
ABSTRACT
The emerging role for CD73 in driving cancer growth and metastasis has presented opportunities to develop anti-CD73 monoclonal antibodies (mAbs) in the treatment of human cancers. Blockade of CD73 by antagonistic CD73 mAbs ameliorates tumor growth and metastasis via the inhibition of enzymatic and non-enzymatic CD73 pathways. In this study, we investigated whether Fc-receptor cross-linking represented a non-redundant mechanism by which anti-CD73 mAbs exert potent suppression of solid tumors and metastases. We engineered four anti-CD73 mAbs, each different in their ability to modulate CD73 enzymatic function and bind Fc receptors. mAbs recognizing a similar epitope of CD73 (CD73–04, TY/23 and 2C5) displayed the greatest antitumor activity. Importantly, we observed that the optimal control of metastasis by anti-CD73 mAbs involved primarily Fc receptor engagement, while suppression of solid tumors required both, enzyme inhibition and activation of Fc receptors. Engagement of Fc-receptors was also essential for optimal anti-metastatic effect in combination with either A2AR inhibitor or anti-PD-1 mAb treatment. The control of experimental metastases relied on the activation of host NK cells and IFNγ, while NK cells, CD8+ T cells and IFNγ were needed for effective antitumor effect in the spontaneous metastases model. These observations advance our understanding of the enzymatic and non-enzymatic functions of anti-CD73 mAbs in solid tumors and metastases. Altogether, these findings will greatly assist in the design of anti-CD73 mAbs to be used as either single agents or in combination with other immunotherapeutic molecules or targeted therapies.
Introduction
The adenosinergic pathway has emerged as a critical immune checkpoint pathway in the treatment of cancers.Citation1,2 Elevated adenosine is commonly identified within the hypoxic tumor microenvironment (TME) and this negatively dictates immune cell recognition and activity, thus favoring the onset and progression of cancers.Citation3 Tumor hypoxia actively stimulates release of extracellular ATP, which is subsequently catabolized to adenosine by the concerted action of two ecto-nucleotidases: CD39 and CD73. Specifically, CD39 and CD73 are surface enzymes that mediate de-phosphorylation of ATP to ADP/AMP and adenosine, respectively. This excess in extracellular adenosine, in turn, binds to, and activates cognate receptors, A2A (A2AR) and A2B (A2BR). Each of these receptors is capable of independently activating signaling cascades and promoting tumor escape via suppression of immune cell functions and induction of angiogenesis.Citation1,2
The enzymatic activities of CD39 and CD73 modify the dynamics of the tumor niche by directing the switch from an ATP-driven inflammatory environment to an adenosine-mediated anti-inflammatory milieu. Several strategies are therefore explored to abrogate this catabolic pathway. In particular, CD73 has garnered much attention owing to its rate-limiting ability to hydrolyze AMP to adenosine, thus representing the bottleneck for this adenosine generation.Citation4 CD73 is expressed on several cancer and immune cells.Citation4 On the host, CD73 promotes immune-evasion, and CD73 gene targeted mice have significantly retarded tumor growth and metastasis.Citation5,6,7 These antitumor effects were largely dependent upon the activation of CD8+ T cells, NK cells and production of IFNγ.Citation5,6,8 Altogether, these observations indicated that the inhibition of the CD73 pathway deterred adenosine accumulation, and thus reversed immune suppression. Indeed, anti-CD73 mAb treatment is effective in inhibiting the growth of primary tumors and their metastases in mice.Citation9,10,11 Anti-CD73 monotherapy has clinical promise, but studies also indicated that these mAbs effectively synergized with anti-PD-1 or anti-CTLA-4 mAbs.Citation12 Additionally, our recent findings identified that co-targeting molecules within the adenosinergic pathway displayed a synergistic reduction in the number of lung metastases. Specifically, the combination blockade of CD73 and A2AR provided improved therapeutic efficacy over monotherapy alone,Citation11 thus reflecting the non-redundant nature of these molecules in mediating antitumor immunity.
Notably, cancer trials (NCT02503774) using anti-human CD73 mAbs are currently underway in the treatment of solid tumors (www.clinicaltrials.gov). We described previously the development of MEDI9447 as an anti-CD73 mAb with several means to affect CD73-associated downstream signaling including enzymatic activity and antibody-mediated internalization. Suppression of syngeneic tumors included the activation of immune-activating cytokines IFNγ and TNF-α, among others.Citation13,14 MEDI9447 was developed to minimize Fc-receptor (FcR) engagement; however, a recent study by Young et al. identified that the anti-metastatic activities of anti-CD73 mAb were largely reliant on the ability of the mAb to engage Fc receptor, in particular, FcγRIV on CD11b+ Gr-1+ myeloid cells.Citation11 Similar studies in the past have been instrumental in deciphering the mechanism underlying the clinical activities of anti-CTLA-4,Citation15,16 and anti-HER2Citation17 mAbs. In the current study, we assessed more extensively whether the control of primary tumors and their metastases by anti-CD73 mAbs always required the activation of Fc-receptors. We engineered mouse cross-reacting anti-CD73 mAbs, each with a different ability to inhibit enzymatic activity, internalize and activate Fc-receptors. The use of these anti-CD73 mAbs in this study has shed insights into the mechanisms by which anti-CD73 mAb activity is mediated during antitumor immunity. Overall, these observations provide new leads and important information for the development of improved anti-CD73 mAbs in the treatment of human cancers.
Results
Binding of anti-CD73 clones to mouse and human CD73
Cross-reacting antibodies have proven to be beneficial for direct cross-species comparisons in antitumor efficacies. To this end, we developed four cross-reacting anti-CD73 mAbs using phage display, namely 2C5, CD73–04, CD73–46 and CD73–69 (). Importantly, clones CD73–04, CD73–46 and CD73–69 were designed such that their Fc portions were triple mutated so as to minimize IgG effector functions. The therapeutic efficacies of these clones are therefore attributed to their ability to bind and block CD73-enzyme functions, but not bind FcR. By contrast, 2C5 was developed with an active Fc region and hence, any differences in the therapeutic efficacies would assist in decoupling direct CD73-mediated antitumor effects from potential FcR-mediated anti-CD73 activity.
Figure 1. Biochemical characterization of anti-CD73 mAbs. (A) Affinity measurements for mouse recombinant CD73 were conducted by immobilizing mouse CD73 on a ProteOn sensor chip. Binding of anti-CD73 clones; 2C5, CD73–04, CD73–46, CD73–69 and Phen0203 (anti-human CD73 mAb) were measured by SPR. The dissociation constant values for anti-CD73 clones binding to (B) mouse or (C) human recombinant CD73 were determined. (D) Binding of anti-CD73 mAb clones to 4T1 mouse mammary carcinoma cells was determined by incubating tumor cells with serially diluted anti-CD73 mAbs or control mAbs (cIg) (1A7 and R347 as controls for mouse and human anti-CD73 mAbs, respectively), followed by incubation with secondary antibody. The median fluorescent intensity (MFI) relative to secondary antibody control is represented. (E) Epitope binning of mouse recombinant CD73 was generated using ForteBio Octet with streptavidin sensors. Biotinylated mouse recombinant CD73 was added along with a 2:1 ratio of TY-23 and anti-CD73 clones: 2C5; CD73–04; CD73–46; and CD73–69. Overlap was calculated as at least 66% drop of signal from the cross-reacting anti-CD73 antibodies.
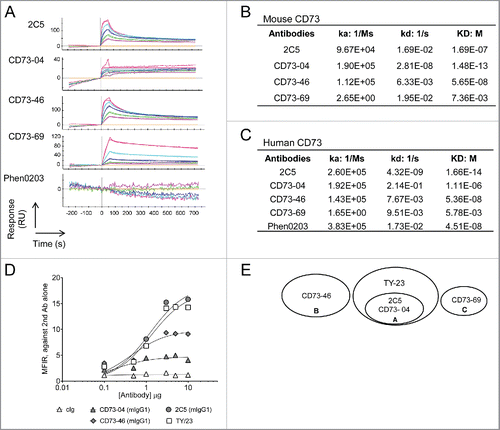
First, we tested the ability of these established clones to bind mouse and human recombinant CD73 protein by Surface Plasmon Resonance (SPR). Clone CD73–04 displayed the strongest binding affinity to recombinant murine CD73 (mouse: Ka−1.90E+05, Kd−2.81E−08)(). Clones CD73–46 (Ka-1.12E+05, Kd−6.33E−03) and 2C5 (Ka−9.67E+04, Kd−1.69E−02) demonstrated similar association constant (Ka) to mouse CD73; however, the dissociation (Kd) for 2C5 was poorer than CD73–46. Clone CD73–69 displayed the lowest affinity to mouse CD73 (Ka−2.65E+00, Kd−1.95E−02) (). In a human CD73 binding assay, binding for 2C5 was superior (human: Ka−2.60E+05, Kd−4.32E−09) compared with the other clones (). CD73–69 showed the least affinity to bind human recombinant CD73 protein ().
Next, we investigated the ability of these clones to bind mouse cancer cells. Mouse 4T1 mammary carcinoma has been previously shown to express CD73.Citation18 The degree to which anti-CD73 clones bound CD73-expressing tumor cells was tested by titrating the concentration of the mAbs and determining the Median Fluorescent Intensity (MFI). The binding affinity for 2C5 (mIgG1) and TY/23 (mIgG1) was the highest, whereas clones CD73–46 (mIgG1) and CD73–04 (mIgG1) showed a moderate level of binding above control Ig (). It was observed that the binding of CD73–69 was abrogated when the Fc domain backbone was changed from human IgG to mouse IgG (Fig. S1). Therefore, in subsequent mouse studies clone CD73–69 was excluded, while mouse and human versions of clones 2C5, CD73–04 and CD73–46 were used to study mouse and human tumor growth, respectively.
Having established the binding affinities for the anti-CD73 clones, we next mapped the epitopes of these clones using the ForteBio Octet high-throughput screening platform. Through this competition assay, we compared various anti-CD73 clones against TY/23 for binding to mouse recombinant CD73. It was observed that CD73 reacting clones belonged to three epitope bins, and only bin A comprising clones CD73–04 and 2C5 overlapped with the known CD73 epitope of antibody, TY/23 (). It was thus predicted that clones CD73–04 and 2C5 might exhibit overlapping functions to the previously characterized mouse anti-CD73 clone, TY/23.
Superior ability of 2C5 to inhibit CD73-enzymatic function and mediate CD73 receptor internalization
Inhibition of CD73 enzymatic function is known to alleviate antitumor functions,Citation6 and therefore we examined the ability of these anti-CD73 mAbs to block CD73-ectonucleotidase activity (). A dose-dependent inhibition was observed for all clones, with the exception of CD73–69 (). This effect was specific to CD73, as evidenced by the absence of inhibition in samples that were incubated with appropriate isotype controls. The IC50 was strongest for clone 2C5 (human IC50−0.1458 nM and mouse IC50−2.483 nM) and this effect was stronger than the previously reported anti-CD73 mAb, Phen0203 (in human studies) () and commonly used anti-CD73 mAb, TY/23 (in mouse) (). CD73–04 and CD73–46 also showed a similar degree of enzyme inhibition activity, albeit several fold reduced compared with 2C5. By contrast, despite eliciting binding to human recombinant CD73, CD73–69 was negligible in its ability to inhibit human CD73 enzymatic function ().
Figure 2. Enzyme activity and internalization ability of anti-CD73 clones. (A) MDA-MB-231 or (B) 4T1 cells (1 × 104 cells each) were incubated with control isotype (cIg) or anti-CD73 mAbs and AMP for (A) 6 h or (B) 8 h, respectively. Enzyme inhibition was measured using Envision after addition of ATP and Cell Titer Glo. IC50 values were determined using GraphPad Prism software. Experiment conducted in (A) included testing of all anti-CD73 mAbs and control isotypes in parallel; however, data are represented across two panels for better clarity of the individual curves. (C) Internalization of anti-CD73 mAbs (hIgG1) on MDA-MB-231 cells was performed by addition of saporin-conjugated antibodies to MDA-MB-231 cells, followed by addition of Cell Titer-Glo. Data are representative of three independent experiments and IC50 values (nM) of internalization on human (MDA-MB-231) and mouse (4T1) mammary tumor cells are shown in the table. (D) B16F10-CD73hi melanoma cells were incubated with Alexa Fluor-647 labeled 2C5 (mIgG1) or 2C5 (mIgG2a) mAbs at 37°C, 5% CO2 across several time-points of 15 min, 30 min or 60 min. Imaging was done using Zeiss AxioScop 2 microscope. Blue = DAPI, Green = Phalloidin and Red/Gray = Alexa Fluor-647 conjugated 2C5 mAb. Magnification = 20×. Scale bar = 20 µm. Images are representative from two independent experiments.
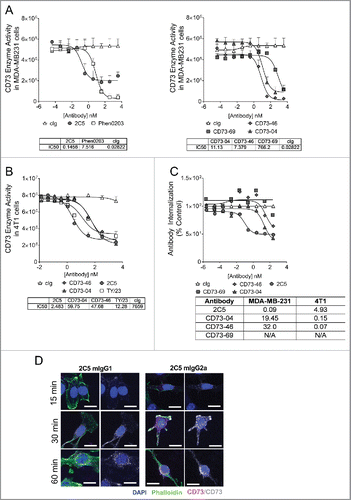
Anti-CD73 mAbs are also capable of inhibiting tumor growth in a manner independent of enzymatic inhibition. This was demonstrated by Terp et al. by use of the human anti-CD73 mAb clone, AD2. AD2 binds to CD73 on tumor cells and induces clustering and internalization of surface CD73. The absence of surface CD73 hampered tumor cell adhesion and invasion. Treatment of mice with AD2 significantly resulted in reduced numbers of MDA-MB-231 metastases.Citation10 Therefore, we tested these newly generated anti-CD73 clones for their ability to kill tumor cells after internalization of the CD73 receptor. 2C5 was observed to better internalize CD73 receptor on MDA-MB231 cells (), than clones CD73–46 and CD73–69. CD73–04 showed moderate receptor internalization on human tumor cells. CD73–69 showed negligible internalization, and was similar to the human control isotype (R347). By contrast, all clones were able to mediate internalization of the CD73 expressed on mouse 4T1 cells (IC50 values for 2C5, CD73–04 and CD73–46 were 4.93 nM, 0.15 nM and 0.07 nM, respectively) ().
Additionally, mouse versions of 2C5 were developed on two different mouse backbones; mIgG1 and mIgG2a to further delineate whether anti-CD73 mAbs required FcR engagement for optimal antitumor activity. We tested these 2C5 isotypes for their ability to internalize upon receptor interaction. Both mouse isotypes of 2C5 were able to undergo CD73 receptor internalization and this occurred as early as 15 min at 37°C ().
Different clones of anti-CD73 mAbs display suppression of solid human and mouse tumors
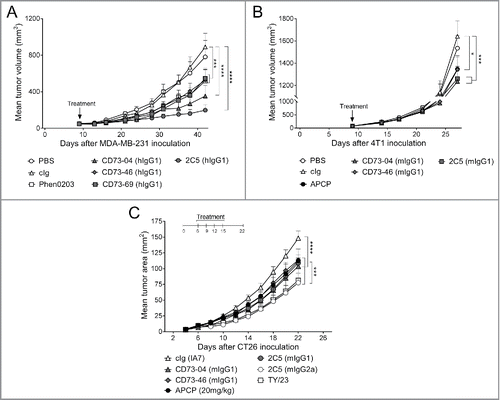
Having established the binding, enzymatic and internalization profile for these anti-CD73 mAbs, we next examined the ability of the anti-CD73 mAbs to suppress solid human and mouse tumors (). The following subcutaneous tumor models were investigated: (i) human MDA-MB-231 xenografts in nude mice, mouse syngeneic (ii) 4T1 mammary carcinoma, and (iii) CT26 colon carcinoma in immunocompetent BALB/c WT mice. All clones were able to significantly suppress the growth of human MDA-MB-231 tumors (), with CD73–04 (hIgG1) and 2C5 (hIgG1) showing the strongest tumor suppression compared with the other anti-CD73 clones (). Despite poor enzymatic inhibition and internalization by CD73–69 (hIgG1), a modest reduction in tumor growth was nonetheless seen. Moreover, this suppression was equivalent to that seen with the human anti-CD73 Ab (Phen0203) (). In the 4T1 tumor model, the effects of clones-CD73–04 (mIgG1) and 2C5 (mIgG1) were similar in suppressing growth of 4T1 tumors. Treatment with CD73–46 (mIgG1) also demonstrated a significant inhibition of 4T1 tumor growth and this effect was similar to the small molecule CD73 inhibitor, APCP (). Similar reduction in CT26 tumor growth was observed in mice receiving CD73–04 (mIgG1), CD73–46 (mIgG1), 2C5 (mIgG1) or APCP (). Importantly, 2C5 (mIgG2a) elicited a stronger suppression of CT26 primary tumor over the other anti-CD73 mAb clones (), suggesting a role for FcR activation in mediating protection to primary tumors.
Figure 3. In vivo efficacy of anti-CD73 clones in the control of primary subcutaneous tumors. (A) Nude mice were treated once by hydrodynamic tail vein injection (HTVI), 9 d after MDA-MB-231 tumor cell implantation when mean tumor volumes was approximately 75 mm3. Treatment consisted of injection of 15 μg of plasmid DNA containing clones CD73–04 (hIgG1), CD73–46 (hIgG1), CD73–69 (hIgG1) and 2C5 (hIgG1) in 100 mL/kg dose volume. (B) BALB/c mice were treated once, by HTVI, 9 d after 4T1 tumor cell implantation when mean tumor volume was approximately 50 mm3. Treatment consisted of injection of 15 μg of plasmid DNA containing clones CD73–04 (mIgG1), CD73–46 (mIgG1) and 2C5 (mIgG1) in 100 mL/kg dose volume. Mice were randomized and sorted into groups based on tumor size and were infused rapidly (∼ seconds) via the tail vein. (C) BALB/c mice were injected subcutaneously with 1 × 105 CT26 colon carcinoma cells. On days 6, 9, 12 and 15 after tumor inoculation, mice were treated with either cIg (IA7, 250 μg i.p.) or anti-CD73 mAbs (CD73–04 (mIgG1), CD73–46 (mIgG1), 2C5 (mIgG1) or 2C5 (mIgG2a), 250 μg each i.p.) or APCP (20 mg/kg, i.p.). Data is shown as mean ± SEM of one experiment with (A–B) 10 mice/group or (C) 5 mice/group. p-values were determined by one-way ANOVA followed by Tukey's multiple comparison test (*p < 0.05; ***p < 0.001, ****p < 0.0001).
Only FcR engaging anti-CD73 mAbs significantly suppress tumor metastases
We next examined metastasis control by anti-CD73 mAbs in the B16F10-CD73hi melanoma model. Previously, we demonstrated that anti-CD73 mAb, TY/23, was able to control B16F10-CD73hi experimental metastasis, and this required CD73 expression on tumors as well as FcγRIV engagement.Citation11 Work by Simpson et al. had also demonstrated that different antibody isotypes can differentially bind FcR and that this influences the nature of antitumor response.Citation19 Given that three of our antibody clones were triple mutated in the Fc region; we investigated how treatment with these clones would impact on metastasis burden (). Particularly, we examined if CD73 enzyme inhibition and internalization capability of these Fc-mutated clones might overcome the loss of FcR engagement in this model. Consistent with previous literature, TY/23 (rat IgG2a) showed significant inhibition of metastasis, and 2C5 (mIgG2a) but not 2C5 (mIgG1) displayed an equivalent capability to reduce B16F10-CD73hi metastatic burden (). By contrast, we observed no apparent single agent activity when mice were treated with Fc-mutated clones, CD73–04 and CD73–46 ( and Fig. S2). These observations were also in concert with our previously published results.Citation11
Figure 4. Suppression of metastasis by anti-CD73 antibodies requires the activation of FcγRIV. (A) C57BL/6 WT mice or (B) C57BL/6 WT, FcϵγR−/− and FcγRIV−/− mice were injected intravenously with 1 × 105 B16F10-CD73hi melanoma cells. On days 0 and 3 after tumor inoculation, mice were treated with either cIg (1A7, 250 μg i.p.) or (A) anti-CD73 clones (TY/23, CD73–04 (mIgG1), CD73–46 (mIgG1), 2C5 (mIgG1), 2C5 (mIgG2a), 250 μg each i.p.) or (B) 2C5 (mIgG2a) (250 μg i.p.). On day 14, lungs were harvested and number of lung metastases was quantified by counting colonies on the lung surface. (C) BALB/c and C57BL/6 WT spleens were harvested from naive mice. Splenocytes were mixed in a 1:1 ratio and plated at 2 × 105 cells/well. These cells were cultured in the presence of cIg (IA7) (triangles) or anti-CD73 clones (TY/23, or 2C5 (mIgG2a)) (squares) at 500 nM and 1,000 nM. After 72 h, supernatants were measured for IFNγ. (D) C57BL/6 WT and perforin gene-targeted mice (B6. pfp−/−) or (E) C57BL/6 WT mice were injected intravenously with 1 × 105 B16F10-CD73hi melanoma cells. On days 0 and 3 after tumor inoculation, mice were treated with (D) cIg (250 μg i.p.) or 2C5 (mIgG2a) (250 μg i.p.) or (E) cIg (250 μg i.p.) or 2C5 (mIgG2a) (250 μg i.p.) or TY/23 (250 μg i.p.). In some experiments (D) IFNγ was neutralized in WT mice by injection with anti-IFNγ (H22, 250 μg i.p.) on days −1, 0, and 7. In (E), mice were depleted of NK cells, CD4+ and CD8+ T cells, or Tregs by treatment with anti-asGM1 (100 μg i.p.), anti-CD4/anti-CD8β (100 μg each i.p.), anti-FR4 (10 μg i.v.), respectively, on days −1, 0 and 7. At day 14, lungs were harvested and metastatic burden was quantified by counting colonies on the lung surface. (A, E) Data represented is pooled from three independent experiments while in (B, D) data are shown as mean ± SEM from one experiment with 5–10 mice/group. One-way ANOVA followed by Tukey's multiple analysis comparison was performed to determine statistical significance in A, B, D and E. In (C) paired t-test was performed. (*p < 0.05; **p < 0.01, ***p < 0.001, ****p < 0.0001).
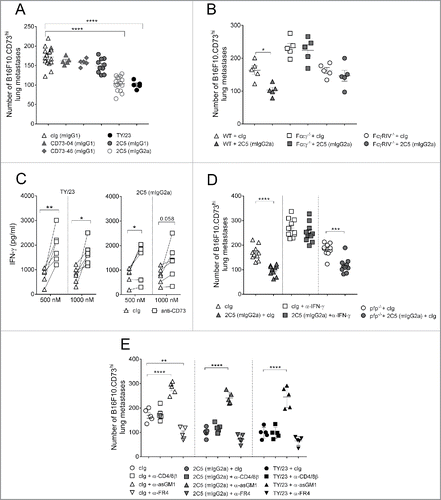
Given this strong anti-metastatic effect by 2C5 (mIgG2a), we next investigated which Fc-receptor cross-links with 2C5 (mIgG2a) to mediate this effect. Treatment of WT mice with 2C5 (mIgG2a) suppressed the number of B16F10-CD73hi lung metastases. By contrast, this therapeutic benefit was lost in the FcϵγR−/− mice (). Similarly, treatment with 2C5 (mIgG2a) did not improve B16F10-CD73hi lung metastases in FcγRIV-deficient mice (), thus implying that like TY/23, 2C5 (mIgG2a) primarily engages FcγRIV for optimal therapeutic activity.
Inhibition of CD73 activity with antagonistic CD73 mAb TY/23 promotes IFNγ secretion within the TME.Citation12 We, therefore, assessed whether blocking with 2C5 (mIgG2a) could similarly induce release of IFNγ. Similar to TY/23, in vitro addition of 2C5 (mIgG2a) to splenocytes significantly increased IFNγ secretion and this increase occurred at 500 nM and 1,000 nM concentrations of the mAb (). Likewise, in vivo, neutralization with anti-IFNγ mAb significantly increased the number of B16F10-CD73hi lung metastases (). Treatment with 2C5 (mIgG2a) showed no effect on lung metastases in these mice, implying that 2C5 (mIgG2a) required IFNγ in conferring protection from metastasis. By contrast, 2C5 mIgG2a was able to suppress lung metastases in perforin-deficient mice ().
NK cells are important in the control of tumor metastasis.Citation20 Therefore, we tested whether the therapeutic effect of 2C5 mIgG2a required NK cells. Consistent with previous studies, we observed an increase in the number of B16F10-CD73hi lung metastases when mice were depleted of NK cells (). Treating NK cell-depleted mice with 2C5 (mIgG2a) or TY/23 did not rescue this high level of metastatic burden (); signifying that efficacy of 2C5 (mIgG2a) was dependent on NK cells. Interestingly, depletion of regulatory T cells (Tregs) using anti-FR4 antibody reduced B16F10-CD73hi metastatic burden (), but the treatment with 2C5 (mIgG2a) or TY/23 did not further improve protection from lung metastasis (). Collectively, these observations indicated that activation of FcγRIV by 2C5 (mIgG2a) was essential for suppressing B16F10-CD73hi lung metastases and this effect was mediated by IFNγ and NK cells.
Co-treatment with anti-CD73 mAb and A2AR inhibitor enhanced suppression of experimental and spontaneous tumor metastases
We next tested whether Fc-engagement was essential for anti-CD73 mAb activity in a combination therapy (). Co-blockade using A2AR inhibitor (A2ARi) SCH58261 and 2C5 (mIgG2a), significantly reduced the number of B16F10-CD73hi lung metastases, compared with either monotherapy alone (). CD73–04 and CD73–46 did not exhibit any single agent activity in suppressing B16F10-CD73hi lung metastases, however, a minor, yet significant inhibition of B16F10-CD73hi lung metastases was observed when CD73–04 or CD73–46 clones were co-treated with SCH58261 (). We next tested whether this benefit only occurs within the adenosinergic pathway or could be extended to other immune checkpoint blockade. Hence, the combination effect of anti-CD73 mAbs with anti-PD-1 mAb was examined. TY/23 and 2C5 (mIgG2a) both showed an additive effect with anti-PD-1 mAb, however, no combinatorial effect was obtained with the Fc-mutated anti-CD73 clones (Fig. S3).
Figure 5. Co-blockade of A2AR and CD73 improves anti-metastatic activity in mice. (A) C57BL/6 WT mice were injected intravenously with 1 × 105 B16F10-CD73hi melanoma cells. On days 0 and 3 after tumor inoculation, mice were treated with either cIg (1A7, 250 μg i.p.) or anti-CD73 clones (CD73–04 (mIgG1), CD73–46 (mIgG1) or 2C5 (mIgG2a), 250 μg/mouse each i.p) or vehicle (DMSO) or A2AR inhibitor (SCH58261, 1 mg/kg i.p). Data is shown as mean ± SEM of 5 mice/group and p-values for panels A–C were determined by one-way ANOVA followed by Tukey's multiple comparison test. (B–D) BALB/c WT mice or (C) BALB/c perforin gene-targeted mice (BALB/c. pfp−/−) were injected in the fourth mammary fat pad with 5 × 104 4T1.2 cells. On day 17, the primary tumor was resected and mice were treated intraperitoneally with either vehicle (DMSO) or A2AR inhibitor (SCH58261, 1 mg/kg i.p.) and cIg (1A7, 250 μg i.p.) or 2C5 (mIgG2a) (250 μg i.p.) on days 18, 21, 24 and 27. In some experiments, (C) WT mice were neutralized for IFNγ by treatment with anti-IFNγ mAb (H22, 250 μg i.p.) or cIg (250 μg i.p.). (D) Depletion of NK cells, CD4+ T cells and/or CD8+ T cells were done by treating mice with anti-asGM1 (100 μg i.p.), anti-CD4 or anti-CD8β (100 μg i.p.) on days 17, 18, 25 and 32. The survival of the mice was monitored and survival analysis performed using Log-rank test. Experiment was conducted once with 5–10 mice/group. (*p < 0.05; **p < 0.01, ***p < 0.001, ****p < 0.0001).
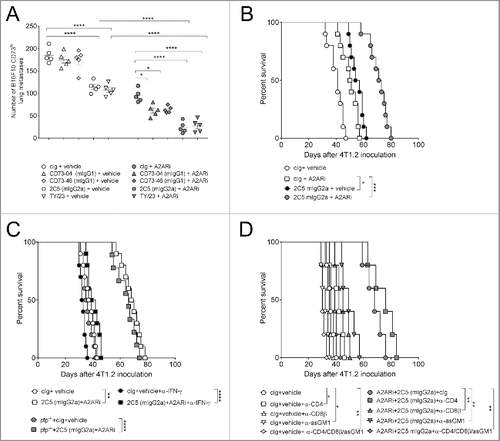
We also studied the anti-metastatic effect of anti-CD73 mAb, 2C5 (mIgG2a) in the 4T1.2 spontaneous metastasis model and identified similar findings to those observed in the B16F10-CD73hi model. Anti-CD73 2C5 (mIgG2a) treatment prolonged survival in mice compared with control IgG and survival was even greater in mice receiving A2ARi and 2C5 (mIgG2a) combination therapy (). This survival benefit with the combination therapy was lost when mice were treated with a neutralizing anti-IFNγ mAb, but not in mice lacking perforin (). Similar to the experimental metastases model, control of 4T1.2 metastases required host NK cells. Interestingly, when combination treated mice were collectively depleted of CD4+ T cells, CD8+ T cells and NK cells; a significant reduction in survival was also observed (). Thus, the combinatorial therapy requires the presence of lymphocytes for effective anti-metastatic immunity in the spontaneous metastasis model.
Discussion
We generated anti-CD73 mAbs, cross reactive with mouse and human CD73 that have varying levels of binding, enzymatic, internalization and FcR cross-linking activity. Our results indicated that anti-CD73 mAbs, clones CD73–04, CD73–46, CD73–69 and 2C5, were capable of binding, internalizing and inhibiting ecto-nucleotidase activity to various degrees. The inhibition of CD73 enzymatic functions and the ability to activate FcR were together essential for effective suppression of primary tumors in mice. Hence, despite loss of Fc function in anti-CD73 mAbs, CD73–04, CD73–46 and CD73–69, these clones were able to, in part, control primary mouse and human tumor growth. By contrast, the optimal suppression of lung metastasis by anti-CD73 mAb was largely reliant on the ability to engage FcR, in particular, FcγRIV. Our data indicated that inhibition of human and mouse solid tumors might thus involve different anti-CD73 mAb mechanisms of action to that of metastases. It is likely that the TME, and in particular the presence of FcγRIV expressing myeloid cells, might be critical in determining how important FcR engagement is to the mechanism. Our data supported the recent description by Young et al. that anti-CD73 mAb promoted anti-metastatic immunity via cross-linking of FcγRIV.Citation11 Additionally, we showed that engagement of Fc-receptors by anti-CD73 mAb was essential for optimal suppression of tumor metastasis in combination with either A2AR inhibitor or anti-PD-1 mAb treatment. Experimental metastases were controlled by host NK cells and IFNγ, while spontaneous metastases were controlled by host NK cells, CD8+ T cells and IFNγ.
Anti-CD73 mAb can mediate antitumor immunity by several mechanisms. These include either inhibition of catalytic activity,Citation6 suppression of cancer cell adhesion, migration and angiogenesis,Citation10,21,22 or by activation of Fc-receptors, mainly FcγRIV.Citation11 CD73–69 (hIgG1) was able to weakly bind the human recombinant CD73, but displayed negligible ATP catalysis and receptor internalization. Despite loss in activity, binding of CD73–69 to CD73 expressing MDA-MB-231 tumors was sufficient to mediate significant suppression of this tumor in mice. Therefore, these findings support a non-enzymatic role for anti-CD73 mAbs in the control of human xenograft MDA-MB-231 tumors. Future experiments are needed to determine how this interaction with CD73 receptor could impact on the growth of MDA-MB-231 tumors. In a previous study by Zhi et al., siRNA knockdown of CD73 expression on MDA-MB-231 cells hampered tumor cell migration, invasion and the ability to adhere to the extracellular matrix, which altogether resulted in poor growth of MDA-MB-231 tumors in vivo.Citation23 Furthermore, CD73 is known to bind to the extracellular matrix molecule, Tenascin-C and this interaction promotes tumor cell adhesion.Citation21 It would be useful to test whether anti-CD73 mAbs abrogate this interaction, thus influencing the growth of MDA-MB-231 tumors. Importantly, clones 2C5 and CD73–04 demonstrated stronger suppression of MDA-MB-231 tumors over the other anti-CD73 mAbs. As both these clones belonged to the same epitope bins, it is likely that the optimal antitumor activity by anti-CD73 mAbs may be epitope specific.
In the 4T1 and CT26 tumor models, treatment with all anti-CD73 mAbs (2C5, CD73–04 and CD73–46) was able to suppress subcutaneous tumor growth. These clones demonstrated superior enzymatic inhibition, and therefore we speculate that tumor control in this model might be mediated partly via enzymatic inhibition. Indeed, the small molecule specific inhibitor of CD73, APCP also inhibited 4T1 and CT26 tumor growth. Importantly, CT26 tumor growth was further suppressed in mice receiving 2C5 (mIgG2a) than those that received 2C5 (mIgG1) or Fc-mutated anti-CD73 mAbs, CD73–04 and CD73–46. These observations signify that the control of primary tumors by anti-CD73 mAbs require both, enzymatic inhibition as well as activating Fc-receptors. Overall, data from human and mouse tumor experiments indicate that anti-CD73 mAbs utilize different mechanisms for suppression of different solid tumors, and this may be due to the species differences or the absence of many lymphocyte subsets in nude mice.
By contrast to solid tumors, anti-CD73 mAbs required FcR cross-linking for optimal anti-metastatic activity. Previously, mouse IgG2a has been shown to bind the activating FcγRIV, and interaction was crucial in controlling the development of several tumors.Citation24 Indeed, we observed significant anti-metastatic effect with 2C5 (mIgG2a), but not 2C5 (mIgG1), and this difference required FcγRIV. The mouse anti-CD73 mAb, TY/23, was also shown to bind FcγRIV primarily on CD11b+Gr-1+ neutrophils and this engagement resulted in the expansion of these myeloid populations.Citation11 Mouse NK cells do not express FcγRIV. Although the role of neutrophils in cancer is controversial, and appears largely dependent upon the tumor niche,Citation25 neutrophils can kill tumor cells via antibody-dependent cell cytotoxicity (ADCC).Citation26 These and/or other FcγRIV+ myeloid cells could therefore represent a potential mechanism underlying the antitumor activity of 2C5 (mIgG2a) function, in addition to its requirement for IFNγ and NK cells in controlling metastasis.
Despite the lack of single agent activity by Fc-mutated clones, their combination with another adenosinergic molecule, A2ARi demonstrated significant anti-metastatic effect. We previously showed that subcutaneously transplanted SM1WT1 melanoma growth in A2AR gene-targeted mice, significantly increased levels of CD73 expression in the TME.Citation11 It is thus predicted that blocking with A2ARi may similarly induce heightened levels of CD73 in the TME. This CD73 then becomes a target for binding of anti-CD73 mAbs that thereby facilitates anti-metastatic effects through processes such as receptor binding, internalization and enzymatic inhibition. This might also explain why co-blocking with Fc-mutated anti-CD73 mAb and anti-PD-1 mAb failed to demonstrate any additional combination effect against metastases.
Collectively, we have demonstrated that anti-CD73 mAb engage several mechanisms for controlling tumor growth and metastasis, and understanding which mechanisms dominate during the development of primary tumors and metastasis is vital. Our data shows that Fc-receptor activation by anti-CD73 mAbs is extremely important for suppression of experimental and spontaneous lung metastases. By contrast, control of solid tumors by anti-CD73 mAbs was also dependent on mechanisms involving binding to CD73 receptor and/or mediating enzymatic inhibition. Further studies are needed to dissect the intracellular signaling pathways involved in the control of primary tumors and metastases. Deciphering these pathways will prove extremely valuable in the design of better anti-CD73 mAbs for the treatment of human cancers.
Materials and methods
Mice
Wild-type (WT) C57BL/6 and BALB/c mice were purchased from Walter and Eliza Hall Institute for Medical Research. Gene-targeted mice for activating FcR (FcϵR−/− and FcγRIV−/− mice) and perforin (pfp−/−) were bred in-house and maintained at the QIMR Berghofer Medical Research Institute. Experiments were conducted as per approved by the QIMR Berghofer Medical Research Institute Animal Ethics Committee. Female Nude mice used in , and female BALB/c mice used in were purchased from Harlan Sprague Dawley (HSD) and experiments were performed at Medimmune in accordance with federal, state and Institutional guidelines in an AAALAC-accredited facility and were approved by the MedImmune Institutional Animal Care and Use Committee.
Cell culture
Mouse B16F10.CD73hi melanoma cells were grown in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% Fetal Calf Serum (Bovogen), 1% Glutamine (Gibco), 1% HEPES (Gibco) and 1% Penicillin/Streptomycin (Gibco). 4T1, 4T1.2 mammary carcinoma cells and CT26 colon carcinoma cell lines were cultured in RPMI 1640, supplemented with 10% Fetal Calf Serum (Bovogen) 1% Glutamine (Gibco) and 1% Penicillin-Streptomycin (Gibco). All cell lines were maintained at 37°C, 5%CO2. Tumor cells were routinely tested for mycoplasma contamination, and mycoplasma-free cell lines were used in the experiments.
Human cells
Human MDA-MB-231 breast adenocarcinoma cells obtained from American Type Culture Collection (ATCC), and grown in Leibovitz L-15 media (Gibco) supplemented with 10% Fetal Bovine Serum (Gibco). The cell line was maintained at 37°C, 5%CO2.
Development of anti-CD73 clones
Anti-CD73 mAb clones were developed from a phage display library as described previously.Citation27 Each anti-CD73 mAb clone was developed in two different backbones, hIgG1 for studies in human MDA-MB-231 cells and mIgG1 backbone for investigations in mouse cancer cell lines. Additionally, clone 2C5 was developed in two mouse isotype backbones, mIgG1 and mIgG2a.
CD73-binding experiments
Binding of anti-CD73 clones to recombinant mouse and human CD73 was measured by SPR using ProteOn Instrument (BioRad) as described previously.Citation13 Data was auto-processed using the ProteOn injection alignment, baseline alignment, and artifact removal and bivalent analyte model fitting.
To test the binding of anti-CD73 mAb clones to mouse tumors, 4T1 cells were dissociated with Accutase (STEMCELL Technologies) and washed thrice with FACS buffer (containing PBS and 4% FBS). Cells were centrifuged and the cell pellet was resuspended in FACS buffer and plated in a 96-well U bottom-plate at a final concentration of 1 × 105 cells/well. Antibodies were serially diluted in FACS buffer and incubated for 1 h on ice. Thereafter, cells were centrifuged for 3 min at 350 g and washed thrice in FACS buffer. This was followed by addition of a goat anti-human Alexa Fluor-488 secondary antibody (Life Technologies) and incubated on ice for 1 h. Cells were washed with FACS buffer and 5 μg/mL of Propidium Iodide (Roche) was used for live/dead staining. Flow cytometry was conducted on an LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo software (Treestar)
Epitope binning assay
Data was generated using a ForteBio Octet with streptavidin sensors: biotinylated mouse recombinant CD73 followed by TY-23 and a 2:1 mixture of TY-23 and indicated antibody. Overlap is calculated as at least 66% drop of signal from the indicated antibody.
Ectonucleotidase activity
4T1 and MDA-MB-231 cells (1 × 104) were plated in serum-free RPMI medium (Invitrogen). Serial concentrations of either mIgG1 (for 4T1) or hIgG1 (for MDA-MB-231) anti-CD73 mAbs or control isotypes (cIg) (IA7 for mouse studies and R347 for human experiments) were added for 10 min. Cells were washed and 200 µM of AMP (Sigma) was added for 8 h (mouse assay) or 6 h (human assay). Plates were centrifuged and cell supernatant was transferred to a white 96-well plate (Costar). This was then followed by addition of 100 µM of ATP (Sigma) and Cell Titer Glo (Promega) at 1:1. CD73 enzyme inhibition was measured using Envision (Perkin Elmer).
Internalization experiments
For internalization experiments using MDA-MD-231 and 4T1 tumor cells, anti-CD73 mAbs (either hIgG1 or mIgG1 backbone, respectively) or cIg were directly conjugated to Saporin. MBA-MD-231 cells and 4T1 cells were plated at a density of 5,000 cells per well into 96-well plates and incubated overnight at 37°C, 5% CO2. Saporin-conjugated antibodies were added to the cells for 72 h, followed by addition of 100 µL of Cell Titer-Glo® (Promega). Internalization was measured on a luminometer (Perkin Elmer).
Internalization assays using 2C5 (mIgG1) and 2C5 (mIgG2a) were done by plating B16F10-CD73hi melanoma cells on matrigel-coated coverslips at a density of 2.5 × 104 cells in a six-well plate. The following day, either Alexa Fluor-647 labeled (Thermo Fisher Scientific) 2C5 (mIgG1) or 2C5 (mIgG2a) antibodies were added and incubated at 37°C, 5% CO2. Internalization was examined at 15 min, 30 min and 60 min. Post-incubation with antibodies; cells were fixed and stained with a nuclear (DAPI) and cytoskeletal (phalloidin) dye. To determine background fluorescence, Alexa-Fluor 647 labeled 2C5 (mIgG2a) and 2C5 (mIgG1) antibodies were incubated with B16F10-CD73hi cells at 4°C for 15 min, 30 min and 1 h.
Allogeneic mixed leukocyte reaction (MLR)
Splenocytes from naïve WT C57BL/6 and BALB/c were harvested, RBC lysed and single-cell suspensions prepared. The cells from the two strains were then mixed in 1:1 ratio and added to a 96-well U Bottom plate at 2 × 105 cells/well. After 72 h, supernatants were harvested and tested for cytokine release using mouse TH1/TH2 CBA kit (BD Biosciences) as per manufacturer's instructions.
Subcutaneous primary tumor experiments
Nude mice or BALB/c mice were treated once by hydrodynamic tail vein injection (HTVI), 9 d after MDA-MB-231 or 4T1 subcutaneous tumor cell implantation, respectively. Treatment consisted of injection of 15 μg of plasmid DNA containing the indicated antibody clones in a dosing volume (adjusted for body weight) of 100 mL/kg. Mice were randomized and sorted into groups based on tumor sizes. Mice were infused rapidly (∼15 sec) via the tail vein.
In CT26 tumor growth experiments, BALB/c mice were injected with 1 × 105 CT26 colon carcinoma cells. Mice were randomized before intraperitoneal (i.p.) treatment with either cIg (IA7, 250 μg) or anti-CD73 mAb clones (250 μg, each) on days 6, 9, 12 and 15 after tumor inoculation.
Experimental tumor metastasis
B16F10-CD73hi melanoma cells (1 × 105) were injected intravenously into the tail vein of C57BL/6 mice. On days, 0 or 3 after tumor inoculation, mice were treated i.p. with cIg (1A7, 250 µg), or anti-CD73 clones (250 µg). In some experiments as indicated in the figure legends, mice were treated i.p. with cIg (1A7, 600 µg), or anti-CD73 clones (600 µg). Depletion of NK cells, CD4+ T cells and/or CD8+ T cells or IFNγ, were done by i.p. treatment on days −1, 0 and 7 with anti-asGM1 (100 µg), anti-CD4 (GK1.5, 100 μg), anti-CD8β (53.5.8, 100 μg) or anti-IFNγ antibody (H22, 250 µg). For depletion of Tregs, anti-FR4 antibody (10 μg) was injected intravenously (i.v.). An appropriate isotype control was also used in these experiments. Lungs were harvested on day 14 and tumor nodules were counted under a dissection microscope.
Spontaneous tumor metastasis
4T1.2 mammary carcinoma cells (5 × 104) were injected into the fourth mammary fat-pad of BALB/c WT mice. On day 17, the primary tumor was surgically removed and wound closed with surgical clips before commencement of treatment. Mice were given vehicle (DMSO) or SCH58261 (1 mg/kg) or cIg (1A7, 250 µg) or anti-CD73 (250 µg) i.p on days 18, 21, 24 and 27. The mice were monitored for survival. Some mice additionally received anti-CD4 (GK1.5, 100 µg), anti-CD8β (53.5.8, 100 µg) or anti-asGM1 (100 µg) or anti-IFNγ antibody (H22, 250 µg) as indicated.
Statistical analysis
Statistical analyses were determined using GraphPad Prism software. For MLR, paired t-tests were performed. In survival experiments, significance was calculated by Log-Rank test. For all other studies, statistical significance was determined by one-way ANOVA followed by Tukey post hoc analysis test to compare across groups. p-value less than or equal to 0.05 were considered significant.
Disclosure of potential conflicts of interest
M.J.S. declares scientific research agreements with Bristol Myers Squibb, Corvus Pharmaceuticals and Aduro Biotech. R.B., E. S., and C.H. are employees of Medimmune, while K.S. is an employee of Astrazeneca. All other authors declare no conflict of interest.
Supplementary_Figures1-3.pdf
Download PDF (995.5 KB)Acknowledgments
We thank Kate Elder and Liam Town for breeding, genotype and maintaining mice during this study. We also thank Arabella Young for helpful discussions and Qihui Huang for technical assistance.
Funding
M.J.S. was supported by a National Health and Medical Research Council of Australia (NHMRC) Senior Research Fellowship (1078671), a NHMRC Project Grant (1120887), and a Research Agreement from Medimmune. M.W.L.T was supported by a NHMRC Project Grant (1120887).
Related Research Data
References
- Ohta A. A Metabolic immune checkpoint: Adenosine in tumor microenvironment. Front Immunol 2016; 7:109; PMID:27066002; https://doi.org/10.3389/fimmu.2016.00109
- Young A, Mittal D, Stagg J, Smyth MJ. Targeting cancer-derived adenosine: New therapeutic approaches. Cancer Discov 2014; 4:879-88; PMID:25035124; https://doi.org/10.1158/2159-8290.CD-14-0341
- Allard B, Beavis PA, Darcy PK, Stagg J. Immunosuppressive activities of adenosine in cancer. Curr Opin Pharmacol 2016; 29:7-16; PMID:27209048; https://doi.org/10.1016/j.coph.2016.04.001
- Allard D, Allard B, Gaudreau PO, Chrobak P, Stagg J. CD73-adenosine: A next-generation target in immuno-oncology. Immunotherapy 2016; 8:145-63; PMID:26808918; https://doi.org/10.2217/imt.15.106
- Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK, Smyth MJ. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res 2011; 71:2892-900; PMID:21292811; https://doi.org/10.1158/0008-5472.CAN-10-4246
- Wang L, Fan J, Thompson LF, Zhang Y, Shin T, Curiel TJ, Zhang B. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Invest 2011; 121:2371-82; PMID:21537079; https://doi.org/10.1172/JCI45559
- Stagg J, Beavis PA, Divisekera U, Liu MC, Moller A, Darcy PK, Smyth MJ. CD73-deficient mice are resistant to carcinogenesis. Cancer Res 2012; 72:2190-6; PMID:22396496; https://doi.org/10.1158/0008-5472.CAN-12-0420
- Yegutkin GG, Marttila-Ichihara F, Karikoski M, Niemela J, Laurila JP, Elima K, Jalkanen S, Salmi M. Altered purinergic signaling in CD73-deficient mice inhibits tumor progression. Eur J Immunol 2011; 41:1231-41; PMID:21469131; https://doi.org/10.1002/eji.201041292
- Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, Dwyer KM, Smyth MJ. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. U. S. A. 2010; 107:1547-52; PMID:20080644; https://doi.org/10.1073/pnas.0908801107
- Terp MG, Olesen KA, Arnspang EC, Lund RR, Lagerholm BC, Ditzel HJ, Leth-Larsen R. Anti-human CD73 monoclonal antibody inhibits metastasis formation in human breast cancer by inducing clustering and internalization of CD73 expressed on the surface of cancer cells. J Immunol 2013; 191:4165-73; PMID:24043904; https://doi.org/10.4049/jimmunol.1301274
- Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, Huang Q, Liu J, Takeda K, Teng MW et al. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell 2016; 30:391-403; PMID:27622332; https://doi.org/10.1016/j.ccell.2016.06.025
- Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin. Cancer. Res. 2013; 19:5626-35; PMID:23983257; https://doi.org/10.1158/1078-0432.CCR-13-0545
- Geoghegan JC, Diedrich G, Lu X, Rosenthal K, Sachsenmeier KF, Wu H, Dall'Acqua WF, Damschroder MM. Inhibition of CD73 AMP hydrolysis by a therapeutic antibody with a dual, non-competitive mechanism of action. MAbs 2016; 8:454-67; PMID:26854859; https://doi.org/10.1080/19420862.2016.1143182
- Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, Hammond SA, Rothstein R, Rios-Doria J, Poon E et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology 2016; 5:e1208875; PMID:27622077; https://doi.org/10.1080/2162402X.2016.1208875
- Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med 2013; 210:1695-710; PMID:23897981; https://doi.org/10.1084/jem.20130579
- Li F, Ravetch JV. Inhibitory Fcgamma receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science 2011; 333:1030-4; PMID:21852502; https://doi.org/10.1126/science.1206954
- Arnould L, Gelly M, Penault-Llorca F, Benoit L, Bonnetain F, Migeon C, Cabaret V, Fermeaux V, Bertheau P, Garnier J et al. Trastuzumab-based treatment of HER2-positive breast cancer: An antibody-dependent cellular cytotoxicity mechanism? Br J Cancer 2006; 94:259-67; PMID:16404427; https://doi.org/10.1038/sj.bjc.6602930
- Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, Dwyer K, Stagg J, Smyth MJ, Darcy PK. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc. Natl. Acad. Sci. USA 2013; 110:14711-6; PMID:23964122; https://doi.org/10.1073/pnas.1308209110
- Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 2008; 8:34-47; PMID:18064051; https://doi.org/10.1038/nri2206
- Krasnova Y, Putz EM, Smyth MJ, Souza-Fonseca-Guimaraes F. Bench to bedside: NK cells and control of metastasis. Clin Immunol 2015; Oct 22. pii: S1521-6616(15)30050-4; PMID:26476139; https://doi.org/10.1016/j.clim.2015.10.001
- Sadej R, Skladanowski AC. Dual, enzymatic and non-enzymatic, function of ecto-5′-nucleotidase (eN, CD73) in migration and invasion of A375 melanoma cells. Acta Biochim Pol 2012; 59:647-52; PMID:23162807
- Airas L, Hellman J, Salmi M, Bono P, Puurunen T, Smith DJ, Jalkanen S. CD73 is involved in lymphocyte binding to the endothelium: Characterization of lymphocyte-vascular adhesion protein 2 identifies it as CD73. J Exp Med 1995; 182:1603-8; PMID:7595232; https://doi.org/10.1084/jem.182.5.1603
- Zhi X, Chen S, Zhou P, Shao Z, Wang L, Ou Z, Yin L. RNA interference of ecto-5′-nucleotidase (CD73) inhibits human breast cancer cell growth and invasion. Clin Exp Metastasis 2007; 24:439-48; PMID:17587186; https://doi.org/10.1007/s10585-007-9081-y
- Nimmerjahn F, Lux A, Albert H, Woigk M, Lehmann C, Dudziak D, Smith P, Ravetch JV. FcgammaRIV deletion reveals its central role for IgG2a and IgG2b activity in vivo. Proc. Natl. Acad. Sci. USA 2010; 107:19396-401; PMID:20974962; https://doi.org/10.1073/pnas.1014515107
- Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: Neutral no more. Nat Rev Cancer 2016; 16:431-46; PMID:27282249; https://doi.org/10.1038/nrc.2016.52
- van Egmond M, Bakema JE. Neutrophils as effector cells for antibody-based immunotherapy of cancer. Semin Cancer Biol 2013; 23:190-9; PMID:23287459; https://doi.org/10.1016/j.semcancer.2012.12.002
- Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, Johnson KS. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol 1996; 14:309-14; PMID:9630891; https://doi.org/10.1038/nbt0396-309