
ABSTRACT
Background: Chondromodulin-I (CHM1) sustains malignancy in Ewing sarcoma (ES). Refractory ES carries a dismal prognosis and patients with bone marrow (BM) metastases do not survive irrespective of therapy. We assessed HLA-A*02:01/CHM1-specific allorestricted T cell receptor (TCR) wild-type and transgenic cytotoxic (CD8+) T cells against ES.
Patients and Methods: Three refractory HLA-A2+ ES patients were treated with HLA-A*02:01/peptide-specific allorepertoire-derived (i.e., allorestricted) CD8+ T cells. Patient #1 received up to 4.8 × 105/kg body weight HLA-A*02:01− allorestricted donor-derived wild-type CD8+ T cells. Patient #2 received up to 8.2 × 106/kg HLA-A*02:01− donor-derived and patient #3 up to 6 × 106/kg autologous allorestricted TCR transgenic CD8+ T cells. All patients were treated with the same TCR complementary determining region 3 allorecognition sequence for CHM1 peptide 319 (CHM1319).
Results: HLA-A*02:01/CHM1319-specific allorestricted CD8+ T cells showed specific in vitro lysis of all patient-derived ES cell lines. Therapy was well tolerated and did not cause graft versus host disease (GvHD). Patients #1 and #3 showed slow progression, whereas patient #2, while having BM involvement, showed partial metastatic regression associated with T cell homing to involved lesions. CHM1319 TCR transgenic T cells could be tracked in his BM for weeks.
Conclusions: CHM1319-TCR transgenic T cells home to affected BM and may cause partial disease regression. HLA-A*02:01/antigen-specific allorestricted T cells proliferate in vivo without causing GvHD.
Introduction
Allorepertoire-derived T cells in cancer immunotherapy are thought to be associated with an increased risk of cross-reactivity as compared with conventional T cells, which in most cases do not eliminate tumors with low mutational burdens. Another obstacle of cancer immunotherapy is the loss of target antigen.Citation1 Thus, efficacious immunotherapy has to address targets presumed to be essential for cancer cell survival. Chondromodulin-I (CHM1) is a direct target of EWS-FLI1 in Ewing sarcoma (ES) and promotes metastatic spread.Citation2 EWS-FLI1 and other EWS-ETS fusion proteins are pathognominic of ES.Citation3 ES patients metastatic to bone marrow (BM) do not survive irrespective of therapy.Citation4 Survival of refractory patients is 6 to 7 mo.Citation5 So far, T cell checkpoint inhibitors have not been successful in tumors with low mutational burden, particularly in childhood malignancies.Citation6-12
Allorecognition is an evolutionary conserved mechanism, highly efficacious in defense against pathogens, which can be exploited in cancer immunotherapy,Citation13 e.g. by major histocompatibility complex (MHC) disparate stem cell transplantation plus treatment with donor-derived T cells.Citation14,15 Allorepertoire-derived T cells were used for the identification of T cell receptors (TCR) recognizing ES-derived peptides in the context of (human leukocyte antigen) HLA-A*02:01; these cells are termed allorestricted peptide-specific T cells. We have described previously tumor control without side effects with allorestricted T cells directed against ES-associated peptides CHM1319 and EZH2666.Citation14,16 Here, we report on the first clinical use of both HLA-A*02:01/CHM1319 TCR transgenic and HLA-A*02:01/CHM1319-peptide directed allorestricted wild-type CD8+ T cells in patients with refractory ES.
Results
Expression of target genes
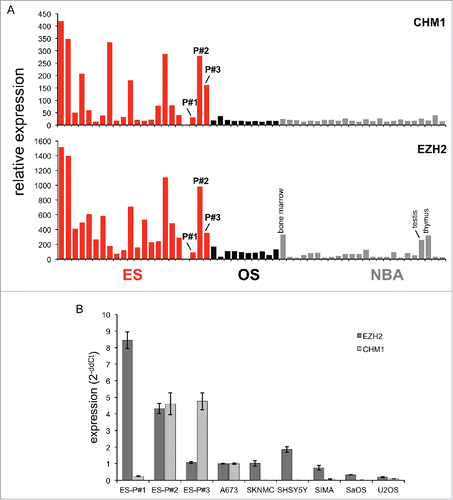
In previous work, we observed overexpression of CHM1 and EZH2 in ES.Citation18 In samples obtained from refractory tumor before adoptive transfer, CHM1 was overexpressed compared to healthy body tissue in patients #2 and #3, and more moderately expressed in patient #1 (). EZH2 was overexpressed in the same samples of patients #1 and #2 and more moderately expressed in patient #3. Furthermore, real-time PCR confirmed strong target upregulation of CHM1 in patient-derived ES cell lines in comparison to controls (). All cell lines remained HLA-A*02:01 positive (Fig. S2).
Figure 1. CHM1 and EZH2 relative gene expression in ES primary tumors and cell lines vs. controls. (A) Expression profile of CHM1 and EZH2 in ES primary samples, patient's-derived HLA-A*02:01+ cell lines ES-P#1, ES-P#2, ES-P#3, osteosarcoma primary samples, and normal body atlas (NBA) analyzed by microarray (GSE45544 and GSE73166). (B) Real-time RT-PCR confirms upregulation of CHM1 in patient-derived HLA-A*02:01+ cell lines ES-P#2, ES-P#3 in comparison to controls. EZH2 is upregulated in ES-P#1, ES-P#2, ES-P#3 in comparison to controls. A673, HLA-A*02:01+ ES cell line; SKNMC, SHSY5Y, and SIMA, neuroblastoma cell lines; SaOS, U2OS, osteosarcoma cell lines were used as controls. Analyses were performed as duplicates. Results were normalized to GAPDH and quantified by the ddCt-method.
Phenotype of transferred T cells
Prior to adoptive T cell transfer in patient #1, more than 88% of transferred CHM1319 multimer positive CD8+ T cells displayed an effector phenotype (CD27dim/CD28−/IL7Ra−/CD62L−/CD45RO+), while 12% displayed a central memory phenotype (CD62L+/CD45RO+). In patient #2, 66% of CD8+ TCR-transgenic T cells displayed the central memory and 34% a naïve phenotype (CD62L+/CD45RA+). In patient #3, 2.5% of CHM1319 multimer positive CD8+ TCR-transgenic T cells displayed a T stem cell memory phenotype (CD62L+/CD45RO+/CD45RA+/CCR7+/CD95+) and 67% displayed a T central memory phenotype (CD62L+/CD45RO+/CD45RA+/CCR7−/CD95+). Also, 30.5% showed an effector phenotype. Differences in phenotypes were most likely due to varying expansion protocols.Citation14,17,19 T cells of patients #1, #2, and #3 were 29.2%, 99.7%, and 45.1% positive for the CHM1319-specific multimer, respectively, and 63.5% positive for the EZH2666 multimer in patient #1 (Fig. S3).
Specificity of transferred T cells
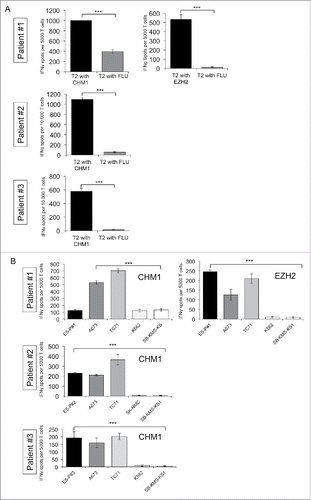
Wild-type HLA-A*02:01/CHM1319 and EZH2666 as well as HLA-A*02:01/CHM1319 TCR transgenic T cells were tested for in vitro functionality. We previously demonstrated processing and transportation of the predicted CHM1319 nonamer to the surface of target cells.Citation19 Wild-type HLA-A*02:01/EZH2666 as well as HLA-A*02:01/CHM1319 TCR transgenic T cells and to a lesser degree wild-type HLA-A*02:01/CHM1319 are specific against respective peptide loaded T2 cells (). HLA-A*02:01/CHM1319 TCR transgenic T cells are specific against HLA-A*02+/CHM1+ ES cell lines (including patient-derived cell lines ES-P#1, ES-P#2 and ES-P#3) in IFNγ ELISpot assays ().
Figure 2. Donor-derived HLA-A*02:01/CHM1319 TCR transgenic T cells specifically recognize HLA-A*02+/CHM1+ cell lines in vitro. (A) CHM1319-TCR-transgenic T cells specifically recognize CHM1319-loaded T2 cells and (B) HLA-A*02:01+/CHM1+ Ewing sarcoma cell lines A673, TC-71, and the patient-derived cell line SB-KMS-GH in contrast to HLA-A*02:01−/CHM1+ SK-NMC, K562, and SB-KMS-KS1 control cell lines in IFNγ ELISpot assays. The E/T ratio for ELISpot assay was 1:4. Error bars represent standard deviation of triplicate experiments. Asterisks indicate significance levels.
In vivo persistence of transferred T cells
In patient #1 no transferred T cells were retrieved from either peripheral blood, BM, lymph-node, or from tumor samples (data not shown).
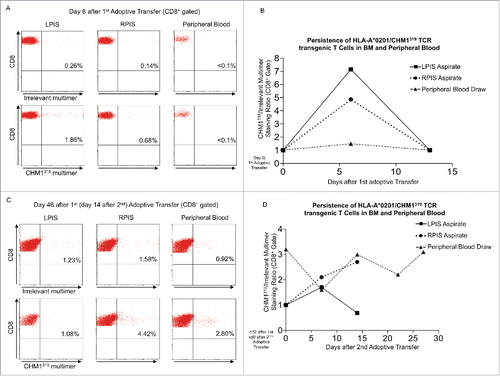
Patient #2 showed a clinical response to T cell therapy. BM aspirates from the posterior iliac spines at day 6 after first adoptive transfer stained 1.9% (left) and 0.7% (right) positive for the CHM1319 multimer, demonstrating homing to affected BM (). At day 14 after first transfer, BM aspirates converted multimer negative (, Fig. S4A). At day 7 after second transfer (day 39 after first), 0.8% (left) and 2.3% (right) aspirates stained positive (Fig. S4B). At day 14 after second transfer, positive cells increased to 1.1% (left) and 4.4% (right). In the same interval, positive staining in peripheral blood cells increased from 0.9 to 2.8% (). HLA-A*02:01/CHM1319 TCR transgenic T cells persisted in the right posterior iliac spine (at least 14 d after second transfer) as well as in the peripheral blood at least 27 d after second transfer ().
Figure 3. Transferred HLA-A*02:01/CHM1319 T cell receptor transgenic T cells home to bone marrow of patient #2 and persist. (A) Bone marrow (BM) aspirates from the left posterior iliacal spine (LPIS) stain positive for the CHM1319 multimer at a stronger rate than BM of the right posterior iliacal spine (RPIS) at day 6 after first adoptive transfer. Peripheral blood (PB) cells do not stain CHM1319 multimer positive. (B) At day 14 after first transfer BM aspirates and peripheral blood cells convert CHM1 multimer negative, demonstrating loss of adoptively transferred T cells after first adoptive transfer (x-axis; CHM1316multimer+/irrelevant multimer+ CD8+ staining ratio). (C) and (D) At day 14 after second adoptive transfer BM aspirates show a stronger CHM1319 multimer flourescence in RPIS but CHM319 loss in LPIS. PB is CHM1319 multimer positive and HLA-A*02:01/CHM1319 T cell receptor transgenic T cells proliferate until last PB draw on day 27 after second (day 56 after first) adoptive transfer. An irrelevant EZH2666/HLA-A*02:01 restricted multimer serves as control (CD8+ gate).
Patient #3: HLA-A*02:01/CHM1319 TCR transgenic T cells were detectable in peripheral blood 42 d after second transfer (68 d after first transfer). In vivo persistence in peripheral blood was shown until 51 d after after second transfer (day 77 first transfer, Fig. S8). Follow-up blood samples were not obtained due to return to the home country.
HLA-A*02:01/CHM1319 TCR transgenic T cell transfer have no side effects and can be associated with partial tumor regression
T cells were well tolerated in all patients. At the time of T cell transfer Patient #1 had progressive disease after refractory fifth relapse 6 y following initial diagnosis. She progressed and died 5 mo after first transfer without any signs of graft versus host disease (GvHD) (Fig. S7A). Survival after last relapse was also 5 mo (Table S1). The time between initial diagnosis and death was 7 y.
Patient #2 had progressive disease after refractory third relapse 2 y after initial diagnosis. He showed partial metastatic regression in positron emission and computed tomography (PET-CT) after adoptive transfer: At day − 1 before first transfer, PET-CT showed a pathological [Citation18F]fluorodeoxyglucose (FDG)-uptake of the BM at the left posterior iliac spine (Fig. S5). Marrow aspirates were positive on the left and the right side, but showed more ES cells on the left side. At day 20 after first transfer the PET-CT FDG-uptake on the left side, as quantified by maximal standardized uptake value (SUVmax), had increased to an SUVmax of 4.1 but remained negative on the right side. On day 21 after second transfer (day 53 after first transfer) immunohistochemistry of the left posterior iliac spine showed CD8+ T cell infiltration in both ES tumor and stromal tissue (Fig. S6A and S6B) while PET-CT revealed a partial regression (). In addition, PET-CT FDG-uptake of the right iliac spine, right acetabular roof, left femoral neck, left acetabular pillar, and the calvarium showed impressive regression with respect to SUVmax ( and ; Table S2). Non-specificity of the PET FDG-uptake was ruled out by focal (vs. diffuse) localization and the interval between the last aplasiogenic therapy and start of T cell transfer (more than 3 mo). ES regression after second adoptive transfer was noted in six of eight metastatic bone/BM sites, mainly in the pelvis. Lung metastases were progressive and 10 new bone/BM metastases emerged with 8 of 10 located in the extremities (). The patient died 2 mo after first adoptive transfer due to pulmonary disease progression without showing any signs of GvHD. Survival after last relapse was 5 mo (Table S1). The time between initial diagnosis and death was 30 mo.
Figure 4. PET-CT FDG-uptake reduction in multiple bone sites after second adoptive transfer. After second adoptive transfer, prior PET-CT positive sites (left panel) at the (A) left iliac spine, (B) right iliac spine and right acetabular roof, as well as (C) left femoral neck, left acetabular piller, and calvarium showed significant regression with respect to metabolic volume and SUVmax (right panel).
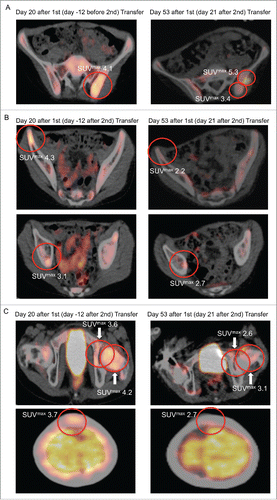
Table 1. Disease course of single PET-CT positive sites after first and second adoptive transfer of HLA-A*02:01/CHM1319 TCR transgenic T cells compared with reference PET-CT before therapy (Patient #2).
Patient #3: This patient had refractory primary disease, progressive 1 y after initial diagnosis. Tumor volume and positron emission tomography magnetic resonance imaging (PET-MRI) FDG-uptake of involved compartments increased continuously after adoptive transfer (Fig. S7B). The patient furthermore had an in-field relapse after delayed first irradiation of the primary (Fig. S7C). CT scans indicated no appearance of new metastases after second transfer until day 43 after second (day 69 after first) transfer. Follow-up PET or CT imaging was not obtained. Two doses of checkpoint inhibitors were administered in 2 instead of the 3 weeks as recommended by the manufacturer. On day 21 after the second dose (day 64 after first, day 38 after second transfer), the patient developed a temporary grade 3 maculo-papular rash with pruritus and grade 1 bullous dermatitis consistent with a known adverse event after checkpoint inhibition (CTCAE v3.1). The rash was successfully treated with two doses of methlyprednisolone (1 mg/kg IV) and skin symptoms regressed completely after 3 weeks. The patient died 4 mo after first adoptive transfer due to lung disease progression (3 mo after second) without receiving any aplasiogenic chemotherapy for 6 mo. Survival after last relapse was 9 mo (Table S1). The time between initial diagnosis and death was 20 mo.
A matched pair analysis with 15 controls from our Cooperative ES Study Group Registry, treated according to best practice (EURO-Ewing 2008, matched for time to first relapse, type of first relapse, primary metastases, age at diagnosis, number of relapses, gender, overall survival and death) revealed a higher mean overall survival of 136 d (816 vs. 680 days) in those patients treated with adoptive T cell transfer. Due to the low number of patients treated with adoptive T cell transfer this difference was not significant.
Discussion
Allorestricted T cells do not undergo thymic educationCitation20 and are generally thought to be more cross-reactive than conventional T cells. While being associated with GvHD, allogeneic donor lymphocyte infusions in general have failed to cause solid tumor regression. Conventional TCR reactivity is low against tumor-derived non-mutated peptides due to thymic education.Citation21,22 One possibility to enhance conventional TCR reactivity is affinity enhancement by random mutagenesis. However, these TCR mutants are also prone to cross-reactivity with unpredictable and fatal off-target effects.Citation23 For efficacious T cell therapy, both, the identification of proteins essential for cancer survival and TCR reactivity have to be addressed.Citation2,14,17,19,24
Utilization of TCRs from the allogeneic repertoire, exploiting a naturally occurring mechanism of self-defense, may circumvent both the requirement for affinity enhancement and anergy of T cells to tumor self-antigens.Citation15,25 Attempts to translate recent achievements of cellular therapy with e.g., chimeric antigen receptor T cells into the treatment of solid tumors, in particular pediatric sarcomas, has had limited success so far.Citation9-11,26,27 Since oncogenic drivers often are not antigenic, additional targets selectively overexpressed and required for malignancy and metastasis are to be identified supporting the application of TCR driven T cells.Citation15,28,29
CHM1 is directly upregulated by the ES driving fusion oncogene EWS-FLI1 and maintains an undifferentiated, invasive phenotype, and metastatic spread. Given its critical role in ES malignancy, CHM1 constitutes an excellent therapeutic target.Citation2 In previous studies, we have demonstrated efficacious T cell responses of clonally expanded wild-type HLA-A*02:01/CHM1319 and HLA-A*02:01/EZH2666 restricted T cells against ES cell lines in vitro and in vivo.Citation19 However, high manufacturing complexity, low cell numbers and rapid T cell exhaustion prompted us to generate large-scale ES-specific TCR transgenic T cells with less differentiated phenotypes off-the-shelf.Citation14,17
To facilitate the generation of highly enriched ES-specific T cells with a central memory-like phenotype, we successfully transduced allogeneic donor cells and autologous cells with CHM1-specific TCRs. We demonstrate here that adoptively transferred haplodisparate HLA-A*02:01/CHM1319-specific allorestricted TCR-driven T cells are well tolerated without GvHD even at doses up to 8 × 106/kg. This may be a key finding in this small series, given common concerns against allorestricted T cells because of their potential for cross-reactivity. The assumption that, because of their 100–1,000-fold higher incidence, allogeneic T cells are more degenerate and/or promiscuous than conventional T cells has never been clearly established experimentally.Citation25 Peptide specificity of our allorestricted cells is indicated by the absence of GvHD, in spite of a cell dose 100 times higher than recommended to avoid GvHD by nonspecific haplodisparate T cells.Citation30
Even in combination with the application of immune checkpoint inhibitors directed against CTLA-4 and PD-1 our haplodisparate T cells were well tolerated. We do not rule out that checkpoint inhibition contributed to tumor regression. Both checkpoint inhibitors may enable in part the transferred HLA-A*02:01/CHM1319 TCR transgenic CD8+ T cells to unfold their antitumor activity leading to in vivo proliferation, trafficking to involved tumor sites and consecutively to execute partial regression of metastatic tumor sites.
Homing of transferred HLA-A*02:01/CHM1319 TCR transgenic T cells to at least two affected BM sites could be demonstrated in patient #2. Moreover, due to persistence of HLA-A2 and CHM1 surface expression, a specific immune response of HLA-A*02:01/peptide+ T cells against all patient-derived ES cell lines was shown. Persistence of HLA-A*02:01 and CHM1 surface expression might be explained by the lack of immunoediting, since the tumor encounters the allorestricted TCR for the first time at therapy. In one of three patients with refractory disseminated disease treated with these T cells, we observed partial tumor regression.
In summary, we demonstrate that HLA-A*02:01/CHM1319 allorestricted TCR transgenic T cells home to affected BM can be associated with disease regression and do not cause GvHD. This is the first report on HLA-A*02:01/CHM1319 allorestricted TCR transgenic T cells in ES patients. Our observations suggest that CHM1316 is a relevant target for a TCR mediated approach. This observation raises hope that patients with advanced ES may benefit from immunotherapy in a state of residual disease in the future. Clinical trials to support this hypothesis are warranted.
Patients and methods
Cell lines, donor PBMC, PCR, expression analyses, ELISpot, identification, and expansion of CHM1316-specific TCR CHM1-4B4, and generation of CHM1319-TCR-transgenic T cells including their immunophenotyping were all used as described previously (Blaeschke et al.,Citation14 Schirmer et al.,Citation17 Supplemental data sheet 1, Table S3).
Tumor staging and detection of antigen-specific T cells
Staging was done by hybrid FDG PET-CT or PET-MRI and BM cytology. Transferred CD8+ T cells were monitored via CD8+/CD3/peptide multimer staining of blood and BM.
Statistics
Descriptive statistics was used to determine mean, standard deviation, and standard error of the mean. Differences were analyzed by unpaired two-tailed student's t-test as indicated using Excel (Microsoft); p-values < 0.05 were considered statistically significant (*p < 0.05; **p < 0.005; ***p < 0.0005). Survival was calculated from the most recent date stating local or systemic relapse, which led to progressive disease, i.e., post-relapse survival.
Patients
All patients and their legal guardians signed informed consent before therapy. Regulatory authorities were notified in advance and therapy was approved by the IRB and the health care ethics committee. All patients underwent the verification of target and HLA-A*02:01 expression ().
Patient #1: In March 2004, a 17 y old girl was diagnosed with localized ES of the left fibula (tumor volume 120 mL). BM aspirates were negative. After treatment with the Euro-Ewing 99 protocol, including local irradiation (44.8 Gy) and busulfan/melphalan-based high-dose chemotherapy with autologous rescue, she suffered a first relapse of the thoracic spine (T VII and T IX) in November 2006. The HLA-A*02:01+ patient received irinotecan and temozolomide relapse therapy followed by involved lesion irradiation (40 Gy), reduced intensity chemotherapy and allotransplant of her haploidentical HLA-A*02:01− brother in July 2007. The second relapse (March 2009, L III) was treated with donor lymphocyte infusions (DLI) up to 3 × 105/kg and local irradiation (36 Gy). The third relapse (September 2009, L I) was treated with local irradiation (50 Gy). The fourth relapse (June 2010, L III and multiple lung metastases) was treated with repetitive DLI up to 2 × 106/kg and local irradiation (18 Gy). She developed acute GvHD °III–IV that was successfully treated with prednisolone, mycofenolate mofetil, and basiliximab. After fifth relapse (multifocal, November 2010, pelvis and left femur), she became refractory and eligible for adoptive therapy with HLA-A*02:01/CHM1319 and HLA-A*02:01/EZH2666 allorestricted cloned wild-type CD8+ T cells from her haploidentical brother. 4 × 104/kg (d0), 2 × 105/kg (d21), and 4.8 × 105/kg (d28) T cells were infused at a 1:1 ratio (Fig. S1A, Table S1).
Patient #2: In April 2013, a 12 y old HLA-A*02:01+ boy with ES of the right femur was started on the Euro-Ewing 2008 protocol. BM aspirates were negative at diagnosis. His first relapse in March 2014 was treated with irinotecan and temozolomide, tandem myeloablative chemotherapy, autologous rescue and reduced intensity conditioning for allotransplant from his haploidentical HLA-A*02:01− mother. Local treatment consisted of MRI-directed involved compartment irradiation to the primary tumor, to osseous metastases (total dose 50–55 Gy) and to the lungs (15 Gy) as well as resection of the right distal femur tumor mass. After third relapse (multifocal, May 2015), he became refractory and eligible for adoptive therapy of HLA-A*02:01/CHM1319 allorestricted TCR transgenic CD8+ T cells from his mother. At this point, BM cytology was positive. 1 × 106/kg donor derived CHM1319-TCR transgenic CD8+ T cells were infused (d0), followed by transfer of 8.2 × 106/kg HLA-A*02:01/CHM1319 T cells (d32 in combination with nivolumab 1 mg/kg and ipilimumab 3 mg/kg, Fig. S1B, Table S1).
Patient #3: In November 2014, a 13 y old boy was diagnosed with ES of the right chest wall without BM involvement. He received chemotherapy similar to Euro-Ewing 2008 without concurrent irradiation. He developed progressive multifocal disease (local relapse plus pulmonary metastases), treated with irinotecan/vincristine. He next failed salvage with topotecan/cyclophosphamide and irinotecan/temozolomide chemotherapy and radiation therapy (total of 50.4 Gy) of the right lung tumor. Autologous stem cell apheresis was impossible due to BM exhaustion. BM aspirates were tumor negative throughout the course. Temporary control of the primary was now achieved by radiation. 3 × 106/kg autologous HLA-A*02:01/CHM1319 allorestricted TCR transgenic CD8+ T cells on day 0 were followed by a second dose of 6 × 106/kg in combination with checkpoint inhibitors nivolumab (1 mg/kg) and ipilimumab (3 mg/kg) on day 26. Prior to each dose, 50 mg of cyclophosphamide was given orally for a total of 7 d. Checkpoint inhibitors were re-administered on day 43 (Fig. S1C, Table S1).
Disclosure of potential conflicts of interest
S. Burdach has an ownership interest in PDL BioPharma and holds intellectual property (EU and US patents) in gene expression analysis. D. Busch holds shares in Juno Therapeutics. The other authors declare no potential conflicts of interest.
Author contributions
UT, SS, AK, MT, DS, FB, OS, RAR, TGPG, and SB did the experiments. UT, SS, IE, AK, MT, DS, FB, OS, RAR, TGPG, GHSR, PHS, and SB analyzed the data. UT, SS, AK, DS, FB, TGPG, GSHR, and SB interpreted the data. SJ, AR, and UD performed the statistical analysis. DHB provided multimer technology. UT, SS, KG, ITvL, and SB fulfilled diagnoses, collected patient information and did the follow up. UT and SB conceived, designed, and supervised the analysis. UT, SS, PHS, and SB drafted and revised the report.
Supplementary_materials.zip
Download Zip (43 MB)Acknowledgments
We thank all patients and families for their participation and support. Anna Hochholzer is acknowledged for technical assistance, Wolfgang Uckert and Matthias Leisegang for supplying the MP-71 vector and Wolfgang Schwingerand Ernst-Christian Urbanfor referral of patient#1.
Funding
This work was supported by grants from the Wilhelm Sander-Stiftung (2006.109.1), Else Kröner–Fresenius–Stiftung (GR & SB; P31/08//A123/07), BMBF (SB, PROVABES 01KT1311), the Deutsche Kinderkrebsstiftung (GR & SB; DKS 2010.07) and the BMBF (GR, UT and SB, TranSarNet 01GM1104B) as well as the Cura Placida Children's Cancer Research Foundation. T.G.P.G. is supported by a grant from ‘Verein zur Förderung von Wissenschaft und Forschung an der Medizinischen Fakultät der LMU München (WiFoMed)’, the Daimler and Benz Foundation in cooperation with the Reinhard Frank Foundation, by LMU Munich's Institutional Strategy LMUexcellent within the framework of the German Excellence Initiative, the ‘Mehr LEBEN für krebskranke Kinder – Bettina-Bräu-Stiftung’, the Fritz-Thyssen Foundation (FTF-40.15.0.030MN), the Friedrich-Baur Foundation, and the German Cancer Aid (DKH-111886 and DKH-70112257).
References
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371(16):1507-17; PMID:25317870; https://doi.org/10.1056/NEJMoa1407222
- von Heyking K, Calzada-Wack J, Göllner S, Neff F, Schmidt O, Hensel T, Schirmer D, Fasan A, Esposito I, Müller-Tidow C, Sorensen PH, Burdach S, Richter GH. The endochondral bone protein CHM1 sustains an undifferentiated, invasive phenotype, promoting lung metastasis in Ewing sarcoma. Mol Oncol. 2017 Mar 20; PMID:28319320; https://doi.org/10.1002/1878-0261.12057
- Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT. A second Ewing's sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet 1994; 6(2):146-51; PMID:8162068; https://doi.org/10.1038/ng0294-146
- Thiel U, Wawer A, von Luettichau I, Bender HU, Blaeschke F, Grunewald TG, Steinborn M, Röper B, Bonig H, Klingebiel T et al. Bone marrow involvement identifies a subgroup of advanced Ewing sarcoma patients with fatal outcome irrespective of therapy in contrast to curable patients with multiple bone metastases but unaffected marrow. Oncotarget. 2016 Oct 25; 7(43):70959-70968; PMID:27486822; https://doi.org/10.18632/oncotarget.10938
- Herzog J, von Klot-Heydenfeldt F, Jabar S, Ranft A, Rossig C, Dirksen U, Van den Brande J, D'Incalci M, von Luettichau I, Grohar PJ et al. Trabectedin followed by irinotecan can stabilize disease in advanced translocation-positive sarcomas with acceptable toxicity. Sarcoma 2016; 2016:7461783; PMID:27843394; https://doi.org/10.1155/2016/7461783
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015; 373(1):23-34; PMID:26027431; https://doi.org/10.1056/NEJMoa1504030
- Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest 2015; 125(9):3413-21; PMID:26258412; https://doi.org/10.1172/JCI80008
- Houot R, Schultz LM, Marabelle A, Kohrt H. T-cell-based immunotherapy: adoptive cell transfer and checkpoint inhibition. Cancer Immunol Res 2015; 3(10):1115-22; PMID:26438444; https://doi.org/10.1158/2326-6066.CIR-15-0190
- Merchant MS, Bernstein D, Amoako M, Baird K, Fleisher TA, Morre M, Steinberg SM, Sabatino M, Stroncek DF, Venkatasan AM et al. Adjuvant immunotherapy to improve outcome in high-risk pediatric sarcomas. Clin Cancer Res 2016; 22(13):3182-91; PMID:26823601; https://doi.org/10.1158/1078-0432.CCR-15-2550
- Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, Yang JC, Dudley ME, Wunderlich JR, Sherry RM et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res 2015; 21(5):1019-27; PMID:25538264; https://doi.org/10.1158/1078-0432.CCR-14-2708
- Rossig C. Cellular immunotherapy strategies for Ewing sarcoma. Immunotherapy 2014; 6(5):611-21; PMID:24896629; https://doi.org/10.2217/imt.14.36
- Orentas RJ, Lee DW, Mackall C. Immunotherapy targets in pediatric cancer. Front Oncol 2012; 2:3; PMID:22645714; http://doi.org/10.3389/fonc.2012.00003
- Kolb HJ, Schmid C, Barrett AJ, Schendel DJ. Graft-versus-leukemia reactions in allogeneic chimeras. Blood 2004; 103(3):767-76; PMID:12958064; https://doi.org/10.1182/blood-2003-02-0342
- Blaeschke F, Thiel U, Kirschner A, Thiede M, Rubio RA, Schirmer D, Kirchner T, Richter , Mall S, Klar R et al. Human HLA-A*02:01/CHM1+ allo-restricted T cell receptor transgenic CD8+ T Cells specifically inhibit Ewing sarcoma growth in vitro and in vivo. Oncotarget 2016; 7(28):43267-80; PMID:27281613; https://doi.org/10.18632/oncotarget.9218
- Burdach S, Kolb HJ. The vigor of defense against non-self: potential superiority of allorestricted T cells in immunotherapy of cancer? Front Oncol 2013; 3:100; PMID:23653891; https://doi.org/10.3389/fonc.2013.00100
- Kirschner A, Thiede M, Blaeschke F, Richter GH, Gerke JS, Baldauf MC, Grünewald TG, Busch DH, Burdach S, Thiel U. Lysosome-associated membrane glycoprotein 1 predicts fratricide amongst T cell receptor transgenic CD8+ T cells directed against tumor-associated antigens. Oncotarget 2016; 7(35):56584-97; PMID:27447745; https://doi.org/10.18632/oncotarget.10647
- Schirmer D, Grunewald TG, Klar R, Schmidt O, Wohlleber D, Rubío RA, Uckert W, Thiel U, Bohne F, Busch DH et al. Transgenic antigen-specific, HLA-A*02:01-allo-restricted cytotoxic T cells recognize tumor-associated target antigen STEAP1 with high specificity. Oncoimmunology 2016; 5(6):e1175795; PMID:27471654; https://doi.org/10.1080/2162402X.2016.1175795
- Staege MS, Hutter C, Neumann I, Foja S, Hattenhorst UE, Hansen G, Afar D, Burdach SE. DNA microarrays reveal relationship of Ewing family tumors to both endothelial and fetal neural crest-derived cells and define novel targets. Cancer Res 2004; 64(22):8213-21; PMID:15548687; https://doi.org/10.1158/0008-5472.CAN-03-4059
- Thiel U, Pirson S, Muller-Spahn C, Conrad H, Busch DH, Bernhard H, Burdach S, Richter GH. Specific recognition and inhibition of Ewing tumour growth by antigen-specific allo-restricted cytotoxic T cells. Br J Cancer 2011; 104(6):948-56; PMID:21407224; https://doi.org/10.1038/bjc.2011.54
- McDonald BD, Bunker JJ, Erickson SA, Oh-Hora M, Bendelac A. Crossreactive alphabeta T cell receptors are the predominant targets of thymocyte negative selection. Immunity 2015; 43(5):859-69; PMID:26522985; https://doi.org/10.1016/j.immuni.2015.09.009
- Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med 2005; 202(7):907-12; PMID:16203864; https://doi.org/10.1084/jem.20050732
- Rammensee HG, Bevan MJ. Evidence from in vitro studies that tolerance to self-antigens is MHC-restricted. Nature 1984; 308(5961):741-4; PMID:6232464; https://doi.org/10.1038/308741a0
- Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, Brewer JE, Gupta M, Plesa G et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med 2013; 5(197):197ra03; PMID:23926201; https://doi.org/10.1126/scitranslmed.3006034
- Richter GH, Plehm S, Fasan A, Rössler S, Unland R, Bennani-Baiti IM, Hotfilder M, Löwel D, von Luettichau I, Mossbrugger I et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc Natl Acad Sci U S A 2009; 106(13):5324-9; PMID:19289832; https://doi.org/10.1073/pnas.0810759106
- Felix NJ, Donermeyer DL, Horvath S, Walters JJ, Gross ML, Suri A, Allen PM. Alloreactive T cells respond specifically to multiple distinct peptide-MHC complexes. Nat Immunol 2007; 8(4):388-97; PMID:17322886; https://doi.org/10.1038/ni1446
- Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, Smithers H, Jensen MC, Riddell SR, Maloney DG et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016; 127(20):2406-10; PMID:26907630; https://doi.org/10.1182/blood-2015-08-665547
- Mackall CL, Merchant MS, Fry TJ. Immune-based therapies for childhood cancer. Nat Rev Clin Oncol 2014; 11(12):693-703; PMID:25348789; https://doi.org/10.1038/nrclinonc.2014.177
- Blank CU, Haanen JB, Ribas A, Schumacher TN. CANCER IMMUNOLOGY. The ‘cancer immunogram’. Science 2016; 352(6286):658-60; PMID:27151852; https://doi.org/10.1126/science.aaf2834
- Crompton BD, Stewart C, Taylor-Weiner A, Alexe G, Kurek KC, Calicchio ML, Kiezun A, Carter SL, Shukla SA, Mehta SS et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov 2014; 4(11):1326-41; PMID:25186949; https://doi.org/10.1158/2159-8290.CD-13-1037
- Klingebiel T, Handgretinger R, Lang P, Bader P, Niethammer D. Haploidentical transplantation for acute lymphoblastic leukemia in childhood. Blood Rev 2004; 18(3):181-92; PMID:15183902; https://doi.org/10.1016/S0268-960X(03)00063-8