.05).
DCs pulsed with sHS-treated tumor cells stimulated antigen-specific CD4+ T cells and induced higher CD8+ T cell activation and proliferation in vitro
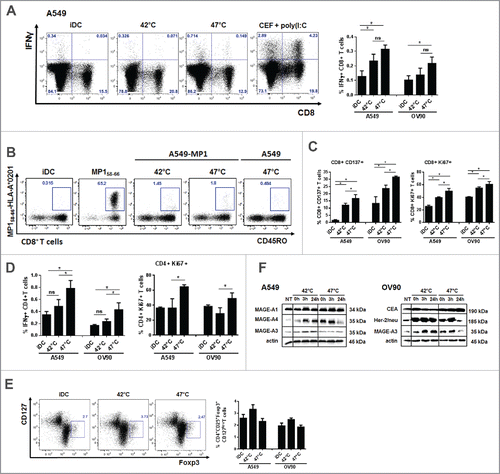
T cell stimulatory capacity of DC pulsed with mHS or sHS-treated tumor cells was tested in autologous antigen unspecific system and autologous antigen-specific system using HS-treated tumor cells that stably express influenza M1 protein as a model antigen.Citation37 DCs loaded with mHS or sHS-treated tumor cells induced similarly high number of IFNγ-producing CD8+ T cells () and MP158–66-specific CD8+ T cells detected by the tetramer staining () than unpulsed iDCs. MP158–66-specific CD8+ T cells displayed a memory phenotype. However, CD8+ T cells induced by DC loaded with sHS-killed tumor cells showed higher proliferation and activation profile (). Also, only these DCs stimulated IFNγ-producing CD4+ T cells that exhibited a high proliferative activity (). There was no induction of CD4+CD25+Foxp3+CD127low T regulatory cells by DCs loaded with either mHS- or sHS-killed tumor cells (). We next analyzed the content of tumor antigens. As shown in , the content of all tested tumor antigens increased in both cell lines after HS treatment. However, in contrast to mHS-treated cells, tumor antigen levels declined back to basal levels of non-treated cells after sHS treatment.
Figure 5. DCs pulsed with sHS-treated tumor cells stimulated antigen-specific CD4+ T cells and induced higher CD8+ T cell activation and proliferation. DCs incubated with sHS or mHS-treated cells for 24 h were added to autologous T lymphocytes at ratio 1:10. The number of IFNγ producing (A) CD8+ and (D) CD4+ T cells, proliferation (Ki67) and activation (CD137) of (C) CD8+ and (D) CD4+ T cells was determined after 7 d after one round of restimulation. (B) The number of MP158–66-specific CD8+ T cells was determined after 9 d. (E) Percentage of Foxp3+CD127low T regulatory cells from CD4+CD25+ T cells. Dotplots are representative of 4–8 donors. Graph shows percentage of IFNγ+CD8+ T cells or IFNγ+CD4+ T cells from CD8+ or CD4+ T cells, respectively and represents means ± SEM of 4–8 donors (*p < 0.05). (F) Immunoblot of tumor antigens after HS treatment. Immunoblots are representative of n = 3.
sHS-treated tumor cells are immunogenic and elicit prophylactic immunity in mice
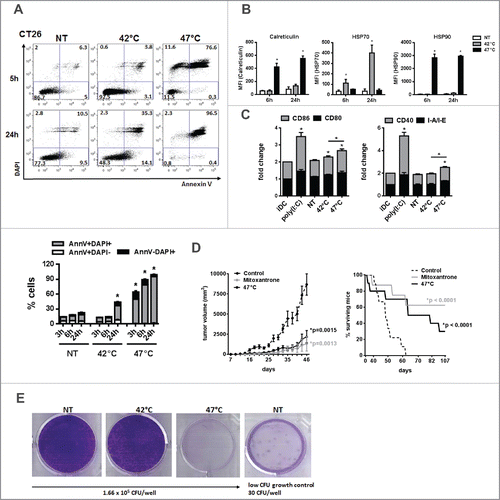
To confirm our data on sHS-treated human tumor cells, we exposed mouse tumor cells CT26 to mHS or sHS treatment and analyzed the exposure of calreticulin, HSP70 and HSP90 and their capacity to induce maturation of BMDCs in vitro (). CT26 seems to be more sensitive to HS-induced cell death than human tumor cells as 80% cells were already dead after 6 h. In line with higher sensitivity of mouse cells to heat treatment, we also observed a significant percentage of dead cells (40%) 24 h after mHS treatment (). However, the exposure of calreticulin and HSP90 was detected only in AnnV+DAPI− cells treated with sHS (). HSP70 was not detectable in sHS-treated tumor cells, but was strongly induced by mHS treatment pointing at possible differences in immunogenic molecules exposure/release in mouse tumor cells under various HS conditions. In line with these data, we observed a higher maturation of DC loaded with sHS-treated tumor cells than DC loaded with mHS-treated tumor cells (). The immunogenicity of sHS-treated CT26 tumor cells was tested in vivo in mouse colorectal cancer model in a prophylactic setting and compared with a known ICD inducer mitoxantrone.Citation7 Mice were vaccinated with sHS- or mitoxantrone-treated CT26 cells at days 0 and 21 and challenged with live CT26 at day 31. A significant tumor size reduction and a survival benefit were observed after vaccination with sHS-treated cells similarly to mice vaccinated with mitoxantrone-treated cells (). The long-term survival of sHS-vaccinated mice after tumor cells challenge (more than 100 d) without any signs of tumor growth was recorded in 3 out of 10 mice. The immunogenicity of mHS-treated cells in vivo was not tested due to a high probability of cell growth at the site of injection. This conclusion was made based on an in vitro colony forming assay prepared from mHS-treated CT26 where a high survival of mHS-treated cells in contrast to sHS-treated cells was observed ().
Figure 6. sHS-treated tumor cells are immunogenic and elicit prophylactic immunity in mice. (A, B) The viability of untreated CT26 cells and cells treated with mHS (42°C, 1 h) or sHS (47°C, 1 h) followed by an incubation at 37°C for 5 h and 24 h. (C) The exposure of HSP70, HSP90 and calreticulin was determined by flow cytometry from AnnexinV+DAPI− cells. Graphs represent means ± SEM of n = 3 (*p < 0.05). Dotplots are representative of n = 3. (D) Mice were vaccinated with sHS or mitoxantrone-treated CT26 cells at days 0 and 21 and challenged with live CT26 at day 31. Tumor growth and survival was monitored in 10 mice per group. Three out of 10 mice were considered long-term survivors after day 100. The results are representative of n = 3. (E) Colony-forming assays were assessed after 14 d.
Discussion
Our data suggest that the immunogenicity of mHS and sHS-treated tumor cells might not share the same determinants. We showed that sHS (47°C), but not mHS (42°C), induces cell death in human cancer cells that display molecular characteristics of ICD described for other ICD inducing agentsCitation38 such as the generation of ROS, release of intracellular Ca2+ and induction of integrated ER stress response, exposure of calreticulin and HSP70 and HSP90, decrease of cell surface CD47 expression and the release of ATP and HMGB1. Severe HS treatment as an ICD inducer might be further supported by the induction of prophylactic immunity by sHS-treated cells in in vivo vaccination cancer model.
The immunogenicity of tumor cells treated with mHS (42°C) has been attributed to HS proteins mainly HSP7020,22. We showed here that mHS treatment induces HSP70, as well as the release of low amounts of other DAMPs such as ATP or HMGB1. We detected activation of ER stress response proteins and caspase-4, however, without detectable ROS generation and intracellular Ca2+ increase. The activation of caspase-4 might also account for detected transient caspase-7 cleavage observed in mHS-treated tumor cellsCitation44 as any other caspases were not cleaved. The low number of dead cells indicates that several stress-mitigating and anti-apoptotic mechanisms were activated to counteract mHS effects.Citation1 Furthermore, we observed that mHS-treated cells were not efficiently phagocytosed by DCs, which might explain the lack of CD4+ T cell stimulatory capacity. As CD47 was present on the cell surface of mHS-treated cells, this and also other “don't eat me” molecules are likely to account for the inefficient phagocytosis.Citation14 On the other hand, mHS-treated cells similarly to sHS-treated cells induced phenotypic DC maturation, which was dependent on the cell–cell contact. However, in these experiments, we cannot exclude the synergy between cell-bound molecules with soluble DAMPs in the DC maturation.Citation45 DCs pulsed with mHS- or sHS-treated tumor cells similarly stimulated IFNγ-producing CD8+ T cells in vitro. This might be explained by a release of sufficient amount of antigenic peptides bound to HSPs from mHS-treated tumor cells, which are then cross-presented by DCs.Citation46 Indeed, we confirmed that HSP70 is secreted into cell culture supernatant (data not shown). This also corresponds the observation that mHS increases the expression of tumor antigens in tumor cell lines. This has been previously shown on mRNA level for human melanoma cells.Citation21 However, despite observed general immunogenicity and no induction of CD4+CD25+Foxp3+CD127low T regulatory cells, the lack of cell death excludes the use of mHS for active cellular immunotherapy in cancer patients.
Most importantly, we showed here that the sHS-induced cell death displays molecular features of ICD characterized by the generation of ROS, intracellular Ca2+ release and induction of integrated ER stress response, exposure of calreticulin and HSP70 and HSP90, decrease of cell surface CD47 expression and the release of ATP and HMGB1 as described for other agents.Citation38 This is in line with a few previous studies showing calreticulin and DAMPs release under sHS in vitro,Citation34,47 and in vivo.Citation33 Since the exposure of calreticulin was not prevented by ROS inhibition as described for HHP,Citation15 it can be speculated that sHS-induced molecular pathways underlying DAMP secretion might exhibit different or possibly unique regulatory features as also documented for various ICD activating treatments.Citation38 We observed rapid activation of intrinsic apoptotic pathway involving the release of cytochrome c, cleavage of caspase-9, −6 and-7, and activation of ER-mediated apoptotic pathway.Citation44,45 The sHS-induced cell death could be diminished by caspase-9 inhibition but not by inhibition of ROS generation. This suggests that caspase-9 plays an important, but not exclusive, role in sHS-mediated cell death. The major point-of-no-return determinants of the cell fate upon sHS treatment would be most likely quick irreversible loss of mitochondrial potential depending on Bax/Bak proteins and the rapid decline in ATP levels under such severe stress concomitantly with the activation of multiple cell-death executional pathways as described for various tumor cell types treated with heat at various thermal dose.Citation17 Interestingly, there was no caspase-8 and −3 cleavage that might be connected to the way how sHS changes membrane fluidics or acts on activity of various membrane proteins, e.g., inactivation of receptor-mediated signaling.Citation1,16 The cleavage of α-fodrin points at the activation of calpains by intracellular Ca2+.Citation48-50 We also observed cleavage of LC3 protein at later time points but no activation of RIP3 kinase (data not shown). The involvement of autophagyCitation51 or necroptosisCitation52 to cell death was not further analyzed in this study.
Human DCs pulsed with sHS-killed tumor cells displayed enhanced immunogenicity in vitro over DC pulsed with mHS-treated tumor cells when analyzed for T-cell stimulation. Despite similar number of IFNγ-producing CD8+ T cells, CD8+ T cells induced by DC pulsed with sHS-killed tumor cells displayed higher activation and proliferation. We showed here that antigen expressed in HS-treated tumor cells is presented to antigen-specific CD8+ T cells using specific MP158–66-HLA-A*0201 tetramers. DCs pulsed with sHS-killed cells, similar to DC loaded with mHS-treated cells, did not enhanced number of CD4+CD25+Foxp3+ T regulatory cells in vitro. The difference between sHS and mHS-treated tumor cells was in the efficiency of phagocytosis by DCs. The phagocytosis of sHS-treated cells was partly dependent on cell surface calreticulin (30%), which suggests possible participation of other “eat-me” molecules in this process. Only DCs pulsed with sHS-treated cells stimulated CD4+ T cells in vitro, which was most likely connected to a high phagocytosis of sHS-treated cells and to availability of tumor antigens. The importance of CD4+ T cells in severe HT-induced antitumor immunity was shown in the study of Yu et al. using microwave thermal ablation in vivo mouse cancer model.Citation33 Whereas CD8+ T cells were essential for antitumor immunity induced by this treatment,Citation33 the depletion of CD4+ T cells diminished the survival of immunized mice by 40% in comparison to 100% without CD4+ T-cell depletion.Citation33 Our data suggest that sHS treatment at 47°C of human cancer cell lines A549 and OV90 might represent the optimal condition to achieve high immunogenicity, high level of tumor antigens and effective cell death, which is required by the regulatory authorities to generate of active cellular cancer immunotherapy. A critical association between the amount of immunogenic signals, specifically calreticulin and HSP90, emitted by dying cells and the clinical efficacy of DC-based vaccine has been documented in indolent B-cell lymphoma patients.Citation47
We have further shown that sHS-treated cells exposed immunogenic DAMPs and induced DC maturation similarly to human cell lines. sHS-treated cells induced protective memory antitumor immunity in mouse colorectal cancer model, which was comparable to the vaccination with mitoxantrone-treated tumor cells, a known ICD inducer.Citation7 Such vaccination assay is considered a gold-standard approach to test the ability of a specific agent to induce ICD.Citation38 Similar studies using high temperature treatment such as magnetic HT in vivo documented the induction of long-lasting antitumor immunity and the advantage of higher therapeutic temperatures (46–55°C) over the mild HT (42–44°C).Citation31,32 Although our findings support severe HS treatment in vitro as an ICD inducer, it remains to be characterized in detail if severe HT-inducing techniques in vivo such as radiofrequency ablation, magnetic HT, microwave ablation or high intensity focused ultrasound induce ICD.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by Sotio a.s. The work was supported by the Ministry of Education, Youth and Sports of the Czech Republic, within the LQ1604 National Sustainability Program II (Project BIOCEV-FAR), by the project “BIOCEV” (CZ.1.05/1.1.00/02.0109) and the Institutional Research Concept RVO 61388971. The work of the Department of Immunology of Charles University is supported by Ministry of Health, Czech Republic-Conceptual Development of Research Organization (University Hospital Motol, Prague, Czech Republic, 00064203).
References
- Hildebrandt B, Wust P, Ahlers O, Dieing A, Sreenivasa G, Kerner T, Felix R, Riess H. The cellular and molecular basis of hyperthermia. Crit Rev Oncol Hematol 2002; 43:33-56; PMID:12098606; https://doi.org/10.1016/S1040-8428(01)00179-2
- Mentre P, Hamraoui L, Hui Bon Hoa G, Debey P. Pressure-sensitivity of endoplasmic reticulum membrane and nucleolus as revealed by electron microscopy. Cell Mol Biol 1999; 45:353-62; PMID:10386792; https://doi.org/10.1088/1742-6596/121/1/112003
- Dudek AM, Garg AD, Krysko DV, De Ruysscher D, Agostinis P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev 2013; 24:319-33; PMID:23391812; https://doi.org/10.1016/j.cytogfr.2013.01.005
- Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Ann Rev Immunol 2013; 31:51-72; PMID:23157435; https://doi.org/10.1146/annurev-immunol-032712-100008
- Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 2012; 12:860-75; PMID:23151605; https://doi.org/10.1038/nrc3380
- Adkins I, Fucikova J, Garg AD, Agostinis P, Spisek R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology 2014; 3:e968434; PMID:25964865; https://doi.org/10.4161/21624011.2014.968434
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007; 13:54-61; PMID:17187072; https://doi.org/10.1038/nm1523
- Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukocyte biol 2007; 81:1-5; PMID:17032697; http://doi.org/10.1189/jlb.0306164
- Matzinger P. The danger model: a renewed sense of self. Science 2002; 296:301-5; PMID:11951032; https://doi.org/10.1126/science.1071059
- Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood 2007; 109:4839-45; PMID:17299090; https://doi.org/10.1182/blood-2006-10-054221
- Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, Séror C, Métivier D, Perfettini JL, Zitvogel L, et al. Chemotherapy induces ATP release from tumor cells. Cell Cycle 2009; 8:3723-8; PMID:19855167; https://doi.org/10.4161/cc.8.22.10026
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002; 418:191-5; PMID:12110890; https://doi.org/10.1038/nature00858
- Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, Kroemer G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ 2008; 15:3-12; PMID:18007663; https://doi.org/10.1038/sj.cdd.4402269
- Kepp O, Galluzzi L, Martins I, Schlemmer F, Adjemian S, Michaud M, Sukkurwala AQ, Menger L, Zitvogel L, Kroemer G. Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer Metastasis Rev 2011; 30:61-9; PMID:21249425; https://doi.org/10.1007/s10555-011-9273-4
- Fucikova J, Moserova I, Truxova I, Hermanova I, Vancurova I, Partlova S, Fialova A, Sojka L, Cartron PF, Houska M et al. High hydrostatic pressure induces immunogenic cell death in human tumor cells. Int J Cancer 2014; 135:1165-77; PMID:24500981; https://doi.org/10.1002/ijc.28766
- Roti Roti JL. Cellular responses to hyperthermia (40–46°C): cell killing and molecular events. Int J Hyperthermia 2008; 24:3-15; PMID:18214765; https://doi.org/10.1080/02656730701769841
- Milleron RS, Bratton SB. 'Heated' debates in apoptosis. Cell Mol Life Sci 2007; 64:2329-33; PMID:17572850; https://doi.org/10.1007/s00018-007-7135-6
- Suto R, Srivastava PK. A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science 1995; 269:1585-8; PMID:7545313; https://doi.org/10.1126/science.7545313
- Melcher A, Todryk S, Hardwick N, Ford M, Jacobson M, Vile RG. Tumor immunogenicity is determined by the mechanism of cell death via induction of heat shock protein expression. Nat Med 1998; 4:581-7; PMID:9585232; https://doi.org/10.1038/nm0598-581
- Frey B, Weiss EM, Rubner Y, Wunderlich R, Ott OJ, Sauer R, Fietkau R, Gaipl US. Old and new facts about hyperthermia-induced modulations of the immune system. Int J Hyperthermia 2012; 28:528-42; PMID:22690925; https://doi.org/10.3109/02656736.2012.677933
- Shi H, Cao T, Connolly JE, Monnet L, Bennett L, Chapel S, Bagnis C, Mannoni P, Davoust J, Palucka AK et al. Hyperthermia enhances CTL cross-priming. J Immunol 2006; 176:2134-41; PMID:18539499; https://doi.org/10.4049/jimmunol.176.4.2134
- Zhang HG, Mehta K, Cohen P, Guha C. Hyperthermia on immune regulation: a temperature's story. Cancer Lett 2008; 271:191-204; PMID:18597930; https://doi.org/10.1016/j.canlet.2008.05.026
- Feng H, Zeng Y, Graner MW, Katsanis E. Stressed apoptotic tumor cells stimulate dendritic cells and induce specific cytotoxic T cells. Blood 2002; 100:4108-15; PMID:12393401; https://doi.org/10.1182/blood-2002-05-1389
- Feng H, Zeng Y, Graner MW, Likhacheva A, Katsanis E. Exogenous stress proteins enhance the immunogenicity of apoptotic tumor cells and stimulate antitumor immunity. Blood 2003; 101:245-52; PMID:12393411; https://doi.org/10.1182/blood-2002-05-1580
- Clark PR, Menoret A. The inducible Hsp70 as a marker of tumor immunogenicity. Cell Stress Chaperones 2001; 6:121-5; PMID:11599573; https://doi.org/10.1379/1466-1268(2001)006<0121:TIHAAM>2.0.CO;2
- Mise K, Kan N, Okino T, Nakanishi M, Satoh K, Teramura Y, Yamasaki S, Ohgaki K, Tobe T. Effect of heat treatment on tumor cells and antitumor effector cells. Cancer Res 1990; 50:6199-202; PMID:2400985; http://doi.org/10.11501/3057478
- Takahashi T, Mitsuhashi N, Sakurai H, Niibe H. Modifications of tumor-associated antigen expression on human lung cancer cells by hyperthermia and cytokine. Anticancer Res 1995; 15:2601-6; PMID:8669832; https://doi.org/10.1186/1756-9966-27-5
- Wong JY, Mivechi NF, Paxton RJ, Williams LE, Beatty BG, Beatty JD, Shively JE. The effects of hyperthermia on tumor carcinoembryonic antigen expression. Int J Radiat Oncol Biol Phys 1989; 17:803-8; PMID:2674083; https://doi.org/10.1016/0360-3016(89)90070-9
- Zerbini A, Pilli M, Penna A, Pelosi G, Schianchi C, Molinari A, Schivazappa S, Zibera C, Fagnoni FF, Ferrari C et al. Radiofrequency thermal ablation of hepatocellular carcinoma liver nodules can activate and enhance tumor-specific T-cell responses. Cancer Res 2006; 66:1139-46; PMID:16424051; https://doi.org/10.1158/0008-5472.CAN-05-2244
- Ito A, Honda H, Kobayashi T. Cancer immunotherapy based on intracellular hyperthermia using magnetite nanoparticles: a novel concept of “heat-controlled necrosis” with heat shock protein expression. Cancer Immunol Immunother 2006; 55:320-8; PMID:16133113; https://doi.org/10.1007/s00262-005-0049-y
- Hu R, Ma S, Li H, Ke X, Wang G, Wei D, Wang W. Effect of magnetic fluid hyperthermia on lung cancer nodules in a murine model. Oncol Lett 2011; 2:1161-4; PMID:22848282; https://doi.org/10.3892/ol.2011.379
- Wang H, Zhang L, Shi Y, Javidiparsijani S, Wang G, Li X, Ouyang W, Zhou J, Zhao L, Wang X et al. Abscopal antitumor immune effects of magnet-mediated hyperthermia at a high therapeutic temperature on Walker-256 carcinosarcomas in rats. Oncol Lett 2014; 7:764-70; PMID:24527084; https://doi.org/10.3892/ol.2014.1803
- Yu Z, Geng J, Zhang M, Zhou Y, Fan Q, Chen J. Treatment of osteosarcoma with microwave thermal ablation to induce immunogenic cell death. Oncotarget 2014; 5:6526-39; PMID:25153727; https://doi.org/10.18632/oncotarget.2310
- Brusa D, Migliore E, Garetto S, Simone M, Matera L. Immunogenicity of 56°C and UVC-treated prostate cancer is associated with release of HSP70 and HMGB1 from necrotic cells. Prostate 2009; 69:1343-52; PMID:19496055; https://doi.org/10.1002/pros.20981
- Adkins I, Koberle M, Grobner S, Autenrieth SE, Bohn E, Borgmann S, Autenrieth IB. Y. enterocolitica inhibits antigen degradation in dendritic cells. Microbes Infect 2008; 10:798-806; PMID:18539499; https://doi.org/10.1016/j.micinf.2008.04.014
- Adkins I, Kamanova J, Kocourkova A, Svedova M, Tomala J, Janova H, Masin J, Chladkova B, Bumba L, Kovar M et al. Bordetella adenylate cyclase toxin differentially modulates toll-like receptor-stimulated activation, migration and T cell stimulatory capacity of dendritic cells. PloS One 2014; 9:e104064; PMID:25084094; https://doi.org/10.1371/journal.pone.0104064
- Hradilova N, Sadilkova L, Palata O, Mysikova D, Mrazkova H, Lischke R, Spisek R, Adkins I. Generation of dendritic cell-based vaccine using high hydrostatic pressure for non-small cell lung cancer immunotherapy. PLoS One 2017; 12:e0171539; PMID:28187172; https://doi.org/10.1371/journal.pone.0171539
- Grobner S, Adkins I, Schulz S, Richter K, Borgmann S, Wesselborg S, Ruckdeschel K, Micheau O, Autenrieth IB. Catalytically active Yersinia outer protein P induces cleavage of RIP and caspase-8 at the level of the DISC independently of death receptors in dendritic cells. Apoptosis 2007; 12:1813-25; PMID:17624595; https://doi.org/10.1007/s10495-007-0100-x
- Fucikova J, Kralikova P, Fialova A, Brtnicky T, Rob L, Bartunkova J, Spísek R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res 2011; 71:4821-33; PMID:21602432; https://doi.org/10.1158/0008-5472.CAN-11-0950
- Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJ et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J 2012; 31:1062-79; PMID:22252128; https://doi.org/10.1038/emboj.2011.497
- Garg AD, Krysko DV, Vandenabeele P, Agostinis P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol Immunother 2012; 61:215-21; PMID:22193987; https://doi.org/10.1007/s00262-011-1184-2
- Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, Durchschlag M, Joza N, Pierron G, van Endert P et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J 2009; 28:578-90; PMID:19165151; https://doi.org/10.1038/emboj.2009.1
- Tufi R, Panaretakis T, Bianchi K, Criollo A, Fazi B, Di Sano F, Tesniere A, Kepp O, Paterlini-Brechot P, Zitvogel L et al. Reduction of endoplasmic reticulum Ca2+ levels favors plasma membrane surface exposure of calreticulin. Cell Death Differ 2008; 15:274-82; PMID:18034188; https://doi.org/10.1038/sj.cdd.4402275
- Shellman YG, Howe WR, Miller LA, Goldstein NB, Pacheco TR, Mahajan RL, LaRue SM, Norris DA. Hyperthermia induces endoplasmic reticulum-mediated apoptosis in melanoma and non-melanoma skin cancer cells. J Invest Dermatol 2008; 128:949-56; PMID:17989736; https://doi.org/10.1038/sj.jid.5701114
- Chen T, Guo J, Han C, Yang M, Cao X. Heat shock protein 70, released from heat-stressed tumor cells, initiates antitumor immunity by inducing tumor cell chemokine production and activating dendritic cells via TLR4 pathway. J Immunol 2009; 182:1449-59; https://doi.org/10.4049/jimmunol.182.3.1449
- Srivastava PK, Udono H, Blachere NE, Li Z. Heat shock proteins transfer peptides during antigen processing and CTL priming. Immunogenetics 1994; 39:93-8; PMID:8276462; https://doi.org/10.1007/BF00188611
- Zappasodi R, Pupa SM, Ghedini GC, Bongarzone I, Magni M, Cabras AD, Colombo MP, Carlo-Stella C, Gianni AM, Di Nicola M. Improved clinical outcome in indolent B-cell lymphoma patients vaccinated with autologous tumor cells experiencing immunogenic death. Cancer Res 2010; 70:9062-72; PMID:20884630; https://doi.org/10.1158/0008-5472.CAN-10-1825
- Bettaieb A, Averill-Bates DA. Thermotolerance induced at a mild temperature of 40°C alleviates heat shock-induced ER stress and apoptosis in HeLa cells. Biochim Biophys Acta 2015; 1853:52-62; PMID:25260982; https://doi.org/10.1016/j.bbamcr.2014.09.016
- Dutta S, Chiu YC, Probert AW, Wang KK. Selective release of calpain produced alphalI-spectrin (alpha-fodrin) breakdown products by acute neuronal cell death. Biol Chem 2002; 383:785-91; PMID:12108543; https://doi.org/10.1515/BC.2002.082
- Vanags DM, Porn-Ares MI, Coppola S, Burgess DH, Orrenius S. Protease involvement in fodrin cleavage and phosphatidylserine exposure in apoptosis. J Biol Chem 1996; 271:31075-85; PMID:8940103; https://doi.org/10.1074/jbc.271.49.31075
- Martins I, Michaud M, Sukkurwala AQ, Adjemian S, Ma Y, Shen S, Kepp O, Menger L, Vacchelli E, Galluzzi L et al. Premortem autophagy determines the immunogenicity of chemotherapy-induced cancer cell death. Autophagy 2012; 8:413-5; PMID:22361584; https://doi.org/10.4161/auto.19009
- Adkins I, Koberle M, Grobner S, Bohn E, Autenrieth IB, Borgmann S. Yersinia outer proteins E, H, P, and T differentially target the cytoskeleton and inhibit phagocytic capacity of dendritic cells. Int J Med Microbiol 2007; 297:235-44; PMID:17462949; https://doi.org/10.1016/j.ijmm.2007.02.005