
ABSTRACT
Tolerance toward tumor antigens, which are shared by normal tissues, have often limited the efficacy of cancer vaccines. However, wild type epitopes can be tweaked to activate cross-reactive T-cell clones, resulting in antitumor activity. The design of these analogs (i.e., heteroclitic peptides) can be difficult and time-consuming since no automated in silico tools are available. Hereby we describe the development of an in silico framework to improve the selection of heteroclitic peptides. The Epitope Discovery and Improvement System (EDIS) was first validated by studying the model antigen SIINFEKL. Based on artificial neural network (ANN) predictions, we selected two mutant analogs that are characterized by an increased MHC-I binding affinity (SIINFAKL) or increased TCR stimulation (SIIWFEKL). Therapeutic vaccination using optimized peptides resulted in enhanced antitumor activity and against B16.OVA melanomas in vivo. The translational potential of the EDIS platform was further demonstrated by studying the melanoma-associated antigen tyrosinase related protein 2 (TRP2). Following therapeutic immunization with the EDIS-derived epitope SVYDFFAWL, a significant reduction in the growth of established B16.F10 tumors was observed, suggesting a break in the tolerance toward the wild type epitope. Finally, we tested a multi vaccine approach, demonstrating that combination of wild type and mutant epitopes targeting both TRP2 and OVA antigens increases the antitumor response.
In conclusion, by taking advantage of available prediction servers and molecular dynamics simulations, we generated an innovative platform for studying the initial sequences and selecting lead candidates with improved immunological features. Taken together, EDIS is the first automated algorithm-driven platform to speed up the design of heteroclitic peptides that can be publicly queried online.
Introduction
Immunotherapy is widely recognized for its potential and several studies have demonstrated that tumor-reactive T-cells are present among the naive repertoire. For this reason, peptide vaccination using cytotoxic T-lymphocyte (CTL) epitopes has been evaluated in different preclinical and clinical studies.Citation1-3
Peptide-based cancer vaccines represent a focused approach that can take into account the inter-patient variability of the neoplastic disease. Nevertheless, finding a candidate target might not be sufficient for a successful therapy.Citation4 While neo-antigens represent the optimal target for cancer immunotherapy, their discovery is not applicable to all clinical settings due to economic and technological limitations. Thus, different classes of tumor antigens have been investigated, such as tumor-associated antigens (TAAs). These proteins are not exclusively found in tumor tissues, hence they can only be targeted by specific T-cells with low-affinity T-cell receptors (TCRs) survive the thymic selection.Citation5 These potential self-reactive clones are kept inactivated by the peripheral tolerance.Citation6 In addition, the tumor cells are known to downregulate the major histocompatibility complex (MHC) molecules to evade immune surveillance.Citation7 The above-mentioned scenario makes TAAs a difficult target for cancer immunotherapy, however, TAAs still represents the largest class of tumor antigens availableCitation8 and they are used into preclinical and clinical applications despite their non-optimal nature as specific targets.Citation9
T-cells are by nature cross-reactive and one specific clone can recognize other highly similar sequences.Citation10-12 In fact, if T-cells were monospecific, an enormous number of lymphocytes would be needed to confer protection against foreign antigens. Mathematical modeling has shed light on the redundancy of this system,Citation13 suggesting that one T-cell might recognize as much as 106 minimal epitopes.Citation14 One of the consequences of this model is that one peptide might be recognized by multiple clones, especially if they feature low-avidity TCRs. This hypothesis is supported by previous studies where modified epitopes (i.e., heteroclitic peptides) were found to be more immunogenic than the original ones. In fact, the presence of amino acid changes in proteins of malignant cells can create epitopes that are able to drive antitumor responses.Citation15 Chen et al. generated mutated forms of the NY-ESO-1 peptide, demonstrating their immunological efficacy.Citation16 Similarly, mutated MHC class-II peptides from the gp100 antigen have been generated and their relative efficacy was studied in humans. The results showed that even minor changes in the sequence of the peptides led to variable responses.Citation17 Recently, Hoppes and colleagues replaced natural amino acids with non-proteinogenic residues and generated improved variants of the SVYDFFVWL peptide derived from the tyrosinase-related protein 2 (TRP2) antigen.Citation18
Although the exact mechanism is not completely understood, the current knowledge suggests that the analogs may have an increased affinity for human leukocyte antigen (HLA) molecules. However, it is not clear whether heteroclitic peptides act by providing a more potent stimulus to a tolerized epitope-specific T-cell, or if they are able to stimulate other clones, such as low avidity ones, that had not been tolerized.
In this study, we describe the development and validation of the Epitope Discovery and Improvement System (EDIS). The advantage of this approach relies on the integration of predictions using several ANN in combination with molecular dynamics simulations (MDSs). While developed to optimize the sequence of tumor associated antigens, overcoming tolerance, this framework could also be used to improve the sequence of peptides.
By studying a classic model peptide, the SIINFEKL epitope from chicken ovalbumin (OVA), we investigated in silico, in vitro and in vivo properties of the two analogs SIINFAKL and SIIWFEKL. In particular, we predicted and modeled their interaction with the murine allele H-2Kb to understand how the mutations affect the binding with MHC-I. Then we evaluated both their therapeutic efficacy upon established B16-OVA melanomas and the immunological response.
While being an optimal strategy, targeting neo-antigens is not always possible, therefore the use of TAAs is widespread into the clinical setting. To mimic this scenario, we decided to study the murine syngeneic tumor antigen TRP2. Interestingly, immunological responses against the TRP2 antigen have been evaluated in different studies on melanoma patients.Citation19 In fact, TRP2-specific CTL clones have been identified among tumor infiltrating lymphocytes.Citation20 In addition, Reynolds and colleagues detected TRP2 reactive T-cells in the peripheral blood of patients.Citation21 Therefore, we decided to study the tumor epitope SVYDFFVWL (TRP2180–188) from this TAA. Among the two heteroclitic peptides that we evaluated, one was able to reduce the growth of established B16F10 tumors more efficiently than the wild type TRP2180–188 epitope. Finally, we demonstrated that targeting TAAs, neo-antigens combination of wild type peptides and their mutated versions results in an increased antitumor efficacy.
In conclusion, a fine equilibrium between the mutation of epitopes and immunological properties needs to be considered when selecting heteroclitic peptides for cancer vaccines. Hereby, we show that the integration of multiple in silico platform can improve the accuracy of the prediction of peptide properties allowing for a more efficient screening and selection of CTL epitope-analogs.
Results
Developing an in-silico platform that predicts the effect of amino acid changes on the immunogenicity of MHC-I epitopes
Prediction servers offer the possibility to estimate several immunological properties of putative MHC-I epitopes. These technologies allow for the screening of sequences obtained from mass-spectrometry analysis of proteins to search for peptides suitable for cancer immunotherapy. This versatile and scalable platform, however, has never been used for the methodic research of heteroclitic peptides.
We started by generating an in silico library of analogs of SIINFEKL, a frequently used model antigen in immunological studies. To this end, each position was mutated with each natural amino acid. Then we run the library into two different prediction servers. The NetMHC 4.0 Server from the Center for Biological Sequence analysis (CBS) was used to predict the binding affinity (IC50 values) of peptides to the murine MHC-I allele H-2Kb.Citation22,23 In addition, the class I immunogenicity prediction server available at the Immune Epitope Database and Analysis Resource (IEDB)Citation24 was used to predict the recognition of the peptide-MHC-I (pMHC-I) complexes by TCRs and immunogenicity scores (IS) were acquired for each analog.
As shown in , the lateral chains of the SIINFEKL peptide (blue) have a specific orientation into space. While residues 1, 2, 3, 5 and 8 are sunk into the MHC-I binding pocket, residues 4, 6 and 7 extrude from the pocket, hence can be recognized by the T-cells. Therefore, we hypothesized that mutations in MHC-I anchors would significantly change the IC50 values, while replacing residues 4, 6 and 7 would not hinder the MHC-I binding affinity. As represented in , all analogs sharing a mutation at the first (XIINFEKL; blue) or second (SXINFEKL; red) position, have a much higher IC50 (i.e., lower affinity). The group of analogs sharing a mutation at the eight residue (dark blue), known to be an important binding anchor,Citation25 feature the highest IC50 mean value, highlighting the importance of such residue in the binding of SIINFEKL peptide to the MHC-I molecule. Consistent with our hypothesis, no significant changes to the IC50 were observed for the analogs sharing a mutation at position 4 (SIIXFEKL; purple) and 7 (SIINFEXL; brown). Interestingly, most of the substitution at position 6 (SIINFXKL; gray) would result in a lower IC50 (i.e., higher affinity). By analyzing the effect of each amino acid as substitute of the native one (Suppl. 1A) we could conclude that, amino acids with big lateral chains, such as glutamic acid (E), aspartic acid (D) or lysine (K) would cause a reduced affinity, no matter which position was used for modifying the native sequence. Regarding the recognition of the epitope by the TCR, the immunogenicity score (IS) did not vary when the first three positions (; 1, 2 and 3 on x axis) were changed, while aa variations at position 4, 5, 6 and 7 had significant effects on the IS. In fact, most of the analogs with a substitution at position 6 (SIINFXKL, gray) showed a lower score, compared with the group of analogs with changes in position 7 (SIINFEXL); variations in position 4 gave mixed results as proven by the wider distribution of the data ().
Figure 1. In silico screening of the mutational library of SIINFEKL. (A) Graphical representation of SIINFEKL peptide (violet) into the binding pocket of the H-2Kb MHC-I molecule (transparent gray). The backbone of the peptide is visualized in green and the lateral groups in blue. Lateral groups facing the MHC-I binding pocket or emerging from the pocket are indicated. In silico prediction of the binding affinity (B) or the immunogenicity score (C) of the analogs of SIIINFEKL. The analogs were grouped according to the position of the mutation (left panels). Alternatively, the analogs were grouped according to the aminoacid used to mutate each position (right panels). The red-dotted line, represents the IC50 and the immunogenicity score of the wild type sequence. (D) The affinity scores of the whole library were plotted and indicating analogs with a higher or lower affinity than the wild type SIINFEKL. The specific peptides SIINFAKL and SIIWFEKL are highlighted and their IC50 values are indicated. (E) The immunogenicity predictions for the H-2Kb allele are displayed for the wild type SIINFEKL and two specific analogs.
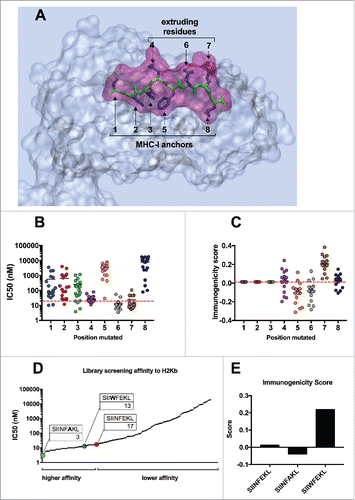
Taken together, these data suggested that the 4th position was not crucial in determining the IC50, but might be very important for the recognition of the TCR. Similarly, changes in position 6 might result in an improved MCH-I affinity, but a generally lower immunogenicity.
To elucidate what is the contribution of each parameter to the immunological response we further evaluated two analogs with different properties. By plotting the IC50 values of all the analogs () we could observe a group of analogs with a higher affinity than SIINFEKL (IC50 < 17 nM). Among these, the SIINFAKL peptide was predicted to bind the MHC-I with the highest affinity, with an IC50 of 3 nM whereas the SIIWFEKL analog also resulted in a lower IC50 value (13 nM) compared with SIINFEKL. As previously discussed, the analog bearing a change in the 6th position (SIINFAKL) showed a lower IS () while the analog with a mutation at the 4th position (SIIWFEKL) showed an improved IS.
Crystal structures of pMHC complexes deliver the most reliable data regarding the conformation and orientation of peptides into the binding pockets. However, resolved structures are not available for most of the pMHC complexes. Hence, we used MDSs to compare the conformation of peptides when inside the MHC binding pocket. As shown in , SIINFAKL (left panel, red solid) is shifted to the bottom of the binding pocket compared with the wild type SIINFEKL (green transparent). This explains the increased predicted binding affinity of this peptide for the H-2Kb molecule. On the contrary, SIIWFEKL () features a large side chain due to the tryptophan in position 4 (4TRP). This allows for a higher extrusion of the peptide from the MHC pocket when compared with either SIINFEKL (central panel, green transparent) or SIINFAKL (right panel, red solid).
Figure 2. Molecular dynamics simulation to unravel conformational changes of epitope-MHC-I complexes. (A) Comparison of spatial conformation epitopes inside the MHC-I binding pocket. SIINFEKL (green transparent), SIINFAKL (red solid) and SIIWFEKL (cyan solid) are compared by superimposition. Molecular dynamics simulations were run for 300 ns, and the most representative states are shown. Left panel: SIINFEKL and SIINFAKL; central panel: SIINFEKL and SIIWFEKL; right panel SIINFAKL and SIIWFEKL. The conformational landscape of the epitope-MHC-I complexes (SIINFEKL, B; SIINFAKL, C; SIIWFEKL, D) are shown from two angles: C-terminal of epitope (left panels) and side views (right panels; epitopes are oriented from N-terminal on the left to C-terminal on the right). The extruding residues, responsible for contacting T-cell receptors are highlighted by yellow captions showing the position and abbreviation for the aminoacid. The structure and orientation of the mutated residue for SIINFAKL and SIIWFEKL peptides are highlighted by the yellow structure of the sidechains.
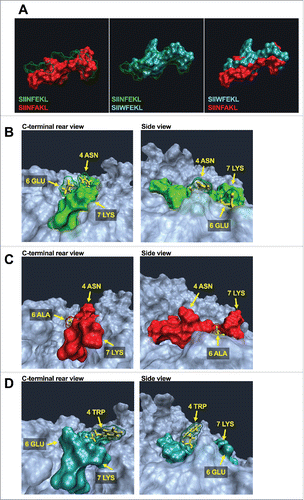
The TCR recognizes the pMHC by binding for one third the peptide and for two thirds the MHC-I. Hence, gathering information about the single residues of the epitope which are extruding from the binding pocket might be poorly predictive. Therefore, we decided to study how the whole portion of pMHC that faces TCR (hereafter referred as the pMHC landscape) changes conformation when using different analogs of the same epitope. The wild type epitope SIINFEKL () features an asparagine in position 4 (4ASN), a glutamic acid in position 6 (6GLU) and a lysine in position 7 (7LYS) which extrude from the binding pocket. The analog SIINFAKL features a smaller alanine in position 6 and this mutation affects the orientation of the other residues as well. As shown in , the whole binding pocket seems to have a more opened conformation; in addition, when comparing the 4ASN and 7LYS present in both the SIINFEKL and SIINFAKL peptides, they seem to extrude more in the latter one. In contrast, the insertion of the tryptophan in the position 4 largely affects the whole pMHC landscape. shows the peptide SIIWFEKL into the binding pocket. The large side chain of the 4TRP residue is evidently extruding from the binding pocket, with the MHC pocket being more closed, thus limiting the TCR access to the peptide ().
In silico properties predicted by EDIS correlate with experimental in vitro data
In our in silico screening both SIINFAKL and SIIWFEKL analogs are predicted to have an improved MHC-I binding affinity, with the latter expected to be better recognized by the TCR. To validate these findings, we performed an in vitro binding assay using RMA-S cells. These cells bear mutations in the antigen-processing machinery, hence they are almost devoid of surface H-2Kb molecules.Citation26 However, the addition of exogenous epitopes can stabilize the α chains and the β2 macroglobulin of the MHC-I molecule, thus the binding affinity is proportional to the amount of H-2Kb on the membrane. We observed a significant increase in the amount of H-2Kb on cells incubated with SIINFAKL and SIIWFEKL at both high () and low concentrations () when compared with the wild type SIINFEKL. This experimentally confirms that the analog peptides do have a higher affinity for MHC-I compared with the native epitope.
Figure 3. Experimental validation of in silico predictions of the MHC-I binding affinity. RMA-S cells were pre-incubated for 1 h at 4°C. Then, 4 × 106 cells were incubated for 2 h with one of the indicated peptides at two different concentrations (A) 1 µg /mL or (B) 0.1 µg/mL in a volume of 1 mL. The presence of H-2Kb molecules on the membrane was measured by flow cytometry and normalized against cells incubated with no peptide (negative control). (C) OT-I splenocytes were labeled with CFSE dye. Then, 3 × 104 cells were incubated with different stimuli for 72 h in a volume of 200 µL of complete media. Then the percentage of proliferating (i.e., CFSE diluted) CD3+CD8+ T-cells was determined by flow cytometry; data was normalized against positive control Concanavalin A (defined as 100% of proliferation). All the data are represented as mean ± SD; Significance was assessed by One-way ANOVA with Tukey's post hoc test; *p < 0.05, ** p <0.01.
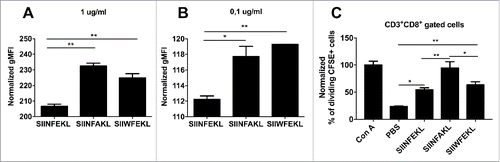
Next, we sought to investigate whether or not the selected analogs would increase the proliferation of T-cells. To this end, CFSE labeled OT-I splenocytes, containing exclusively SIINFEKL-specific CD8+ T-cells, were incubated with the three peptides. After 3 d of incubation, we determined the amount of proliferating CD3+CD8+ T-cells (i.e., with a diluted CSFE fluorescence) by flow cytometry. As expected both the SIINFEKL and the SIIWFEKL peptides were able to stimulate the proliferation of the cells significantly more than the negative control. Interestingly, incubation with SIINFAKL resulted in a significantly increased proliferation of OT-I T-cells compared with the other peptides. This finding confirms the presence of cross-reactivity and the ability of the mutant analogs to stimulate the population of lymphocytes recognizing the native epitope, which is a crucial property for any heteroclitic peptide.
Analogs selected with the EDIS framework show improved antitumor activity in vivo compared with the native epitope
We then investigated whether the two mutant analogs would be immunologically active in a therapeutic cancer vaccine approach. To this end, we treated 9-d established B16-OVA tumors with the three peptides using the previously described cancer vaccine platform PeptiCRAd.Citation27
In these settings, the therapeutic intra-tumor vaccination with SIINFEKL resulted in a slightly reduced tumor growth () compared with mice treated with saline solution or the adjuvant alone. In contrast, a superior antitumor efficacy was achieved by therapeutic vaccination with either SIIWFEKL (p < 0.05) or SIINFAKL (p < 0.01) as can be appreciated also by the single tumor curves (Fig. S2A) and by the increased doubling time of tumors treated with these peptides (Fig. S2B).
Figure 4. Antitumor response and in vivo efficacy of SIINFEKL mutated analogs. (A) B16OVA cells were injected into both flanks of female C57BL6/J mice (2.5 × 105 cells/flanks). Mice (eight per group) were treated with PBS (Mock), Ad5D24-CpG human adenovirus (Adjuvant), and either SIINFEKL, SIINFAKL or SIIWFEKL peptides complexed to the virus-adjuvant. Intra-tumor injections were made on day 9, 11, 13 as represented by the asterisks on the x axis. Tumor volumes were measured every 2–3 d by a digital caliper. Tumor volumes normalized against the values on the 9th day are presented as the mean ± SEM; significance was calculated by Two-way ANOVA with Tukey's post hoc test. Spleens, tumors and draining lymph nodes were collected to determine to percentage of (B) total CD8+ T-cells (CD19−CD3+CD8+) or (C) SIINFEKL-specific T-cells (double positive Pentamer+CD8+ gated on CD19−CD3+). (D) A correlation analysis was performed by plotting for each mouse the % of antigen-specific T-cells against the intensity of the signal (gMFI) of such population. Data from spleens (empty triangles), draining lymph nodes (empty squares) and tumors (black dots) are presented. Log transformed values were used to compute the Pearson's correlation coefficiencies; p-value for the correlation is 0.0002.
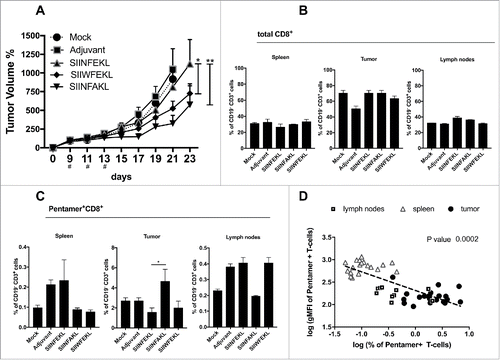
Flow cytometry analysis revealed no major differences in the number of CD19−CD3+CD8+ CTLs in secondary lymphoid organs or tumors (). However, a significant increase in the amount of SIINFEKL-specific CTLs (i.e., antitumor T-cells) was observed in the tumors of mice treated with the SIINFAKL peptide (). This suggested a beneficial cross-response and the expansion of SIINFEKL-specific cells that infiltrated the tumors.
Following the hypothesis that clones with low-avidity TCRs might be crucial in the antitumor responses avoiding tolerance, we performed an inter-organ correlation analysis among all the groups by comparing the percentage of SIINFEKL-specific T-cells with their avidity for the pMHC-I complexes. The avidity of T-cells for the pMHC-I complexes was measured by considering the affinity of TCRs for SIINFEKL-Pentamers, since high avidity clones would capture more Pentamers and have stronger fluorescence signal, i.e., a higher geometrical mean fluorescence intensity (gMFI). Interestingly, we observed a clear negative correlation (): larger populations of SIINFEKL-specific CTLs showed also the lowest fluorescence intensity of staining (Pearson's correlation analysis; p = 0.0002). These data suggests that low-avidity T-cell clones are attracted into the tumor () in larger numbers and this mechanism could be exploited in different settings to circumvent the tolerance when targeting different tumor antigens such as TAAs.
Taken together these findings demonstrate that therapeutic vaccination with heteroclitic peptides significantly improves immune response resulting in a reduced growth of aggressive B16OVA tumors. In addition, Pentamer staining suggested that SIINFEKL-specific T-cell clones can be cross-stimulated by using the SIINFAKL mutant analog. Finally, the avidity of stimulated CTL clones seems to negatively correlate with their expansion/proliferation.
The heteroclitic analogs SIINFAKL and SIIWFEKL can cross-stimulate a response against the native epitope SIINFEKL
To achieve antitumor activity, the clones stimulated by immunization using heteroclitic peptides must recognize the native epitope which is displayed by the tumor cells. For this reason, an analysis of cross-reactivity of the immune response was performed by IFNγ ELISPOT assay on samples collected from animals described in the previous experiment.
As expected, splenocytes from mice treated with SIINFEKL reacted ex vivo to SIINFEKL (), with two mice showing a hyper-response (>500 SFU/well). A certain degree of cross-response toward the native SIINFEKL was observed in mice treated with SIIWFEKL also (). Mice immunized with SIINFAKL also cross-responded to SIIWFEKL peptide (). Consistently to our previous observations, splenocytes of mice treated with SIINFAKL cross-responded to SIINFEKL peptide () and this explains its antitumor efficacy.
Figure 5. Cross-reactivity of the immunological responses elicited by the mutated analogs of SIINFEKL. Splenocytes from mice treated with SIINFEKL, SIINFAKL or SIIWFEKL (presented in , here indicated on the x axis), were assayed for cross-responses to the peptides. 3 × 105 cells were incubated with 100 ng of each indicated peptide for 72 h and the IFNγ response was determined by ELISPOT assay. (A) The number of Spot-Forming Units (SFU) is normalized to 1 × 106 cells. The red-dotted line represents the background of the assay (splenocytes incubated with PBS). (B) The cumulative size of the spots from mice of the same group (same column) in response to different stimuli (bars of different colors) is shown. Data is shown as mean ± SEM. C) The average spot size across all the groups in response to each peptide is represented. Data is shown as the mean ± SEM; One-way ANOVA with Tukey's post hoc test was used to calculate the statistical significance.
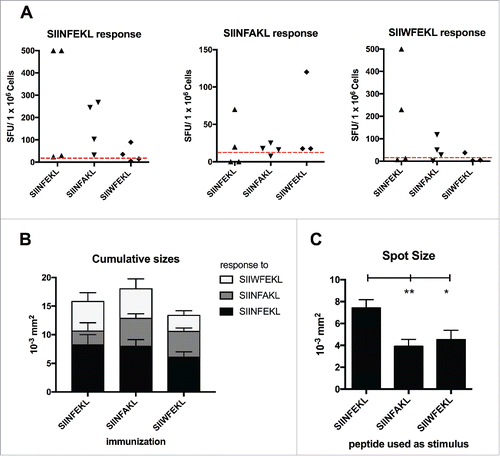
Next, we analyzed the size of the spots as an indirect measure of the amount of IFNγ produced by the single activated T-cells in response to the different MHC-I epitopes. Splenocytes of mice immunized with SIINFAKL produced large spots when re-pulsed with all the peptides, demonstrating a broad response (). Nevertheless, as shown in , when using SIINFEKL to re-stimulate the splenocytes (independently from the initial immunization) we observed the largest spots, suggesting that the efficacy of SIINFAKL is not achieved by hyper-stimulation of the TCR.
An heteroclitic peptide designed with EDIS was able to break the tolerance toward B16F10 melanomas
One of the main challenge that cancer vaccines strategies face is the paucity of antigens which are exclusively present on tumor cells. Melanoma-associated antigen TRP2 features epitopes that can bind with high affinity to the MHC-I; however, either peripheral tolerance or absence of T-cell clones with TRP2-high avidity TCRs prevents rejection of tumors upon therapeutic vaccination with the SVYDFFVWL epitope.
First, we studied the properties of the wild type epitope in silico. As described for SIINFEKL, we changed each position with one of the other natural amino acids, and grouped the analogs based on the mutated position (for example group 1: XVYDFFVWL; group 2: SXYDFFVWL; group 3: SVXDFFVWL; etc.). This provided useful insights in the contribution of each position to the stability of this specific pMHC-I complex and its immunogenicity. As shown in , most of the analogs with a mutation in one of the first three positions display an inferior affinity toward H-2Kb (i.e., higher IC50) compared with the WT peptide (represented by a dashed line). According to the predictions, mutations at either the 7th or 8th residue result in increase of binding affinity, while changes at the 9th position results in decreased binding in 100% of the cases, as shown by a higher IC50 of all these peptides (SVYDFFVWX group; 9th on x axis). Interestingly, position 4 seems to not affect the overall binding. The central residues are essential for TCR recognition, and this is proved by the overall lower predicted immunogenicity for most of the analogs sharing a mutation in these positions when compared with the natural epitope (). Only few analogs display a higher immunogenicity score than TRP2 WT (dashed line). As shown also in Fig. S1D (red dots), only 12 analogs from a total of 162 were predicted to have a better immunogenicity. This observation suggested us that the sequence may be already optimized for TCR recognition and that poor TCR stimulation might be caused by the tolerance toward this peptide. Therefore, we focused on contacting other low-affinity TCR that survived the thymic selection. For this reason, we choose two analogs with an increased predicted affinity for H-2Kb but avoiding mutations in the central region: SAYDFFVWL (TRP2 2A) and SVYDFFAWL (TRP2 7A). These analogs displayed a lower IC50 () and were predicted to be recognized as efficiently as the TRP2 WT epitope (). Consistent with the in silico analysis, both analogs stabilized the MHC-I molecules on the surface of RMA-S cells at both low and high concentrations ().
Figure 6. In silico study of the mutational library of the epitope TRP2180–188. The mutational library of the epitope SVYDFFVWL (TRP2 WT) was screened in silico for MHC-I binding affinity (A) or immunogenicity (B) for the allele H-2Kb. The analogs were grouped according to the position of the mutation (left panels) or according to the aminoacid used to mutate each position (right panels). C) The IC50 (left) and Immunogenicity score (right) of the analogs SVYDFFAWL (TRP2 7A) and SAYDFFVWL (TRP2 2A) predicted in silico are represented. (D) RMA-S cells were pre-incubated for 1 h at 4°C. Then, 4 × 106 cells were incubated for 2 h with one of the indicated peptides at two different concentrations 0.1 µg/mL (left) or 1 µg/mL (right) in a volume of 1 mL. The presence of H-2Kb molecules on the membrane was measured by flow cytometry and normalized against cells incubated with no peptide (negative control). Data is represented as the mean ± SEM; Unpaired student's t-test, *p < 0.05, **p < 0.01. E) Comparison of spatial conformation epitopes inside the MHC-I binding pocket: TRP2 WT (orange), TRP2 7A (green) and TRP2 2A (purple). Molecular dynamics simulations were run for 300 ns, and the most representative states are shown. ¾ view (left panel) and side view (right panel). The mutated residues are colored in red. (F) The conformational landscapes of the epitope-MHC-I complexes are shown from peptide C-terminal perspective. The extruding residues, responsible for contacting T-cell receptors are highlighted by yellow captions showing the position and abbreviation for the aminoacid. The α chain of the MHC is represented in transparent white.
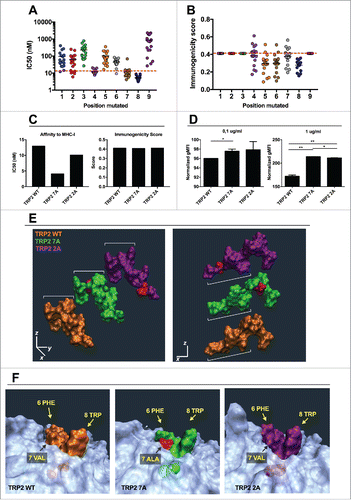
Molecular dynamics simulation of these analogs revealed that the mutations would change the conformation of the peptides (). The central residues extruding from the MHC-I molecule have a different orientation when comparing TRP2 WT, TRP2 7A and TRP2 2A peptides (). In addition, the portion of the peptide which is in deep contact with the MHC-I pocket () has a different conformation, especially when considering the TRP2 2A peptide. These spatial changes result into differences of the pMHC structure which is supposed to be in contact with the TCR (). For instance, the presence of the small alanine in position 7 of the TRP2 7A analog, affects the spatial orientation of phenylalanine and tryptophan at positions 6 and 8, respectively (). No major spatial changes were observed for the pMHC complex when we simulated the presence of the TRP2 2A peptide into the binding pocket of the H-2Kb molecule when compared with the TRP2 WT peptide ().
Next, we used the aggressive melanoma model B16F10 to test whether the selected variants would elicit an antitumor response. As expected, immunization with TRP2 WT did not affect the tumor growth (). Similarly, treatment with TRP2 2A did not result in any significant change in tumor progression. Interestingly, mice immunized with TRP2 7A peptide showed a significant slower tumor growth when compared with mock-treated mice (p = 0.0015), or mice treated with either TRP2 WT (p < 0.0001) or TRP2 2A (p < 0.0001).
Figure 7. Breaking the tolerance to TRP2 tumor antigen and evaluation of multivaccine therapy. (A) B16-F10 melanoma cells were injected into both flanks of female C57BL6/J mice (1.5 × 105 cells/flank). Mice (eight per group) were treated with PBS (Mock), Ad5D24-CpG human adenovirus (Adjuvant), and either TRP2 WT, TRP2 7A or TRP2 2A peptides complexed to the virus-adjuvant. Intra-tumor injections were made on day 9, 11 and 13 (asterisks on the x axis). Tumor volumes normalized against the values on the 9th day and are presented as the mean ± SEM (data from two independent experiments); significance was calculated by Two-way ANOVA with Tukey's post hoc test. (B) Splenocytes were collected from mice and they were assayed for cross-responses to the peptides. 4 × 105 cells were incubated with 100 ng of indicated stimuli for 72 h and the IFNγ response was determined by ELISPOT assay. The number of Spot-Forming Units (SFU) is normalized to 1 × 106 cells. The red-dotted line represents the background of the assay (splenocytes incubated with PBS). Data is presented as mean ± SEM; Significance was calculated by using One-way ANOVA with Tukey's post hoc test; *p < 0.05,**p < 0.01, ***p < 0.001. (C) B16OVA bearing mice were treated on day 10, 12 and 14 with PBS (mock), a combination of TRP2 WT and 7A peptides, a combination of SIINFEKL and SIINFAKL peptides and a multitherapy featuring all four different epitopes together. Tumor volumes normalized against the values on the 10th day. A threshold of 400% of initial volume on day 10 was chosen to distinguish between fast (solid line) and slow (dotted line) progressing tumors. Percentages of slow progressing tumors are indicated near the graph of each group.
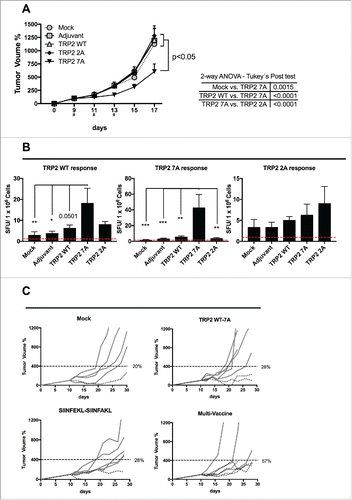
Subsequently, we studied the immunological responses in treated mice. As shown in (left panel), treatment with native peptide (i.e., TRP2 WT) weakly increased the immune response toward the antigen compared with mock-treated mice, confirming a degree of tolerance toward this sequence. Immunization with the TRP2 2A peptide did not result in any major response. Interestingly, mice immunized with TRP 7A showed a significantly higher cross-response against the TRP2 WT epitope.
Taken together these data demonstrated that TRP2 7A was able to break the tolerance toward the WT peptide. In addition, we also observed a high direct-response against TRP2 7A in mice treated with TRP2 7A (). This suggested the absence of tolerance toward this peptide.
Finally, we investigated the efficacy of a multi-peptide vaccine, composed of wild type and modified epitopes targeting TAAs and neo-antigens. In particular, we asked whether or not targeting both classes of antigens would be beneficial, when using heteroclitic peptides and wild type epitopes. Therefore, we chose a challenging model by treating 10-d established B16.OVA tumors with either TRP2 WT + 7A analog peptides, SIINFEKL + SIINFAKL peptides or a combination of all four peptides (multi-vaccine). The study groups that received the therapeutic vaccines showed varying degrees of response (Fig. S3A). As shown by the individual tumor growth curves in , the percentage of slowly progressing tumors was higher in the group that received the multi vaccine therapy. In fact, 57% of mice in the multi-peptide vaccine group had tumor volumes that were below 400% of the starting volume. The analysis of the growth of the tumors (by using the area under the curve) revealed that all the study groups performed significantly better than mock (Fig. S3B). This observation suggests that a multi-targeted approach that takes into account clone variability and downregulation of specific antigens may be more effective when compared with narrow treatments targeting single antigens.
Discussion
Tumor antigens can be generally divided into tumor-specific antigens (TSAs) or TAAs. TSAs comprise antigens that are exclusively expressed by tumors cells, while TAAs are expressed also in healthy tissue. Identification of unique TSAs is difficult to achieve, hence targeting TAAs represents a more feasible approach. Most of TAAs, such as MAGE-A1Citation28 or TRP-2,Citation29 are self-antigens present in healthy tissues, thus tolerance mechanisms prevent immune responses against them. Mutated forms of wild-type epitopes (i.e., heteroclitic peptides), have proved to be a valuable tool to break the tolerance or hyperstimulate tolerant T-cells.Citation5,16,30 Careful theoretical design together with in silico and in vitro screening is necessary to evaluate the mutated analogs, making the process time-consuming and cumbersome. Previous studies have reported contradictory findings where the correlation between the in vitro and the in vivo observations resulted to be poor.Citation31 Therefore, new approaches to optimize the screening process are needed. In this study we established an in silico framework by using CBS and IEDB prediction servers, which are usually used to find MHC epitopes inside protein sequences.Citation32 Hereby, we propose an innovative use of this tools to screen mutational libraries of defined MHC-I epitopes. In addition, we implemented molecular dynamic simulations in our pipeline, to further validate the initial data.
By grouping the analogs based on the mutated amino acid we evaluated the contribution of each position to the overall stability the pMHC-I–TCR interactions. This visualization of data allows for an easy and fast comparison of the native sequence (SIINFEKL or SVYDFFVWL) with all the analogs. Using this approach, we demonstrated that very few amino acids in very specific positions led to an increased MHC-I affinity (i.e., lower IC50; red dots in Fig. S1A and C), while the insertion of amino acids with large lateral chains (E, F, I or W) resulted into analogs with improved immunogenicity scores (Fig. S1B and C; red dots). Our data highlights that modifications of the TCR-facing residues, such as positions 4 or 6, led to heteroclitic peptides with improved MHC-I affinity. These observations widen the classical model where non-anchor residues would poorly affect the overall IC50.Citation25,33,34 In fact, both SIINFAKL and SIIWFEKL analogs showed an improved affinity for MHC in silico and in vitro. It is worth noticing that pMHC-I complexes can change their structure after binding the TCR. Downstream signals seem to amplify small discrepancies between similar peptides, according to the kinetic proof-reading model of T-cell activation.Citation10 This leads to a cross-reactivity model where one TCR can recognize different peptides and one peptide can be recognized by many TCRs.
An innovative aspect of our work consists in the integration of data from different in silico techniques. However, in our experience data from immunogenicity prediction is much more difficult to interpretate. In fact, we did not observe a strong correlation between the predicted immunogenicity and the degree of proliferation of antigen specific lymphocytes in vitro. This finding suggests that the prediction of the immunogenicity is much more challenging since in silico prediction algorithms do not take into account the poly-clonality of T-cell responses. In fact, in vivo validations demonstrated that both analogs were able to reduce the growth of B16.OVA tumors. Consistent with our in vitro results, the best MHC-I binder (i.e., SIINFAKL) proved to be the most effective in vivo (Fig. S2C). We speculate that peptides with increased MHC-I affinity were able to interact with a wider variety of T-cell clones. This hypothesis is supported by the fact that immunization with SIINFAKL peptide resulted in an increased cross-reactivity. Interestingly, despite not observing any hyper-stimulation of TCRs, we report an increased proliferation of OT-1 cells. We believe that this effect is mediated by the higher affinity of the peptide for the MHC-I, hence a prolonged availability on the surface of APCs. We suggest that the beneficial responses achieved by treatment with heteroclitic peptides may derive from their ability to evoke a poly-clonal response rather than hyper-stimulation of the specific T-cell clone against the native epitope. Messaoudi and colleagues have previously investigated some analog epitopes in their work, such as SIINFAKL, showing that the different pMHC-I conformation has important effects on the recognition of the complex by specific TCR-like antibodies.Citation35 However, they did not investigate the immunological consequences. In lack of crystal structures of analogs into the MHC binding pocket, we performed MDSs. We could appreciate differences into the orientation of the peptides into the binding pocket, and most importantly different conformational changes of the pMHC complexes. Consistently, the SIIWFEKL peptide, which did not show any priming of OT-I T-cells, was the analog showing markedly different pMHC conformation when compared with SIINFEKL. This finding, supports the use of molecular dynamics simulation as a tool to understand immunological properties of heteroclitic peptides.
In this manuscript, we highlight the importance of considering both the MHC-I affinity and TCR recognition during the in silico design of heteroclitic peptides. This is also supported by the work of Romero and colleagues who studied two epitopes from the MART-1/Melan-A antigen. In particular, by placing a strong anchor at position 2 of the natural epitopes, they were able to increase the affinity for HLA-A02. However, while one peptide was also better recognized by specific CTL clones, the other peptide lost the ability to engage TCRs of the same clones.Citation36
Subsequently, we applied this in silico analysis to the antigen TRP2. This self-protein is a melanocyte lineage marker; therefore, a degree of tolerance limits its use as cancer vaccine target. Other groups have previously described mutated forms of the main H-2KbH-2Kb restricted epitope TRP2180–188 (TRP2 WT). While immunological responses were achieved, as proved by the expansion of TRP2 WT-specific T-cells, tumor growth was not affected; this suggests a strong tolerance against this self-peptide.Citation31 By using the EDIS framework we selected two analogs predicted to have an improved MHC-I affinity and a similar immunogenicity score compared with the WT epitope. The 7A and 2A analogs were studied by MDSs and the 7A-MHC complex resulted to be similar to the WT-MHC. In fact, therapeutic vaccination with TRP2 7A led to a reduced tumor growth and an increased cross-response. Targeting TAAs is challenging as most of the TAAs-specific T-cells recognize sub-dominant epitopes, while very few T-cell clones recognize the dominant epitopes. In addition, these clones are characterized by a lower affinity for the pMHC complex and they might be kept in a tolerogenic state.Citation4 Our findings demonstrate that heteroclitic peptides target low-affinity T-cell clones through cross-stimulation of their TCRs.
Finally, we investigated whether combining wild type and mutated forms of the same epitope could increase the efficacy of therapeutic vaccination. To this end we directly compared TRP2 WT + 7A therapy with SIINFEKL + SIINFAKL therapy; in addition, we also tested a combination of all four peptides. Responses could be observed in all the treatment groups, however, by targeting both TRP2 and SIINFEKL antigens with combination therapy (Multi-vaccine group) we achieved a response in a higher percentage of treated animals. This suggests that targeting both TAAs and neo-antigens within the same therapeutic window, may increase the efficacy of cancer vaccines. This represents a convenient approach to limit immune evasion due to antigen loss and maximize the efficacy of the treatment.
In conclusion, we demonstrated that combination of the available prediction servers and MDSs provides useful insights into different aspects of the MHC-I-epitope–TCRs interactions. Considering the reciprocal influence between MHC-I affinity and TCR recognition of any given epitope is becoming increasingly important. In particular, our study provides an open innovative framework that can be used by other researchers as starting point for an improved design of mutant epitopes with higher clinical efficacy. The in silico platform in its version 1.0 can be freely used by following the link https://github.com/aniketsh/EDIS_platform.git.
Materials and methods
In silico analysis of heteroclitic peptides
The wild type sequence of the epitope was substituted in each position with all other natural amino acids. The virtual library of analogs was then run into ANN based prediction servers to estimate the MHC-I binding affinity (NetMHC 4.0 Server from the Center for Biological Sequence analysisCitation22,23) and the class I immunogenicity (Immune Epitope Database and Analysis ResourceCitation24). The results were ranked according to MHC-I binding affinity and binders with an IC50 lower than the one displayed by the wild type epitope were considered for further studies. Data on class I immunogenicity was taken into account when considering peptides with similar MHC-I affinity. Lead-candidates were then studied by molecular dynamics simulation by simulating their interaction with the MHC-I binding pocket (in the α-chain) for 300 ns. The most represented structure was considered the most stable and chosen as the most predictive conformation of the peptide-MHC-I complex.
Cell lines and peptide reagents
The mouse melanoma cell line B16-F10 was purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). The cell line B16.OVA,Citation37 a mouse melanoma cell line expressing chicken OVA, was kindly provided by Prof. Richard Vile (Mayo Clinic, Rochester, MN, USA). RMA-S cell lineCitation38 was a kind gift of Prof. Toos Daemen (University of Groningen, Groningen, Netherlands). All cell lines were cultured under appropriate conditions.
SIINFEKL (OVA257–264), SIINFAKL, SIIWFEKL, SVYDFFVWL (TRP-2180–188), SVYDFFAWL, SAYDFFVWL and all their poly-K extended versions were purchased from Zhejiang Ontores Biotechnologies Co. (Zhejiang, China) at a purity of >90%.
Molecular histocompatbility complex (MHC) class-I stabilization assay
The antigen processing-defective mutant cell line RMA-S express a very limited amount of MHC-I molecules on their surface, and they are devoid of peptides. The addition of exogenous epitopes can stabilize the processing machinery and increase the amount of H-2Kb on the membrane. 2 × 106 RMA-S cells were incubated with indicated peptides at a concentration of 0.1 of 1 µg/µL in 2 mL of total volume. After 1.5 h of incubation at +37°C, cells were washed with PBS and fixed in 4% formalin for 15 min at +4°C. Cells were then washed again with PBS and centrifuged at 1,200 rpm for 5 min. The pellet was stained with a PE-labeled anti-H-2Kb antibody (Biolegend, San Diego, CA, USA). Cells were washed two times and suspended in 400 µL of PBS. The amount of H-2Kb on the membrane was quantified by a Gallios flow cytometer (Beckman Coulter). gMFI was normalized to the negative control (RMA-S cells incubated without peptide).
Carboxyfluorescein succinimidyl ester (CFSE) proliferation assay with OT-I splenocytes
The CD8+ T-cells of C57BL/6-Tg(TcraTcrb)1100Mjb/Crl (OT-I) mice (Charles River) have a T-cell receptor specific for the OVA-derived class I epitope SIINFEKL.Citation39 Spleens were collected from OT-I mice and were gently disrupted through 70 µM cell strainers and single cell suspensions were frozen in 10% DMSO at −80°C. On the day of the assay, splenocytes were thawed, washed and resuspended in PBS. Then, splenocytes were added with CFSE (Sigma-Aldrich) diluted 1:100 in a volume of 1.1 mL of PBS for 10 min at +37°C. Labeled cells were washed two times in 10 mL of PBS and then resuspended in complete media. CFSE-labeled OT-I splenocytes were incubated for 72 h with indicated stimuli (13 µM for peptides and 0.1 µg /µL for Concanavalin A) in 230 µL in 96 multiwell plates (V-bottom shaped). Samples were washed and incubated with Fc Block (Biolegend) and then stained with APC-labeled anti-CD8+ and PE-labeled anti-CD3 antibodies. Cells were washed two times, resuspended in 400 µL and analyzed by flow cytometry.
Animal experiments and immunizations
C57BL/6J (H-2Kb and H-2Db) mice have been obtained from Scanbur (Denmark) at 4–6 weeks of age. Mice have been kept in air-isolated cages with unlimited access to food. All procedures have been carried in a level 2 biosafety cabinet under sterile conditions. Mice have been anesthetized using isofluorane vaporizers. Subcutaneous tumor models have been made by injecting 2.5 × 105 B16.OVA or 1.5 × 105 B16-F10 tumor cells (when 80% confluent in T175 flasks) on the flanks of each mouse in 100 µL of non-supplemented RPMI-1640 media. When the tumors reached an average diameter of 3–5 mm (usually 9–10 d) treatments were initiated after blind randomization of mice. Details about each treatment schedule are fully given in the figure legends. All animal experiments were reviewed and approved by the Experimental Animal Committee of the University of Helsinki and the Provincial Government of Southern Finland.
For therapeutic immunization, we used immunogenic oncolytic human adenoviruses serotype 5 (unable to replicate in murine tumors) as adjuvants for enhanced toll-like receptor stimulation.Citation27,40,41 Briefly, MHC-I epitope sequences were elongated at the N-terminus with additional lysines to increase their net charge. This modification does not hinder the cross-presentation of the epitopes.Citation27 Then 20 µg of peptide was mixed with 1 × 109 viral particles of negatively charged adenoviruses to allow the formation of virus-peptide complexes. Then the peptide-virus complex was administered intra-tumorally in 50 µL of natrium chloride solution.
Enzyme linked immunospot (ELISPOT) IFNγ assay
Spleens have been collected from mice, disrupted and passed through a 70 μm cell strainer. They were stored in −80°C in 10% DMSO. On the day of the assay, splenocytes were quickly thawed at +37°C. Samples were transferred into falcon tubes and 8 mL of complete media was slowly added. Cells were centrifuged to eliminate residual DMSO and resuspended in serum free CTL media supplemented with 1% L-glutamine and benzonase (50 U/mL). Samples were counted and viability was assayed by Trypan-Blue staining. Samples viability ranged from 80% up to 95%. Splenocytes were plated at a concentration of 2.5 × 105 or 3 × 105 cells/well (refer to figure legends). Cells have been stimulated with 50 or 200 ng of peptides of interest in 200 µL of final volume. PBS + 1% DMSO and Concanavalin A have been used as negative and positive controls respectively.
Stimulation was performed for 72 h in incubators (37°C, 5% CO2). Afterwards, the plates have been processed according to manufacturer's instructions (CTL Immunospot, Bonn, Germany). The analysis of the plates (digital images production and spot evaluation) has been outsourced to CTL Immunospot.
Statistical analysis
Statistical significance was determined using GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA). A detailed description of the statistical methods used to analyze the data from each experiment can be found in each figure legend.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1319028_supplementary_data.zip
Download Zip (2.1 MB)Acknowledgments
We would like to thank all our colleagues that helped us in performing the experiments and the interpretation of data. In addition, we would like to thank Prof. Toos Daemen for kindly providing the RMA-S cell line.
Funding
This study was supported by NOVO A/S (Novo Nordisk) and the European Research Council (ERC) consolidator grant.
References
- Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich PY, Mendrzyk R et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med 2012 Aug; 18(8):1254–61; PMID: 22842478; https://doi.org/10.1038/nm.2883
- Vacchelli E, Martins I, Eggermont A, Fridman WH, Galon J, Sautès-Fridman C, Tartour E, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: peptide vaccines in cancer therapy. Oncoimmunology 2012; 1(9):1557-76; PMID: 23264902; https://doi.org/10.4161/onci.22428
- Chianese-Bullock KA, Pressley J, Garbee C, Hibbitts S, Murphy C, Yamshchikov G, Petroni GR, Bissonette EA, Neese PY, Grosh WW et al. MAGE-A1-, MAGE-A10-, and gp100-derived peptides are immunogenic when combined with granulocyte-macrophage colony-stimulating factor and montanide ISA-51 adjuvant and administered as part of a multipeptide vaccine for melanoma. J Immunol 2005; 174(5):3080-6; PMID: 15728523; https://doi.org/10.4049/jimmunol.174.5.3080
- Menez-Jamet J, Gallou C, Rougeot A, Kosmatopoulos K. Optimized tumor cryptic peptides: the basis for universal neo-antigen-like tumor vaccines. Ann Transl Med 2016; 4(14):266; PMID: 27563653; https://doi.org/10.21037/atm.2016.05.15
- Hebeisen M, Allard M, Gannon PO, Schmidt J, Speiser DE, Rufer N. Identifying individual T Cell receptors of optimal avidity for tumor antigens. Front Immunol 2015; 6:582; PMID: 26635796; https://doi.org/10.3389/fimmu.2015.00582
- Daniel C, Nolting J, von Boehmer H. Mechanisms of self-nonself discrimination and possible clinical relevance. Immunotherapy 2009; 1(4):631-44; PMID: 20582233; https://doi.org/10.2217/imt.09.29
- Yutoku M, Fuji H, Grossberg AL, Pressman D. An experimental model for evaluation of factors in tumor escape from immunological attack. Cancer Res 1975; 35(3):734-9; PMID: 1116132
- Bright RK, Bright JD, Byrne JA. Overexpressed oncogenic tumor-self antigens. Hum Vaccin Immunother 2014; 10(11):3297-305; PMID: 25483660; https://doi.org/10.4161/hv.29475
- Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 2012; 12(4):269-81; PMID: 22437939; https://doi.org/10.1038/nri3191
- Regner M. Cross-reactivity in T-cell antigen recognition. Immunol Cell Biol 2001; 79(2):91-100; PMID: 11349650; https://doi.org/10.1046/j.1440-1711.2001.00994.x
- Chen HD, Fraire AE, Joris I, Brehm MA, Welsh RM, Selin LK. Memory CD8+ T cells in heterologous antiviral immunity and immunopathology in the lung. Nat Immunol 2001; 2(11):1067-76; PMID: 11668342; https://doi.org/10.1038/ni727
- Selin LK, Cornberg M, Brehm MA, Kim SK, Calcagno C, Ghersi D, Puzone R, Celada F, Welsh RM. CD8 memory T cells: cross-reactivity and heterologous immunity. Semin Immunol 2004; 16(5):335-47; PMID: 15528078; https://doi.org/10.1016/j.smim.2004.08.014
- Zarnitsyna VI, Evavold BD, Schoettle LN, Blattman JN, Antia R. Estimating the diversity, completeness, and cross-reactivity of the T cell repertoire. Front Immunol 2013; 4:485; PMID: 24421780; https://doi.org/10.3389/fimmu.2013.00485
- Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today 1998; 19(9):395-404; PMID: 9745202; https://doi.org/10.1016/S0167-5699(98)01299-7
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015; 520(7549):692-6; PMID: 25901682; https://doi.org/10.1038/nature14426
- Chen JL, Dunbar PR, Gileadi U, Jäger E, Gnjatic S, Nagata Y, Stockert E, Panicali DL, Chen YT, Knuth A et al. Identification of NY-ESO-1 peptide analogues capable of improved stimulation of tumor-reactive CTL. J Immunol 2000; 165(2):948-55; PMID: 10878370; https://doi.org/10.4049/jimmunol.165.2.948
- Chen S, Li Y, Depontieu FR, McMiller TL, English AM, Shabanowitz J, Kos F, Sidney J, Sette A, Rosenberg SA et al. Structure-based design of altered MHC Class II-restricted peptide ligands with heterogeneous immunogenicity. J Immunol 2013 Nov 15; 191(10), 5097-106; PMID:24108701; https://doi.org/10.4049/jimmunol.1300467
- Hoppes R, Oostvogels R, Luimstra JJ, Wals K, Toebes M, Bies L, Ekkebus R, Rijal P, Celie PH, Huang JH et al. Altered peptide ligands revisited: vaccine design through chemically modified HLA-A2-restricted T cell epitopes. J Immunol 2014; 193(10):4803-13; PMID: 25311806; https://doi.org/10.4049/jimmunol.1400800
- Boasberg PD, Hoon DS, Piro LD, Martin MA, Fujimoto A, Kristedja TS, Bhachu S, Ye X, Deck RR, O'Day SJ. Enhanced survival associated with vitiligo expression during maintenance biotherapy for metastatic melanoma. J Invest Dermatol 2006; 126(12):2658-63; PMID: 16946711; https://doi.org/10.1038/sj.jid.5700545
- Wang RF, Appella E, Kawakami Y, Kang X, Rosenberg SA. Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J Exp Med 1996; 184(6):2207-16; PMID: 8976176; https://doi.org/10.1084/jem.184.6.2207
- Reynolds SR, Celis E, Sette A, Oratz R, Shapiro RL, Johnston D, Fotino M, Bystryn JC. HLA-independent heterogeneity of CD8+ T cell responses to MAGE-3, Melan-A/MART-1, gp100, tyrosinase, MC1R, and TRP-2 in vaccine-treated melanoma patients. J Immunol 1998; 161(12):6970-6; PMID: 9862732
- Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med 2016; 8(1):33; PMID: 27029192; https://doi.org/10.1186/s13073-016-0288-x
- Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 2009; 61(1):1-13; PMID: 19002680; https://doi.org/10.1007/s00251-008-0341-z
- Calis JJ, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD, Sette A, Keşmir C, Peters B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol 2013; 9(10):e1003266; PMID: 24204222; https://doi.org/10.1371/journal.pcbi.1003266
- Rammensee HG, Falk K, Rotzschke O. Peptides naturally presented by MHC Class-I molecules. Annu Rev Immunol 1993; 11:213-44; PMID: 8476560; https://doi.org/10.1146/annurev.iy.11.040193.001241
- Esquivel F, Yewdell J, Bennink J. RMA/S cells present endogenously synthesized cytosolic proteins to class I-restricted cytotoxic T lymphocytes. J Exp Med 1992; 175(1):163-8; PMID: 1309852; https://doi.org/10.1084/jem.175.1.163
- Capasso C, Hirvinen M, Garofalo M, Romaniuk D, Kuryk L, Sarvela T, Vitale A, Antopolsky M, Magarkar A, Viitala T et al. Oncolytic adenoviruses coated with MHC-I tumor epitopes increase the antitumor immunity and efficacy against melanoma. Oncoimmunology 2016; 5(4):e1105429; PMID: 27141389; https://doi.org/10.1080/2162402X.2015.1105429
- Novellino L, Castelli C, Parmiani G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother 2005; 54(3):187-207; PMID: 15309328; https://doi.org/10.1007/s00262-004-0560-6
- Wang R-F, Appella E, Kawakami Y, Kang X, Rosenberg SA. Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J Exp Med 1996; 184(6):2207-16; PMID: 8976176; https://doi.org/10.1084/jem.184.6.2207
- Dyall R, Bowne WB, Weber LW, LeMaoult J, Szabo P, Moroi Y, Piskun G, Lewis JJ, Houghton AN, Nikolić-Zugić J. Heteroclitic immunization induces tumor immunity. J Exp Med 1998; 188(9):1553-61; PMID: 9802967; https://doi.org/10.1084/jem.188.9.1553
- McWilliams JA, McGurran SM, Dow SW, Slansky JE, Kedl RM. A modified tyrosinase-related protein 2 epitope generates high-affinity tumor-specific T cells but does not mediate therapeutic efficacy in an intradermal tumor model. J Immunol 2006; 177(1):155-61; PMID: 16785510; https://doi.org/10.4049/jimmunol.177.1.155
- Brusic V, Bajic VB, Petrovsky N. Computational methods for prediction of T-cell epitopes—a framework for modelling, testing, and applications. Methods 2004; 34(4):436-43; PMID: 15542369; https://doi.org/10.1016/j.ymeth.2004.06.006
- Cole DK, Edwards ES, Wynn KK, Clement M, Miles JJ, Ladell K, Ekeruche J, Gostick E, Adams KJ, Skowera A et al. Modification of MHC anchor residues generates heteroclitic peptides that alter TCR binding and T cell recognition. J Immunol 2010; 185(4):2600-10; PMID: 20639478; https://doi.org/10.4049/jimmunol.1000629
- Koch CP, Perna AM, Pillong M, Todoroff NK, Wrede P, Folkers G, Hiss JA, Schneider G. Scrutinizing MHC-I binding peptides and their limits of variation. PLoS Comput Biol 2013; 9(6):e1003088; PMID: 23754940; https://doi.org/10.1371/journal.pcbi.1003088
- Messaoudi I, LeMaoult J, Nikolic-Zugic J. The mode of ligand recognition by two peptide: MHC class I-specific monoclonal antibodies. J Immunol 1999; 163(6):3286-94; PMID: 10477598
- Romero P, Valmori D, Pittet MJ, Zippelius A, Rimoldi D, Lévy F, Dutoit V, Ayyoub M, Rubio-Godoy V, Michielin O et al. Antigenicity and immunogenicity of Melan-A/MART-1 derived peptides as targets for tumor reactive CTL in human melanoma. Immunol Rev 2002; 188:81-96; PMID: 12445283; https://doi.org/10.1034/j.1600-065X.2002.18808.x
- Moore MW, Carbone FR, Bevan MJ. Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell 1988; 54(6):777-85; PMID: 3261634; https://doi.org/10.1016/S0092-8674(88)91043-4
- De Bruijn ML, Schumacher TN, Nieland JD, Ploegh HL, Kast WM, Melief CJ. Peptide loading of empty major histocompatibility complex molecules on RMA-S cells allows the induction of primary cytotoxic T lymphocyte responses. Eur J Immunol 1991; 21(12):2963-70; PMID: 1660811; https://doi.org/10.1002/eji.1830211210
- Didierlaurent AM, Collignon C, Bourguignon P, Wouters S, Fierens K, Fochesato M, Dendouga N, Langlet C, Malissen B, Lambrecht BN et al. Enhancement of adaptive immunity by the human vaccine adjuvant AS01 depends on activated dendritic cells. J Immunol 2014; 193(4):1920-30; PMID: 25024381; https://doi.org/10.4049/jimmunol.1400948
- Cerullo V, Seiler MP, Mane V, Brunetti-Pierri N, Clarke C, Bertin TK, Rodgers JR, Lee B. Toll-like receptor 9 triggers an innate immune response to helper-dependent adenoviral vectors. Mol Ther 2007; 15(2):378-85; PMID: 17235317; https://doi.org/10.1038/sj.mt.6300031
- Cerullo V, Diaconu I, Romano V, Hirvinen M, Ugolini M, Escutenaire S, Holm SL, Kipar A, Kanerva A, Hemminki A. An oncolytic adenovirus enhanced for toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol Ther 2012; 20(11):2076-86; PMID: 22828500; https://doi.org/10.1038/mt.2012.137