
ABSTRACT
An oral sustained-release formulation of Interleukin-10 suppressed tumor growth and enhanced survival in the APCmin/+/Bacteroides fragilis spontaneous colon cancer model. Therapeutic benefit was associated with a 5-fold reduction in CD4+RORγt+Foxp3−IL-17+ T-helper cell, CD4+RORγt+Foxp3+IL-17+ pathogenic T-regulatory cell and CD4+RORγt−Foxp3+IL-17− conventional T-regulatory cell numbers and a concurrent 2-fold enhancement in CD8+ T-cell activity in the colon. Selective subset depletion and functional blockade studies demonstrated that at steady-state CD4+RORγt+IL-17+ T-cell subsets and CD4+Foxp3+ cTreg supported tumorigenesis, whereas CD8+ cytotoxic T-lymphocytes impeded tumor progression following IL-10 therapy. Suppression of tumor growth by CD8+ T-cells was associated with enhanced tumor infiltration and cytotoxic granule exocytosis. These findings establish the utility of oral IL-10 as a potential new therapeutic in the management of colon cancer and shed light on the cellular mechanisms that underlie its antitumor activity.
Abbreviations
| ANOVA | = | Analysis of variance |
| APCmin/+ | = | adenomatous polyposis coli multiple intestinal neoplasia |
| BSA | = | Bovine serum albumin |
| CD | = | Cluster of differentiation |
| CTL | = | Cytotoxic T-lymphocyte |
| cTreg | = | conventional T-regulatory cell |
| FCS | = | Fetal calf serum |
| Foxp3 | = | Forkhead box P3 |
| IEL | = | Intra-epithelial lymphocyte |
| IL | = | Interleukin |
| IL-23R | = | IL-23 receptor |
| LP | = | Lamina propria |
| LPL | = | Lamina propria lymphocyte |
| MDSC | = | Myeloid-derived suppressor cell |
| MLN | = | Mesenteric lymph node |
| OCT | = | Optimal cutting temperature |
| PBS | = | Phophate-buffered saline |
| pgTreg | = | pathogenic T-regulatory cell |
| PIN | = | Phase inversion nanoencapsulation |
| PP | = | Peyer's patch |
| RORγt | = | Retinoic acid receptor gamma t |
| TH | = | T-helper |
Introduction
The link between chronic inflammation and colon cancer is well-established.Citation1 Specifically, danger signals that are produced by hyperproliferative dysplastic enterocytes, and those released when commensal bacteria infiltrate the damaged epithelium are thought to induce tumor-promoting inflammatory activity.Citation1,2 Early stages of inflammation involve the production of innate cytokines such as IL-1, IL-6 and IL-23 by infiltrating myeloid cells leading to, among other pathologies, TH17-like immune activity.Citation1,2 Numerous studies have established a strong link between enhanced IL-17A (IL-17) production and tumorigenesis, particularly in the GI tract.Citation3-5 Blockade of IL-17, IL-22 or IL-23R can halt this process in pre-clinical models establishing the myeloid cell-TH17 axis as a major driver of inflammatory cancer.Citation3-5 Pro-tumorigenic activity of the type 17 response is mediated by immune cell-dependent as well as independent mechanisms. For example, IL-17 has been shown to be chemotactic for MDSC and is thought to enhance tumor growth in part via increasing myeloid-cell mediated suppression of antitumor effector cell activity in the tumor microenvironment.Citation6,7 Separately, others have shown that the TH17 cytokines IL-17 and IL-22 can directly stimulate epithelial cell proliferation and tumor progression.Citation3,4 Taken together these findings suggest that the myeloid cell-T-cell axis represents an attractive target for therapy of inflammatory cancer.
T-cell-produced IL-10 is critical to maintaining steady-state homeostasis in the gut.Citation8,9 IL-10 converts immature blood monocytes to tolerogenic macrophages,Citation8,9 directly suppresses TH17 cell proliferation,Citation10,11 and conversely enhances Treg function.Citation11 The potential utility of recombinant IL-10 in the treatment of inflammatory diseases of the gastrointestinal tract, including cancer has been discussed.Citation12 However, the promise of IL-10 as an immune therapeutic has not been realized.Citation13 Whether this is due to the inability of the cytokine to effectively reverse established inflammation and/or the failure to reach physiologically-relevant levels in the disease microenvironment following bolus systemic delivery is not known.Citation13 The finding that adoptive transfer of gut-homing Treg can ameliorate inflammatory disease and suppress colon carcinogenesis in an IL-10-dependent manner is consistent with the latter.Citation14,15
We recently demonstrated that oral administration of a particulate sustained-release formulation of IL-10 resulted in rapid uptake in the Peyer's Patches (PP) and the mesenteric lymph nodes (MLN), suppressed intestinal polyposis and ameliorated systemic symptoms of disease in the APCmin/+ mice.Citation16 Analysis of cellular mechanisms revealed that IL-10 mediated its therapeutic effect via preferential neutralization of CD4+Foxp3+RORγt+IL-17+ pathogenic T-regulatory cell (pgTreg) activity in the intestinal lamina propria (LP).Citation16 In the subsequent studies, we obtained preliminary evidence that treatment could also block adenoma to adenocarcinoma transition in the APCmin/+/Bacteroides fragilis (B. fragilis) model of colon cancer.Citation17 However, whether IL-10 would be effective in the long-term and how it suppresses the genesis and the progression of bona fide colon cancer were not investigated. In this manuscript, we report that oral IL-10 can promote effective long-term suppression of carcinoma in the APCmin/+/B. fragilis model and that the therapeutic effect is associated with the pleiotropic activity of IL-10 on multiple CD4+ T-cell subsets and cytotoxic CD8+ T-cells in the colonic LP.
Results
Oral IL-10 suppresses colon tumor growth in a CD4+ T-cell-dependent manner
Previous studies demonstrated that oral IL-10 suppressed intestinal polyposis in the APCmin/+ mice.Citation16 We wanted to determine whether this approach would be effective against colon cancer. This notion was tested in the compound APCmin/+/B. fragilis model in which infection of mice with an enterotoxic strain of the bacterium results in the development of bona fide adenocarcinomas in the colon.Citation5 To assess antitumor efficacy of oral IL-10, therapy was initiated one week after inoculation of bacteria and continued for 3 weeks. Analysis of colon tumor burden at the end of treatment revealed that IL-10 promoted a 3-fold reduction in tumor burden (). Next, a survival study was performed to determine whether chronic oral IL-10 administration would provide long-term benefit. The data demonstrate that long-term treatment resulted in a significant 25 d extension of survival ().
Figure 1. Oral IL-10 suppresses colon tumor growth and extends survival via its activity on CD4+ T-cells. (A) Short-term therapy. Age-matched mice were either left untreated (WT, APCmin/+) or treated with blank control particles, IL-10 particles or antibiotics (APCmin/+ + B. fragilis) as described in section Materials and methods. Colon tumor burden was quantified at the end of the study. Error bars = SEM, n = 8–22 per group. (B) Survival. Mice were administered B. fragilis and were treated starting 1 week after bacterial inoculation until euthanasia. Significance: p = 0.018 (Log-Rank), n = 11 per group. (C) Role of CD4+ T-cells in IL-10-mediated amelioration of disease. B. fragilis-inoculated mice were treated either with anti-CD4+ depletion antibody, oral IL-10, a combination of both or antibiotics as described in section Materials and methods. Colon tumor burden was analyzed at the end of the 3 week treatment period. Error bars = SEM, n = 5–8 per group. Significance: #,##,###denote p ≤ 0.05, 0.01 or 0.001, respectively.
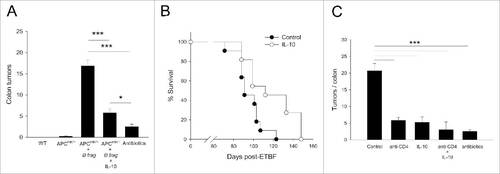
Inflammatory CD4+ T-cells, particularly the TH17 subset, have been implicated in colon tumorigenesis.Citation1,2,5 Separately, the ability of IL-10 to modulate the activity and proliferation of different CD4+ T-cell subsets is well-established.Citation18 We therefore wanted to determine whether the IL-10-CD4+ T-cell axis was responsible for tumor suppression in the colon. To this end, CD4+ T-cell depletion was performed in untreated controls as well as in IL-10 treated mice, and tumor burden was determined. Treatment again resulted in a robust 4-fold decline in the number of colon tumors (). In addition, CD4+ T-cell depletion alone was just as effective as IL-10 treatment in suppressing tumor growth. Importantly, combined treatment, i.e. IL-10 plus CD4+ T-cell depletion, was no more effective than either treatment alone suggesting that the therapeutic activity of IL-10 was primarily mediated via its activity on CD4+ T-cells.
IL-10 eliminates pro-tumorigenic CD4+RORγt+IL-17+ T-cell subsets from the colonic LP
IL-17 is critical to tumorigenesis in the APCmin/+/B. fragilis model.Citation5 We next investigated whether the activity of IL-10 on CD4+ T-cells specifically involved the IL-17+ subset. We first analyzed IL-17-producing CD4+ T-cells as a whole in control and treated mice. The data demonstrate a nearly 10-fold increase in the numbers of CD4+IL-17+ T-cells in the colonic LP of B. fragilis-infected animals in comparison to naive APCmin/+ mice () confirming the previously-reported link between CD4+IL-17+ cells and tumorigenesis.Citation5 Importantly, treatment resulted in a 3-fold decline in the CD4+IL-17+ cell prevalence consistent with a role for the IL-10-CD4+IL-17+ subset axis in tumor suppression (). Analysis of the MLN populations revealed similar but less dramatic changes establishing that the therapeutic effect primarily manifested in the colon (Fig. S1A). Separately, analysis of colonic LP γδT-cells, which contribute to tumor growth in the APCmin/+/B. fragilis model,Citation19 revealed a 5-fold reduction in IL-17-producing γδT-cell numbers in treated animals demonstrating that the activity of IL-10 extended to non-CD4+ T-cells (Fig. S2). At the same time, since data in revealed that CD4+ T-cells were the major driver of colon tumorigenesis subsequent studies focused on this population.
Figure 2. IL-10-mediated decline in the prevalence of colonic LP CD4+IL-17+ T-cell subsets is partially responsible for tumor suppression. (A) CD4+IL-17+ cell numbers. CD4+ T-cells were gated on and analyzed for IL-17 production by intracellular staining. Representative flow panels and quantitative data for total colonic LP CD4+IL-17+ T-cell numbers are shown. Error bars = SEM, n = 4–5 per group. (B) CD4+ T-cell subset numbers. CD4+ T-cells were gated on and analyzed for expression of Foxp3 and RORγt. Representative flow panels and quantitative data are shown for TH17 (CD4+RORγt+Foxp3−) and pgTreg (CD4+RORγt+Foxp3+). Error bars = SEM, n = 4 per group. (C) IL-17 production by CD4+ T-cell subsets. LP TH17 and pgTreg subsets in control and IL-10-treated mice were analyzed for IL-17 production by intracellular staining. Representative data are shown. (D) RORγt inhibition. Mice were either treated with vehicle, RORγt-inhibitor SR1001, IL-10 or SR1001 + IL-10 and colon tumor burden was analyzed. Error bars = SEM, n = 5–11 per group. Significance: #,##,###denote p < 0.05, 0.01 or 0.001, respectively.
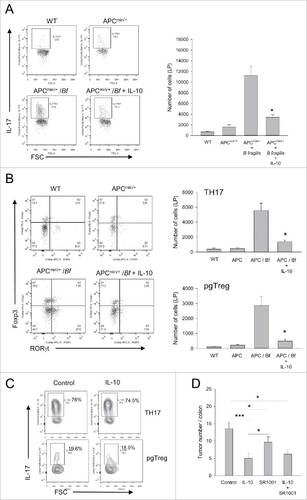
Our previous study in the conventional APCmin/+ mice had revealed that IL-10 preferentially targeted IL-17-producing pgTreg in the intestinal LP.Citation16 To determine whether a similar selective effect was responsible for suppression of colon tumors, subset-specific analysis was performed. The data showed a dramatic 16 to 20-fold increase in both pgTreg and TH17 cells in the colons of B. fragilis infected APCmin/+ mice as compared with APCmin/+ controls (). Importantly, oral IL-10 achieved a 4 to 6-fold reduction in both subsets in the infected mice demonstrating broad activity on all CD4+IL-17+ T-cell populations (). Analysis of MLN revealed a similar overall pattern except that the changes were more modest (Fig. S1B). We then analyzed the percent of IL-17+ cells within each subset before and after treatment to determine whether IL-10 also engendered a qualitative effect. We found that IL-10 did not affect the ability of persisting LP cells to produce IL-17 as distinct from our previous findings in the intestine ().
To determine whether the therapeutic activity of IL-10 was strictly associated with a diminished type 17 response, mice were treated with a highly-specific small molecule inhibitor of RORγt, SR1001. This study demonstrated that while SR1001 was able to achieve significant tumor suppression it was not as effective as oral IL-10 suggesting that IL-10 modulated additional IL-17-independent yet CD4+ T-cell-dependent mechanisms ().
IL-10 eliminates pro-tumorigenic CD4+Foxp3+RORγt− conventional T-regulatory cells from the colonic LP
A recent study demonstrated that conventional Treg (cTreg) displayed pro-tumorigenic function in the APCmin/+/B. fragilis model.Citation20 Specifically, the authors found that expanding cTreg act as a sink for IL-2, which results in preferential expansion of TH17 cells at the expense of other helper subsets ultimately leading to enhanced tumor growth.Citation20 Consistent with this observation, analysis of CD4+Foxp3+RORγt− cTreg revealed a 4 to 5-fold increase in their numbers in the colons of diseased animals (). We then examined whether oral IL-10 impacted cTreg presence in the colonic LP. Unexpectedly, IL-10 treatment resulted in a dramatic decline in cTreg numbers to background levels (). This finding is in contrast to our previous study, which showed a modest increase in cTreg in the intestinal LP of IL-10-treated APCmin/+ mice, again suggesting that the biology of dysplasia and how IL-10 modulates its course differ in the APCmin/+ vs APCmin/+/B. fragilis models.
Figure 3. IL-10 diminishes tumor-promoting cTreg, the absence of which results in reduced CD4+IL-17+ T-cell prevalence and increased CD8+ T-cell activity. (A) Conventional Treg numbers. Age-matched mice were either left untreated (WT, APCmin/+) or treated with blank control particles, IL-10 particles or antibiotics (APCmin/+ + B. fragilis) as described in section Materials and methods. Colon LP cTreg were quantified at the end of the study. Error bars = SEM, n = 4 per group. (B) Conventional Treg depletion. B. fragilis colonized mice were treated with PBS or anti-CD25 antibody for 3 weeks starting 1 week after bacterial inoculation. Colon tumor burden was quantified at the end of treatment. Error bars = SEM, n = 10–12 per group. (C) Effect of cTreg depletion on CD4+ and CD8+ T-cells. Percentage of IL-17+ CD4+ T-cells and IFNγ+ CD8+ T-cells were quantified in mice treated either with PBS or anti-CD25 as in Panel B study. Error bars = SEM, n = 3 per group. Significance: #, ##, ###denote p < 0.05, 0.01 or 0.001, respectively.
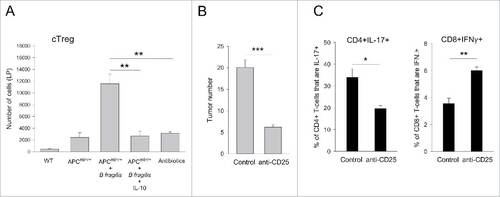
The observation that disease amelioration is accompanied with a major decline in LP cTreg numbers suggested that loss of IL-17-producing cells in treated mice could be secondary to cTreg elimination. This notion was supported by a depletion study in which administration of an anti-CD25 antibody to mice resulted not only in the suppression of tumorigenesis () but also in a reduction of IL-17-producing CD4+ T-cells in the colon (). Furthermore, cTreg-depletion enhanced IFNγ-production by CD8+ T-cells in the colon revealing a dual effect by cTreg on CD4+IL-17+ T-cell prevalence and CD8+ T-cell activity ().
IL-10 enhances antitumor CD8+ T-cell activity
While the above data suggest that the pleiotropic activity of IL-10 on the cTreg-TH17 axis was responsible for the therapeutic benefit, the inability to duplicate this effect with direct suppression of RORγt left open the possibility that loss of cTreg could have additional, TH17-independent, antitumor effects. It is well-known that cTreg can enhance tumor growth via suppression of antitumor effector T-cell function. Consistent with this, depletion of cTreg resulted in enhanced CD8+ T-cell activity (). Therefore, we hypothesized that in our system IL-10 could promote CD8+ CTL cytotoxicity via preferential elimination of cTreg. To test this, we first quantified the CD8+ T-cell/cTreg vs TH17/cTreg ratios in control vs treated mice. The data demonstrate that IL-10 promoted a preferential increase in the CD8+ T-cell/cTreg but not in the TH17/cTreg ratio in the colonic LP (). A similar increase in TH1 to cTreg ratio was also observed (Fig. S3). These changes were primarily driven by the dramatic plunge in cTreg as the overall numbers of TH1 and CD8+ T-cells also declined by ∼2-fold after IL-10 therapy (Fig. S3B).
Figure 4. IL-10-mediated tumor suppression is in part dependent on cytotoxic CD8+ T-cell activation. (A) Effector T-cell/cTreg ratios. The ratio of LP CD8+ and CD4+IL-17+ T-cells to cTreg were quantified in WT, APCmin/+, control-treated APCmin/+/B. fragilis and IL-10-treated APCmin/+/B. fragilis mice. Error bars = SEM, n = 4–6 per group. (B) Role of CD8+ T-cells in tumor suppression. B. fragilis-colonized APCmin/+ mice were either treated with blank or control particles with or without CD8+ T-cell depletion as described in section Materials and methods. Colon tumor burden was determined at the end of the study. Error bars = SEM, n = 5–12 per group. (C) CD8+ T-cell activation. Single cell suspensions were prepared from the colonic LP. CD8+ T-cells were gated on and were analyzed for intracellular IFNγ and membrane CD107a expression. Representative flow panels and quantitative data are shown. Error bars = SEM, n = 4 per group. Significance: #, ##, ###denote p < 0.05, 0.01 or 0.001, respectively. (D) Histological analysis of CD8+ T-cells in colon tumors. Tumor sections from control and IL-10-treated mice were stained for CD8+ (blue) and CD107a (red). The arrows point to CD8+/CD107a double-positive cells. Bar = 50 μm.
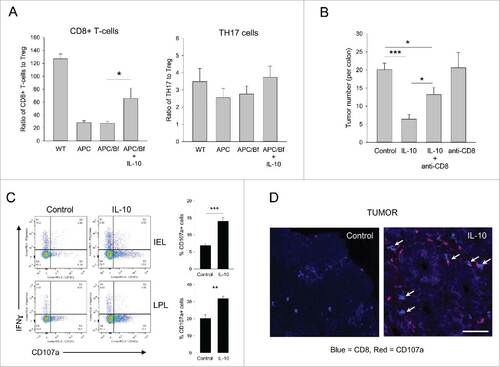
The above observations suggested that enhanced activation of pre-existing TH1 and/or CTL could in part be responsible for tumor suppression. Depletion of CD8+ T-cells in IL-10-treated mice significantly diminished therapeutic efficacy (), consistent with a role for CTL activation in post-therapy tumor suppression. Furthermore, CD8+ T-cell elimination in the absence of IL-10 treatment did not affect tumor growth suggesting that IL-10-dependent activation of CTL was necessary for tumor suppression. To this end, analysis of colonic IEL/LPL CD8+ T-cells in control vs IL-10-treated mice revealed a 2-fold increase in CD107a expression in the IL-10 group, particularly within the IEL population, demonstrating that treatment enhanced the cytotoxic activity of CD8+ T-cells (). In contrast, IL-10 did not enhance, but in fact, slightly diminished IFNγ production in CD8+ T-cells (, Fig. S3C). Similarly, neither the overall numbers nor the percent of IFNγ+ CD4+ T-cells increased in treated mice suggesting a minimal role for TH1 cell activity in IL-10-mediated tumor suppression (Fig. S3B and C).
Next, histological analysis of tumors and colons was performed to further define treatment-induced changes in the cellular landscape in situ. Laser-scanning confocal imaging of control tumors revealed a general lack of lymphocytic infiltrates except for a few intensely-staining myeloid cell/CD4+ T-cell nests with rare CD8+ T-cells dispersed throughout (Fig. S4, tumor). IL-10 treatment resulted in the disappearance of the myeloid cell/CD4+ T-cell nests and conversely enhanced CD8+ T-cell presence in the tumor (Fig. S4, tumor, expanded view). Importantly, and consistent with the data shown in , tumor-infiltrating CD8+ T-cells in IL-10-treated mice displayed robust CD107a expression while those found in control tumors were CD107a-negative (). Analysis of non-tumor tissue revealed few CD8+ T-cells but intense CD4+ T and CD11b+ myeloid cell infiltration in the colonic LP of control animals (Fig. S4A). IL-10 treatment on the other hand, significantly reduced CD11b+ myeloid and CD4+ T-cell presence (Fig. S4, colon). Collectively the histological findings in IL-10 treated mice, i.e., an overall loss of CD4+ T-cells from the tumors/colonic LP with concurrent activation of tumor-associated CTL, were in agreement with the quantitative data shown in .
Discussion
We have found that oral IL-10 inhibited colon tumorigenesis in the APCmin/+/B. fragilis model. Tumor suppression was associated with the pleiotropic activity of IL-10 on CD4+ T−, γδT− and CD8+ T-cells. Specifically, treatment diminished IL-17-producing CD4+RORγt+Foxp3− TH17, CD4+RORγt+Foxp3+ pgTreg and γδT-cells in the colon. Unexpectedly, IL-10 also induced the elimination of CD4+Foxp3+RORγt− cTreg, which are known to enhance tumor growth in the APCmin/+/B. fragilis model.Citation20 Finally, our data revealed that the elimination of pathogenic CD4+ T-cell subsets was accompanied with enhanced CD8+ T-cell cytotoxicity, which contributed to tumor suppression. These findings confirm the potential of oral IL-10 as a novel cancer therapeutic and illuminate the cellular mechanisms that underlie its antitumor activity in the inflamed colon.
The across-the-board reduction in the numbers of IL-17-producing CD4+ T-cells was consistent with the known properties of IL-10.Citation10,11 Phenotypic analysis of post-therapy T-cell populations revealed that despite the dramatic decline in quantity, the persisting TH17 and pgTreg continued to produce IL-17 supporting the notion that elimination of the cells was due to the ability of IL-10 to block their expansion as opposed to direct inhibition of IL-17 production. The mechanism that underlies the ability of IL-10 to modulate IL-17+ γδT-cell activity is less well-defined. It is known that IL-10 can directly suppress IFNγ-production in γδT-cells.Citation21 A similar effect on IL-17 secretion may, on the other hand, be indirect and require cTreg involvement.Citation22,23
The dramatic loss of cTreg in treated mice was unexpected as IL-10 is traditionally thought to promote cTreg activity. To this end, a recent study reported a similar effect by PEG-IL-10 in a murine transplantable mammary tumor model.Citation24 How exactly IL-10 diminishes cTreg prevalence remains unknown. One possibility is that it antagonizes cTreg recruitment via suppression of chemokine production in the inflamed mucosa.Citation25 Whereas the mechanism underlying this observation remains to be defined, loss of cTreg may, at least in part, explain the concurrent disappearance of TH17 cells. As discussed above expanding cTreg can act as a sink for IL-2 and thus re-direct T-helper cell responses toward a type 17 profile in the colon.Citation20 It is therefore conceivable that the overall decline in IL-17-producing cells is, at least in part, an indirect consequence of cTreg loss.
The studies described here revealed differences between the intestinal polyposis and the colon cancer models. Briefly, in the intestinal polyposis model the overall increase in CD4+ T-cell populations is modest and IL-10 treatment does not affect CD4+ T-cell numbers.Citation16 IL-10 does suppress IL-17 production by intestinal CD4+ T-cells and this effect is due, primarily, to a preferential elimination of IL-17 producing pgTreg.Citation16 In addition, IL-10 treatment does not reduce relative cTreg prevalence in the intestine.Citation16 In contrast, disease induction is accompanied with a surge in multiple IL-17-producing T-cell subsets and cTreg in the inflamed colonic LP and IL-10 promotes a global reduction in all CD4+ T-cell populations. The intestine and the colon are not only different structurally but also with respect to T-cell composition and bacterial burden.Citation26 Therefore it is not surprising that the T-cell biology of intestinal polyposis, a process that is likely more sterile in nature, is significantly different than that of B. fragilis-driven colon cancer in the APCmin/+ mouse. Regardless, oral IL-10 suppressed pathology in both settings.
Elimination of pro-tumorigenic CD4+ T-cells was accompanied with the enhancement of CD8+ T-cell cytotoxicity. Two distinct but non-mutually exclusive mechanisms can account for this finding. Loss of cTreg, which can suppress CTL cytotoxicity, is one potential indirect mechanism. Alternatively, IL-10 can directly restore cytotoxic function to tumor-associated CD8+ T-cells.Citation27-29 Separately, whereas IL-10 enhanced cytotoxic granule exocytosis, it had an antagonistic effect on IFNγ production. These data are consistent with others' observations that IL-10 can stimulate cytotoxic effector molecule production in antigen-experienced cells independent of IFNγ.Citation29
While our study focused on delineating the effects of oral IL-10 on T-cell populations, histological analysis demonstrated significant changes in CD11b+ myeloid cell infiltrates in the colon. Specifically, in control mice the LP was heavily infiltrated by myeloid cells, which upon treatment were mostly eliminated. How oral IL-10 alters CD11b+ cell prevalence in treated mice is yet to be determined. It is known that IL-10 can suppress chemokine production by stromal cells and therefore could potentially abrogate myeloid cell recruitment.Citation30 A separate but related question that was not addressed here is the primary cellular target of IL-10 in the colon. Whether IL-10 mediated its therapeutic activity via direct signaling into pathogenic T-cells, and/or through its upstream activity on myeloid cells remain to be determined.
Collectively, our results reveal a dual functional role for exogenous IL-10 in the dysplastic colon, i.e., abrogation of pro-tumorigenic CD4+ T-cell activity accompanied with an induction of antitumor CD8+ T-cell cytotoxicity. In this context, it is tempting to speculate that IL-10 could be particularly effective in sensitizing immune-resistant colon tumors to immune checkpoint inhibitors.Citation31,32
Materials and methods
Mice and the tumor model
C57BL/6 (B6) and C57BL/6J-ApcMin/J (APCmin/+) mice were purchased from Jackson Laboratory. Enterotoxic B. fragilis strain 86-5443-2-2 was a kind gift from Dr. Cynthia L Sears (Johns Hopkins University School of Medicine, Baltimore, Maryland). For colonization with B. fragilis, 5–6 week old APCMin+ mice were administered clindamycin (0.1 g/L) and streptomycin (5 g/L) for 3–5 d before oral gavage (∼5 × 107 bacteria in PBS) essentially as described.Citation5 All studies were conducted in accordance with guidelines set forth by the Institutional Animal Care and Use Committee at the University of Louisville (Louisville, KY).
Reagents and treatments
Two particle formulations were produced using a modified phase inversion nanoencapsulation (PIN) process33: (i) control (no cytokine) and (ii) recombinant murine IL-10-encapsulated (Peprotech, Inc.) with a loading of 0.5 μg cytokine/mg of particles. The PIN process provides >90% encapsulation efficiency for cytokines with an average particle size of 0.8 microns (range of 0.1–5 micron) and minimal (< 5%) batch to batch variation in physicochemical properties (data not shown andCitation33). IL-10 or blank particles were administered via oral gavage (1 mg particles in 0.2 mL PBS) starting 1 week after B. fragilis inoculation 3 times per week for 3 weeks. Treatment resulted in a 2 to 3-fold increase in IL-10 levels in the small intestinal LP, but not in the colon or serum, within 6 h of administration (Fig. S5). Antibiotic treatment was initiated 2 weeks after B. fragilis inoculation. Mice were administered ampicillin (A; 1 g/L; Goldbio), vancomycin (V; 500 mg/L; Goldbio), neomycin sulfate (N; 1 g/L; Goldbio) and metronidazole (M; 1 g/L; VWR) in drinking water for 3 weeks as described.Citation34 For survival analysis, mice were treated until they reached the IACUC-approved euthanasia score as described previously by us.Citation16
Flow cytometry
MLN were processed into single cell suspensions and colons were digested and fractionated into intraepithelial (IEL) and lamina propria lymphocytes (LPL), as described.Citation35 For experiments requiring detection of intracellular antigens, cell suspensions were cultured in the presence of Golgistop (5 μL/mL; BD), phorbol myristate acetate (50 ng/mL; Sigma) and ionomycin (1mg/mL Sigma). Cells were then permeabilized and fixed using an intracellular staining kit (eBioscience). The following antibodies were used: CD4+ (RM4-5, eBioscience), CD8α (53–6.72, Biolegend), CD45R/B220 (RA3-6B2, BD PharMingen), CD16/CD32 (2.4G2, eBioscience), CD107a (1D4B, Biolgend), IL-17A (TC11-18H10,Biolegend), Foxp3 (FJK-16s, eBioscience), RORγt (Q31-378, BD PharMingen), IFNγ (XMG1.2, BD PharMingen), γδ-TCR (eBioGL3, eBioscience).
Lymphocyte depletion and functional blockade studies
Anti-CD4+ (GK1.5, BioXCell) and anti-CD8+ (53–6.72, BioXCell) were given intraperitoneally to APCmin/+ mice (0.2mg, three times per week for 3 weeks) to deplete CD4+ and CD8+ T lymphocytes, respectively. Anti-mouse CD25 (7D4, BioXCell) was injected ip (0.2 mg, three times per week for 3 weeks). Depletion efficiencies were ≥90% and ≥80% for anti-CD4+ and anti-CD25 antibodies, respectively. For RORγt inhibition, SR1001 was administered essentially as described.Citation36 Briefly, animals were treated with 25 mg/kg SR1001 twice per day ip for 3 weeks.Citation36 All treatments were initiated 1 week after bacterial inoculations.
Histology
MLN, colon and tumor tissues were harvested from mice, embedded in Tissue-Plus Optimal Cutting Temperature (OCT) Compound (Fisher HealthCare, Houston, TX, USA) and snap-frozen in liquid nitrogen. Serial cryosections (14 μm) were prepared with a Cryostar NX70, Thermo Scientific cryostat at −19 °C (Kalamazoo, MI, USA). Cryosections were kept at room temperature for at least 24 h before staining. As described previously, immunostaining protocol was used with modifications.Citation37 For analysis of CD8+ T-cell CD107a expression, staining antibodies were added sequentially in the following order: CD8α Alexafluor 647 (BD PharMingen) and CD107a (Biolegend). CD8α diluted with 2% fetal calf serum (FCS) in 1X PBS pH 7.4 to 1:5 and incubated at 37 °C for 40 min. Sections were then blocked with 1X PBS that contained 10% Goat serum, 1% BSA, 0.3M glycine and 0.1% Tween 20 for 1 h at 37 °C. A 1:20 dilution of CD107a was added and incubated at 4 °C overnight. Sections were washed with 1X PBS-T and processed for imaging. For analysis of colon sections, staining antibodies were added sequentially in the following order: CD4+ PE (eBioscience), CD11b Alexa 488 (eBioscience), and CD8α Alexafluor 647 (BD PharMingen). Antibodies were diluted with 2% fetal calf serum (FCS) in 1X PBS pH 7.4 to 1:5 for CD4+, 1:10 for CD11b and 1:5 for CD8α. Each antibody was sequentially incubated at 37 °C for 40 min. Images were captured using a Leica SP5 confocal laser scanning microscope (Leica, Wetzlar, Germany) and processed using Fiji Software.Citation38 Panels containing confocal images were generated using Adobe Photoshop version 13.0 × 32. Images were marked using the drawing tools to highlight the results and to provide orientation of the tissues.
Statistical analysis
Student's t-test was used to determine the significance of the differences between control and experimental groups in pairwise comparisons. In experiments with multiple groups homogeneity of inter-group variance was analyzed by one-way ANOVA. Log-rank test was used for analysis of survival studies. p values of 0.05 or less were considered statistically significant.
Disclosure of potential conflicts of interest
N.K.E has ownership interest in TherapyX, Inc., which is developing PIN particle technology for commercial application. The remaining authors declare no competing financial interests.
KONI_A_1319027_supplemental_data.zip
Download Zip (223.8 KB)Funding
The work was supported by the US National Institutes of Health (AI092133 and CA100656 to N.K.E) and the University at Albany and Wadsworth Center, New York State Department of Health start-up funds (M.D.J.).
Related Research Data
References
- Wang K, Karin M. Tumor-elicited inflammation and colorectal cancer. Adv Cancer Res 2015; 128:173-96; PMID:26216633; https://doi.org/https://doi.org/10.1016/bs.acr.2015.04.014
- Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012; 491:254-8; PMID:23034650; https://doi.org/https://doi.org/10.1038/mi.2016.53
- Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, Harrison O, Powrie F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med 2013; 210:917-31; PMID:23589566; https://doi.org/https://doi.org/10.1084/jem.20122308
- Wang K, Kim MK, Di Caro G, Wong J, Shalapour S, Wan J, Zhang W, Zhong Z, Sanchez-Lopez E, Wu LW et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity 2014; 41:1052-63; PMID:25526314; https://doi.org/https://doi.org/10.1016/j.immuni.2014.11.009
- Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 2009; 15:1016-22; PMID:19701202; https://doi.org/https://doi.org/10.1038/nm.2015
- He D, Li H, Yusuf N, Elmets CA, Li J, Mountz JD, Xu H. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J Immunol 2010; 184:2281-8; PMID:20118280; https://doi.org/https://doi.org/10.4049/jimmunol.0902574
- Thiele Orberg E, Fan H, Tam AJ, Dejea CM, Destefano Shields CE, Wu S, Chung L, Finard BB, Wu X, Fathi P et al. The myeloid immune signature of enterotoxigenic Bacteroides fragilis-induced murine colon tumorigenesis. Mucosal Immunol 2016; 10(2):421-33; PMID:27301879; https://doi.org/https://doi.org/10.1038/mi.2016.53
- Shouval DS, Biswas A, Goettel JA, McCann K, Conaway E, Redhu NS, Mascanfroni ID, Al Adham Z, Lavoie S, Ibourk M et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity 2014; 40:706-19; PMID:24792912; https://doi.org/https://doi.org/10.1016/j.immuni.2014.03.011
- Zigmond E, Bernshtein B, Friedlander G, Walker CR, Yona S, Kim KW, Brenner O, Krauthgamer R, Varol C, Muller W et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity 2014; 40:720-33; PMID:24792913; https://doi.org/https://doi.org/10.1016/j.immuni.2014.03.012
- Huber S, Gagliani N, Esplugues E, O'Connor W, Jr., Huber FJ, Chaudhry A, Kamanaka M, Kobayashi Y, Booth CJ, Rudensky AY et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3(−) and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 2011; 34:554-65; PMID:21511184; https://doi.org/https://doi.org/10.1016/j.immuni.2011.01.020
- Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, Jack RS, Wunderlich FT, Bruning JC, Muller W et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 2011; 34:566-78; PMID:21511185; https://doi.org/https://doi.org/10.1016/j.immuni.2011.03.018
- O'Garra A, Barrat FJ, Castro AG, Vicari A, Hawrylowicz C. Strategies for use of IL-10 or its antagonists in human disease. Immunol Rev 2008; 223:114-31; PMID:18613832; https://doi.org/https://doi.org/10.1111/j.1600-065X.2008.00635.x
- Marlow GJ, van Gent D, Ferguson LR. Why interleukin-10 supplementation does not work in Crohn's disease patients. World J Gastroenterol 2013; 19:3931-41; PMID:23840137; https://doi.org/https://doi.org/10.3748/wjg.v19.i25.3931
- Erdman SE, Rao VP, Poutahidis T, Ihrig MM, Ge Z, Feng Y, Tomczak M, Rogers AB, Horwitz BH, Fox JG. CD4(+)CD25(+) regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res 2003; 63:6042-50; PMID:14522933
- Erdman SE, Sohn JJ, Rao VP, Nambiar PR, Ge Z, Fox JG, Schauer DB. CD4+CD25+ regulatory lymphocytes induce regression of intestinal tumors in ApcMin/+ mice. Cancer Res 2005; 65:3998-4004; PMID:15899788; https://doi.org/https://doi.org/10.1158/0008-5472.CAN-04-3104
- Chung AY, Li Q, Blair SJ, De Jesus M, Dennis KL, LeVea C, Yao J, Sun Y, Conway TF, Virtuoso LP et al. Oral interleukin-10 alleviates polyposis via neutralization of pathogenic T-regulatory cells. Cancer Res 2014; 74:5377-85; PMID:25228656; https://doi.org/https://doi.org/10.1158/0008-5472.CAN-14-0918
- Ozbilge H, LeVea C, Chung AY, Li Q, Egilmez NK. Modulating gut immunity and neoplasia with oral cytokine adjuvants. Oncoimmunology 2015; 4:e1002724; PMID:26137401; https://doi.org/https://doi.org/10.1080/2162402X.2014.1002724
- Sabat R, Grutz G, Warszawska K, Kirsch S, Witte E, Wolk K, Geginat J. Biology of interleukin-10. Cytokine Growth Factor Rev 2010; 21:331-44; PMID:21115385; https://doi.org/https://doi.org/10.1016/j.cytogfr.2010.09.002
- Housseau F, Wu S, Wick EC, Fan H, Wu X, Llosa NJ, Smith KN, Tam A, Ganguly S, Wanyiri JW et al. Redundant innate and adaptive sources of IL17 production drive colon tumorigenesis. Cancer Res 2016; 76:2115-24; PMID:26880802; https://doi.org/https://doi.org/10.1158/0008-5472.CAN-15-0749
- Geis AL, Fan H, Wu X, Wu S, Huso DL, Wolfe JL, Sears CL, Pardoll DM, Housseau F. Regulatory T-cell response to enterotoxigenic Bacteroides fragilis colonization triggers IL17-dependent colon carcinogenesis. Cancer Discov 2015; 5:1098-109; PMID:26201900; https://doi.org/https://doi.org/10.1158/2159-8290.CD-15-0447
- Rojas RE, Balaji KN, Subramanian A, Boom WH. Regulation of human CD4(+) alphabeta T-cell-receptor-positive (TCR(+)) and gammadelta TCR(+) T-cell responses to Mycobacterium tuberculosis by interleukin-10 and transforming growth factor beta. Infect Immun 1999; 67:6461-72; PMID:10569764
- Zhou W, Zhou X, Gaowa S, Meng Q, Zhan Z, Liu J, Li J, Fan H, Liu Z. The critical role of induced CD4+ FoxP3+ regulatory cells in suppression of interleukin-17 production and attenuation of mouse orthotopic lung allograft rejection. Transplantation 2015; 99:1356-64; PMID:25856405; https://doi.org/https://doi.org/10.1097/TP.0000000000000526
- Tao J, Kamanaka M, Hao J, Hao Z, Jiang X, Craft JE, Flavell RA, Wu Z, Hong Z, Zhao L et al. IL-10 signaling in CD4+ T cells is critical for the pathogenesis of collagen-induced arthritis. Arthritis Res Ther 2011; 13:R212; PMID:22192790; https://doi.org/https://doi.org/10.1186/ar3545
- Chan IH, Wu V, Bilardello M, Jorgenson B, Bal H, McCauley S, Van Vlasselaer P, Mumm JB. PEG-rIL-10 treatment decreases FoxP3(+) Tregs despite upregulation of intratumoral IDO. Oncoimmunology 2016; 5:e1197458; PMID:27622052; https://doi.org/https://doi.org/10.1080/2162402X.2016.1197458
- Loiseau C, Requena M, Mavigner M, Cazabat M, Carrere N, Suc B, Barange K, Alric L, Marchou B, Massip P et al. CCR6(−) regulatory T cells blunt the restoration of gut Th17 cells along the CCR6-CCL20 axis in treated HIV-1-infected individuals. Mucosal Immunol 2016; 9:1137-50; PMID:26883727; https://doi.org/https://doi.org/10.1038/mi.2016.7
- Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol 2014; 14:667-85; PMID:25234148; https://doi.org/https://doi.org/10.1038/nri3738
- Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, Blaisdell S, Basham B, Dai J, Grein J et al. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell 2011; 20:781-96; https://doi.org/https://doi.org/10.1016/j.ccr.2011.11.003
- Emmerich J, Mumm JB, Chan IH, LaFace D, Truong H, McClanahan T, Gorman DM, Oft M. IL-10 directly activates and expands tumor-resident CD8(+) T cells without de novo infiltration from secondary lymphoid organs. Cancer Res 2012; 72:3570-81; PMID:22581824; https://doi.org/https://doi.org/10.1158/0008-5472.CAN-12-0721
- Chan IH, Wu V, Bilardello M, Mar E, Oft M, Van Vlasselaer P, Mumm JB. The potentiation of IFN-gamma and induction of cytotoxic proteins by pegylated IL-10 in human CD8 T cells. J Interferon Cytokine Res 2015; 35:948-55; PMID:26309093; https://doi.org/https://doi.org/10.1089/jir.2014.0221
- Woo JI, Kil SH, Oh S, Lee YJ, Park R, Lim DJ, Moon SK. IL-10/HMOX1 signaling modulates cochlear inflammation via negative regulation of MCP-1/CCL2 expression in cochlear fibrocytes. J Immunol 2015; 194:3953-61; PMID:25780042; https://doi.org/https://doi.org/10.4049/jimmunol.1402751
- Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov 2015; 5:43-51; PMID:25358689; https://doi.org/https://doi.org/10.1158/2159-8290.CD-14-0863
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015; 372:2509-20; PMID:26028255; https://doi.org/https://doi.org/10.1056/NEJMoa1500596
- Egilmez NK, Jong YS, Mathiowitz E, Bankert RB. Tumor vaccination with cytokine-encapsulated microspheres. Methods Mol Med 2003; 75:687-96; PMID:12407772
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004; 118:229-41; PMID:15260992; https://doi.org/https://doi.org/10.1016/j.cell.2004.07.002
- Weigmann B, Tubbe I, Seidel D, Nicolaev A, Becker C, Neurath MF. Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat Protoc 2007; 2:2307-11; PMID:17947970; https://doi.org/https://doi.org/10.1038/nprot.2007.315
- Solt LA, Kumar N, Nuhant P, Wang Y, Lauer JL, Liu J, Istrate MA, Kamenecka TM, Roush WR, Vidovic D et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 2011; 472:491-4; PMID:21499262; https://doi.org/https://doi.org/10.1038/nature10075
- De Jesus M, Ahlawat S, Mantis NJ. Isolating and immunostaining lymphocytes and dendritic cells from murine Peyer's patches. J Vis Exp 2013(73):e50167; PMID:23525039; https://doi.org/https://doi.org/10.3791/50167
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012; 9:676-82; PMID:22743772; https://doi.org/https://doi.org/10.1038/nmeth.2019