
ABSTRACT
Inflammation and infection are key promoters of colon cancer but the molecular interplay between these events is largely unknown. Mice deficient in leukotriene B4 receptor1 (BLT1) are protected in inflammatory disease models of arthritis, asthma and atherosclerosis. In this study, we show that BLT1−/− mice when bred onto a spontaneous tumor (ApcMin/+) model displayed an increase in the rate of intestinal tumor development and mortality. A paradoxical increase in inflammation in the tumors from the BLT1−/−ApcMin/+ mice is coincidental with defective host response to infection. Germ-free BLT1−/−ApcMin/+ mice are free from colon tumors that reappeared upon fecal transplantation. Analysis of microbiota showed defective host response in BLT1−/− ApcMin/+ mice reshapes the gut microbiota to promote colon tumor development. The BLT1−/−MyD88−/− double deficient mice are susceptible to lethal neonatal infections. Broad-spectrum antibiotic treatment eliminated neonatal lethality in BLT1−/−MyD88−/− mice and the BLT1−/−MyD88−/−ApcMin+ mice are protected from colon tumor development. These results identify a novel interplay between the Toll-like receptor mediated microbial sensing mechanisms and BLT1-mediated host response in the control of colon tumor development.
Introduction
Inflammation and infection are key promoters of cancer but the molecular interplay between these events are unclear.Citation1-4 Recent studies have highlighted the importance of microbiota in modulating immunity, inflammation and cancer.Citation5-7 Dysbiosis-mediated inflammation has been shown to the promote colon cancer progression and various mechanisms that link bacteria to tumor growth are beginning to emerge.Citation6,8-10 Clinical studies outlined an association of pathogenic gut bacteria such as Streptococcus bovisCitation11-15 Helicobacter pyroliCitation16, Bacteroides fragilisCitation17, Escherichia coliCitation18, PrevotellaCitation8 and Fusobacterium sppCitation19 to progression of colorectal cancer (CRC). Adenomatous polyposis coli (APC) is a well-characterized tumor suppressor and mutations in the APC gene are associated with both hereditary and sporadic colon cancers in humans.Citation20 ApcMin/+ mice carrying a germ-line mutation in the APC gene develop multiple polyps in the small intestine and are a widely used model for intestinal cancers.Citation21 It is known that inflammation mediated by Toll-like receptors in a MyD88 dependent manner promotes intestinal tumor development in ApcMin/+ mice.Citation22
Leukotriene B4 (LTB4), a proinflammatory mediator produced by the rapid sequential actions of 5-lipoxygenase and leukotriene A4 hydrolase on arachidonic acid is a potent leukocyte chemoattractant.Citation23 LTB4 mediates its effects through two G-Protein coupled receptors (GPCRs), BLT1 and BLT2.Citation24-26 Absence of BLT1, the high affinity receptor for LTB4, protects mice from developing inflammatory arthritis, airway hyper responsiveness and delays the progression of atherosclerosis.Citation24,26-30 Recently, BLT2 was shown to mediate chemotherapy resistance in many cancer types.Citation31 Since colon cancer is considered to be strongly promoted by inflammation, we crossed the BLT1−/− miceCitation32 onto the ApcMin/+ backgroundCitation21 to examine the role of LTB4/BLT1 axis in the development of intestinal tumors. As with the other chronic inflammation promoted disease models, we anticipated protection from tumor development in BLT1−/−ApcMin/+ mice. However, a paradoxical increase in inflammation and intestinal tumor development in the BLT1−/−ApcMin/+ mice was observed. In a series of experiments, we identified that defective host response in BLT1−/−ApcMin/+ mice translate into altered gut microbiota, increased MyD88 dependent inflammation and enhanced intestinal tumor development. The BLT1−/− mice also displayed enhanced tumor burden in carcinogen (azoxymethane)-induced inflammation (dextran sodium sulfate) promoted colon tumor model. Using germ-free mice, we demonstrated that colon tumor development in the BLT1−/−ApcMin/+ is dependent on gut microbiota.
Methods
Mice
Previously described BLT1 deficient mice (> F9 on C57 BL/6 background)Citation32 were crossed with C57BL/6-ApcMin/+ obtained from the Jackson Laboratory (Bar Harbor, ME) to generate BLT1+/+, BLT+/− and BLT1−/− in ApcMin/+ background. MyD88−/− and MyD88+/− mice in the background of BLT1−/− and BLT1−/−ApcMin/+ mice were also generated using standard breeding protocols. Mice with MyD88 deletion displayed neonatal lethality that was further exacerbated with BLT1 deletion. Breeders harbouring a MyD88−/− allele (MyD88−/−, BLT1−/−MyD88−/−, MyD88−/−ApcMin/+ and BLT1−/−MyD88−/−ApcMin/+) were maintained on Enrofloxacin (Baytril) at 165 mg/L in drinking water that gave a daily dosage of ∼25 mg/kg until the pups are weaned between 21 and 28 d of age. All the experimental mice were maintained on regular autoclaved water and housed in ventilated cages in barrier facility under specific pathogen free conditions at the research resources center of the University of Louisville. All the experimental protocols have been approved by the Institutional Animal Care and Use Committee (IACUC) at University of Louisville.
Genotyping
DNA was extracted from tail-snips of mice using a Direct PCR lysis reagent (Viagen Biotech) according to the manufacturer's instructions. Genotyping PCR for ApcMin/+ was performed according to Jackson Laboratories. The genotyping PCR for BLT1 and MyD88 were performed as described previously.Citation32,33
Survival of mice
Mice (ApcMin/+, BLT1+/−ApcMin/+, BLT1−/−ApcMin/+) were passively followed for long-term survival. Significance of differences in survival was determined by the Mantel-Haenszel/ Log-rank test. The survival rates of the BLT1−/−MyD88−/− mice were calculated using data from several rounds of breeding between BLT1−/−MyD88+/− mice. All pups were genotyped on day 3 and percent survival rates for 3 different genotypes BLT1−/−MyD88+/+, BLT1−/−MyD88+/−, BLT1−/−MyD88−/− were calculated on day 15. Statistical analysis was performed by using the Mann-Whitney U test with Graph Pad Prism 4 software.
Measurement of haematocrits
The retro orbital eye bleeds (200 μL) were collected into heparin coated microvettes (Sarstedt) and analyzed using the Hemavet-950 Haematology System (Drew Scientific) at the indicated ages. Statistical analysis was performed using the Mann-Whitney U test with Graph Pad Prism 4.0 software.
Micro positron emission tomography (PET) imaging
Age matched (∼100 d of age) ApcMin/+, BLT1−/−ApcMin/+ and BLT1+/−ApcMin/+ were used for Micro Positron Emission Tomography (PET) imaging studies. The mice were fasted overnight (16 hours) and then injected 100 µCi of 9-(4-(18)F-Fluoro-3-[hydroxymethyl]butyl)guanine ([18F]FHBG) at 37°C. The images of these mice were collected using microPET R4 small animal and rodent PET scanner for both temporal profile of radioactivity uptake and total activity profile at the end of 1 hour. The “Acquisition Sinogram Image Processing using IDL's VirtualMachine” (ASIPro VM™) was used for image reconstruction and data analysis. We uniformly (Intensity ranging from 0 to 100) reconstructed transverse, coronal and sagittal images from the image file. We measured the maximum, minimum, mean, median and total activity levels in a fixed (25 pixel width x 25 pixel height) Region of Interest (ROI) in all the 3 planes.
Histopathology
Mice were killed at 2 different time points (40 d and 110 days). The entire intestinal tract was removed and flushed with 1X PBS using a blunt-end syringe to remove fecal material. The cleaned intestine divided into 4 parts consisting of the large intestine (including colon and cecum) and 3 equal length sections of the small intestine as proximal, middle and distal parts. Tissues were fixed with 10% neutral formalin and paraffin embedded. The sections were cut at 5 μm thickness and one of the sections used for hematoxylin and eosin (H&E) and others for immunohistochemical analysis.
Enumeration of polyps
The small intestines and colons were dissected and washed with 1X PBS, longitudinally opened and spread on the filter paper and fixed in the 10% formalin for 16 hrs followed by transferring into 70% alcohol. The tumors were counted and sized under stereomicroscope containing reticule eyepiece. Polyps were categorized as <1 mm, 1–2 mm, 2–3 mm and >3 mm size. The mean number of tumors/mouse ± SEM, and the mean tumor diameter (mm) in the group ± SEM were calculated for the small intestine and colon separately. Statistical analysis was performed using the Mann-Whitney U test using Graph Pad Prism 4.0 software. Microadenomas in the intestines of 40 day old mice were counted and sized using Swiss-roll H&E sections under calibrated Aperio imageScope.
Immunohistochemistry (IHC)
BrdU
The proliferative activity of the cells in the polyps was determined by measuring the incorporation of 5 ′-bromo-2 ′-deoxyuridine (BrdU). Briefly, 1 ml of BrdU (1 mg/ml) was given i.p. and mice were killed after 2 hrs and intestines isolated and paraffin embedded for sectioning. IHC for the detection of BrdU was performed using BrdU In Situ-Detection kit from BD Pharmigen™ according to manufacturer's instructions. BrdU-positive nuclei (brown nuclei) cells were manually counted per field at 200x magnification. Statistical analysis was performed using the Mann-Whitney U test.
β-catenin and COX-2 immunohistochemistry
The β-catenin-specific mouse monoclonal antibody (Clone E-5, 1:100) and COX-2 specific Goat polyclonal antibody (clone M-19, 1:50) were used for IHC. After 1 hr incubation with the primary antibody at room temperature, the slides were washed twice with 1X PBS (5 min per wash), and then incubated with the secondary antibody solution (biotinylated goat anti-mouse IgG 1:200 for β-catenin, biotinylated donkey anti-goat IgG 1:200 for COX2) for 30 min at room temperature. Visualization of β-catenin was done by using ABC staining system (Santa Cruz Biotechnology, California). Negative controls for all staining were done by omitting primary antibodies or including isotype controls.
TUNEL
Apoptotic cells in the colon tumors were determined using terminal deoxynucleotidyl transferase-mediated nick end-labeling (TUNEL) assay. The apoptotic cells were detected using In Situ Cell Death Detection Kit, Fluorescein (Roche) according to manufacturer's instructions. The green fluorescence images were captured using Nikon Eclipse TE300 fluorescence microscope. TUNEL-positive cells were manually counted per field at 200 x magnification.
mRNA expression analysis by microarrays and real-time PCR
Total RNA was prepared from small intestine and colon tissues of WT and BLT1−/− mice as well as from size matched tumors of BLT1−/−ApcMin/+ and ApcMin/+ mice (105–110 d age) using TriZol and followed by RNeasy Minikit from Qiagen. The RNA was treated with DNase using Turbo DNase kit, Ambion Inc. Microarray analysis of these samples was performed using whole mouse genome chip (Mouse 430 2.0 array, Affymetrix) according to standard protocols. Experimental and sample preparation variations were standardized by applying the global scaling procedure to all absolute analysis data using constant global target intensity. The data was analyzed using Affymetrix' MAS 5 algorithm for probe set summarization and followed by pair wise comparison. For quantitative real-time PCR, 1 µg of total RNA was reverse transcribed in 50 µl reaction using TaqMan reverse transcription reagents (Applied Biosystems) using random hexamer primers. 2 µl of cDNA and the 1 µM real time PCR primers (Real Time Primers, LLC, Elkins Park, PA) were used in a final 20 µl qPCR reaction with ‘power SYBR-green master mix' (Applied Biosystems). Real time qPCR was performed in ABI-Prism 7900 sequence detect system (Applied Biosytems). Expression of the target genes was normalized to β-actin and displayed as fold change relative to the wild type sample. Data are representative of tumors/tissues isolated from at least 5 different mice for each genotype.
Fluorescence in situ hybridization (FISH)
To detect bacteria in the lungs of BLT1−/−MyD88−/−, the paraffin embedded cross sections of whole lungs were first de-paraffinized and de-hydrated by sequential changes of xylene (3 times) and alcohol (100% 1 time; 95% 1 time; 70% 1 time) for 5 min. The slides were air-dried and incubated with oligonucleotide probe (5′-GCT GCC TCC CGT AGG AGT-3′) complementary to a region of the 16 S rRNA, a highly conserved domain in Bacteria. The probe is labeled with Cy3 fluorophore at 5′ end (Integrated DNA Technologies, CA). The hybridization was performed in the presence of 50 µl of hybridization buffer (100 mM Tris-HCl, pH 8.0, 0.9 M NaCl, 35% formamide) containing Eub338 probe (5 ng/µl). A large coverslip was placed on the slides and carefully pressed until the hybridization solution was evenly distributed over the respective section and incubated for 1 hr in humidified chamber at 46°C. The coverslip was carefully removed and the slides were rinsed with distilled water and incubated with DAPI solution (0.5 µg/ml) for 10 min followed by 5 min 1X PBS wash (3 times) at room temperature. The slides were mounted with aqueous anti-fade mounting media (BioMedia, CA). The fluorescence images were captured using Nikon Eclipse 80i with Texas Red/DAPI emission filter sets.
Dextran sodium sulfate (DSS) induced Colitis
Mice received 3% DSS (36–50 kD; MP Biomedicals) in drinking water ad libitum for the duration of the experiment. The weight of the mouse was measured on Day 0 of the experiment and was used as 100% for calculating weight loss. Mice were killed and blood was removed by intracardiac puncture. The spleen, liver, mesenteric lymph nodes, and the colon including cecum were aseptically removed. The spleen, liver and mesenteric lymph nodes were stored on ice in sterile 1X PBS. The length of the colon was measured. The colon washed with 1X PBS to remove fecal matter and divided into 3 equal parts. The medial colon was opened longitudinally and washed with sterile 1X PBS and blotted to remove excess water. Each tissue was homogenized with an ultra-turrax T8 homogenizer (Ika-Werke) and serially diluted samples were plated on tryptic soy agar plates and allowed to incubate for 24 h. Colonies were counted and total CFUs were determined.
AOM/ DSS -induced colon cancer
Mice were given azoxymethane (AOM) (10 mg/kg) i.p. 7 d before administration of DSS. 2% DSS was given in drinking water in 3 cycles of 7 d on DSS followed by 14 d on normal drinking water with an additional 13 d of drinking water alone in the last cycle. Mice were killed, colons removed and the length measured. Colons were opened longitudinally and examined under a dissecting stereo microscope for tumor counting.
Generation of germ-free mice
Germ free mice (BLT1−/− and BLT1−/−ApcMin/+) were generated and maintained at Taconic, Germantown, NY. The mice at the age of 40 d and 110 d were killed and the polyp numbers counted and compared with specific pathogen free (SPF) animal facility at University of Louisville.
Microbiota analysis
The fecal bacterial DNA was isolated using QiAmp stool DNA isolation kit from wild type (n = 5), BLT1−/− (n = 5), MyD88−/− (n = 5), BLT1−/−MyD8−/− (n = 5), ApcMin/+ (n = 8), BLT1−/−ApcMin/+ (n = 10), MyD88−/−ApcMin/+ (n = 5), BLT1−/−MyD8−/−ApcMin/+ (n = 5) mice. The 16 S rRNA gene V1-V3 regions was amplified using primers, 27f (AGAGTTTGATCCTGGCTCAG) and 534 r (ATTACCGCGGCTGCTGG). These primers were anchored with adapters and Multiplex Identifiers (MIDs) for 454 sequencing to distinguish various samples in a single 454 sequencing reaction. The PCR cycling conditions were 95°C for 5 min, followed by 30 cycles of 94°C for 30 seconds, 56°C for 30 seconds, and 72°C for 1 min and 30 seconds with a final extension period of 8 min at 72°C. The quadruplicate PCR amplicons were purified using AMPure magnetic bead kit and quantified using Quat-iT Picogreen kit. The amplicons were divided into 6 groups, where each group contains 10 individual samples containing unique MIDs incorporated into primer sequences. The pooled amplicons sequenced using 454/Roche GS FLX system or 454Roche Jr according to manufacturer's protocols. The microbiota data analysis was performed using QIIME platform scripts (www.qiime.org). The sequences were rarefied at randomly selected 2000 sequences and further downstream analysis was performed. The microbial classification was performed using GreenGenes reference database (gg_13_8_otus) using QIIME tools (www.qiime.org).Citation34 The sequences reference picked into Operational Taxonomic Units (OTUs) by clustering 97% sequence similarity (uclust) using the GreenGenes reference data set gg_13_8_otus and classified at various taxonomic ranks (phylum, order, class, family, genus, and species). We generated the α-diversity plots (which describe the richness and/or evenness of taxa in a single sample) and the β diversity principle coordinate plots generated using phylogenetic metrics generated using UniFrac distances using QIIME pipeline.Citation35
Semi-quantification of Akkermansia muciniphila by real time PCR
The real time PCR primers targeting variable regions of 16 S rRNA gene of Akkermansia muciniphila (FP: 5′ GCCTCAGCGTCAGTTAATGT 3′, RP: 5′ AGGCTGTTTCGTAAGTCGTG 3′) were synthesized. Universal primers (FP: 5′ TGCAYGGYYGTCSTCAGCTCGTG 3′ RP: 5′ TGACGTCYTCCRCYCCTTCCTC 3′) were used to normalize the levels of total bacteria in the sample. Total fecal DNA (1 ng) was used as template in 20 µl SyBR (Applied Biosystems) reaction mixture and the RT PCR was performed at 50°C for 2 min and then 95°C for 10 min and followed by 40 cycles at 95°C for 10 seconds and 58°C for 45 seconds. The PCR reactions were subjected to heat dissociation protocol present in ABI prism 7900 sequence detection system to confirm the presence of single and unique PCR product. Reaction mixtures without genomic DNA were also used as negative controls to confirm the absence of primer-dimer formation. The relative amount of DNA is compared with control (average of 4 samples of ApcMin/+ fecal DNA) using 2−ΔΔCt methodCitation36 by normalizing with universal primer PCR product in all the experiments. The real time PCR products were purified and sequenced to confirm the authenticity of products belongs to intended bacteria.
Intestinal permeability assay
The gut barrier integrity was assessed by intestinal permeability assay using FITC-dextran (MWav = 4000; FD4). The mice were fasted overnight. The FD4 (80 mg/mL PBS) was administered by oral gavage (600 mg/kg bodyweight) to fasted mice. The plasma was collected after 2 h and the FD4 leakage was measured on a fluorescence plate reader.
Results
Accelerated mortality and tumorigenesis in BLT1−/−ApcMin/+ mice
To examine the function of BLT1 in intestinal tumorigenesis, BLT1−/− mice were crossed to ApcMin/+ mice to generate compound mice. The BLT1−/−ApcMin/+ mice on an identical genetic background and housing conditions displayed a significant increase in mortality relative to ApcMin/+ mice (). Littermate heterozygous mice (BLT1+/−ApcMin/+) showed intermediate survival patterns suggesting a gene dosage dependent effect. The life expectancy of BLT1−/−ApcMin/+ at 102 d was ∼30% less than the 156 d for ApcMin/+ mice. Progression of intestinal tumors is associated with severe anemia in ApcMin/+ mice. Analyses of haematocrit showed that age matched BLT1−/−ApcMin/+ mice were severely anemic relative to ApcMin/+ mice (). Micro Positron Emission Tomography (MicroPET) imaging indicated higher levels of metabolic activity in the intestines of BLT1−/−ApcMin/+ compared with ApcMin/+ mice (Supplementary Fig. 1). Age matched (∼110 days) BLT1−/−ApcMin/+ mice showed significantly increased number of tumors compared with ApcMin/+ mice in the small intestine and the colon ( and Supplementary Figure 2A, B). Most notably, there is a predominance of larger tumors both in small intestines (> 1 mm) and in colons (> 3 mm) of the BLT1−/−ApcMin/+ mice ( and Supplementary Figure 2C). Several large tumors are clearly visible in the colons of BLT1−/−ApcMin/+ mice with many of them nearly occluding the lumen (). Thus, it is evident that tumors are initiated at much higher rate and grew larger in BLT1−/−ApcMin/+ mice. In ApcMin/+ mice intestinal tumors are initiated by the loss of heterozygosity during the first few weeks of life.Citation37 To examine the role of BLT1 at early stage of tumorigenesis, we assessed tumor formation at the age of 40 d in ApcMin/+ and BLT1−/−ApcMin/+. There is detectable anemia, an indirect marker for tumor burden in these mice that is significantly more in BLT1−/−ApcMin/+ mice relative to ApcMin/+ mice (Supplementary Figure 3A). Consistent with this finding, histopathological examination showed the presence of more micro adenomas in the small intestines of BLT1−/−ApcMin/+ mice (Supplementary Figure 3B, D) compared with ApcMin/+ mice. While there were no detectable colon tumors in ApcMin/+ mice at this stage, several colon tumors including some large tumors were observed in BLT1−/−ApcMin/+ mice (Supplementary Figure 3C, E).
Figure 1. Increased mortality and tumorigenesis in BLT1−/−ApcMin+ mice. (A) Kaplan-Meier plot of survival from ApcMin/+ (n = 27) (blue), BLT1+/−ApcMin/+ (n = 29) (green), and BLT1−/−ApcMin/+ mice (n = 29) (red) are shown. Difference in survival of ApcMin/+ mice compared with other 2 strains was determined to be significant by the Mantel-Haenszel/Log-rank test (P< 0.0001). (Mean survival for ApcMin/+ (156 days), BLT1+/−ApcMin/+ (126 days), and BLT1−/−ApcMin/+ (102 days). (B) Severe anemia in BLT1−/−ApcMin/+ mice. Hematocrit values were determined for the indicated mice at the age of ∼110 d. Statistical analysis was performed using Mann-Whitney U test (*** = P < 0.001). (C-F) Increased intestinal tumor burden in BLT1−/−ApcMin/+ mice. Total number of polyps in the small intestine (C) and colon (D) were quantified by stereoscopic microscopy in age matched (100–110 d old) ApcMin/+ (n = 18) and BLT1−/−ApcMin/+ (n = 12) mice using longitudinally opened tissue sections. (E) The size distribution of colon tumors in ApcMin/+ (blue bar) and BLT1−/−ApcMin/+ (red bar) was measured. There was a significant increase in the large sized tumors (> 2 and >3 mm) in BLT1−/−ApcMin/+ when compared with ApcMin/+. Statistical analysis was performed using Mann-Whitney U test. Error bars, ± SEM. *, P < 0.05; **, P < 0.01; and ***, P < 0.001. (F) Representative cross section images of colons stained with Hematoxylin and Eosin (H&E) shows increased number and size of tumors in BLT1−/−ApcMin/+ compared with ApcMin/+. Images were captured using Aperio Image scope, Aperio Technologies Inc.
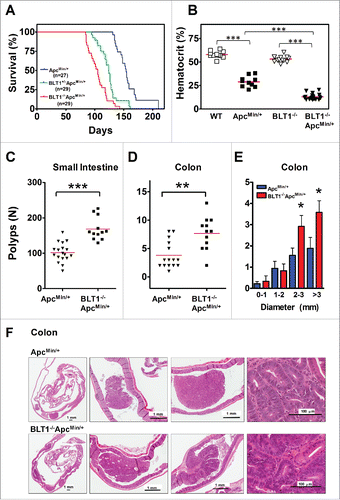
Figure 2. Analysis of β-catenin localization, proliferation and apoptosis in BLT1−/−ApcMin/+ tumors. (A) IHC analysis of β-catenin staining of colon adenomas of ApcMin/+ and BLT1−/−ApcMin/+ mice. The polyps from the BLT1−/−ApcMin/+ mice showing more intense nuclear localization of β-catenin relative to the colon polyps ApcMin/+. (B) 5-bromo-2-deoxyuridine (5-BrdU) injected into mice 2 hrs before sacrificing the mice. The cross section of colons stained with BrdU antibody. Immunohistochemical (IHC) analysis of 5-BrdU-incorporation showed a significant increase in the number of proliferating cells in colonic polyps of BLT1−/−ApcMin/+ mice. (C) Terminal deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling (TUNEL) stain performed on colons of ApcMin/+ and BLT1−/−ApcMin/+ mice. The number of TUNEL positive apoptotic cells are significantly decreased in BLT1−/−ApcMin/+ tumors compared with ApcMin/+ mice. Statistical analysis was performed using Mann-Whitney U test. Error bars, ± SEM and ***, P < 0.001.
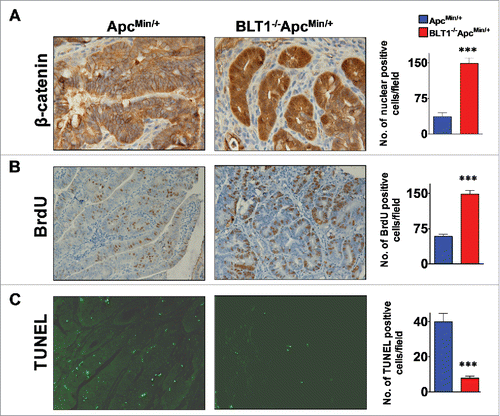
Figure 3. BLT1−/− mice are more susceptible to DSS induced colitis and AOM/DSS induced colon tumorigenesis. Wild type (WT) and BLT1−/− mice received 3% DSS in drinking water ad libitum for the duration of the experiment. (A) The bodyweights of wild type (n = 10) and BLT1−/− (n = 8) mice were measured every 2 d for 2 weeks using weight at Day 0 of the experiment as 100% for calculating weight loss. (B) The mice were killed at day 14 and colon length of BLT1−/− (n = 10) was significantly reduced compared with wild type mice (n = 11). (C) Tissue bacterial loads of DSS treated Wild type (n = 8) and BLT1−/− (n = 8) mice. The spleen, liver, mesenteric lymph nodes, and the colon were aseptically removed and each tissue was then homogenized in sterile 1X PBS and was serially diluted and plated on tryptic soy agar plates and incubated for 24 hrs at 37°C. Colonies were counted and total CFUs were determined based on the total volume of the specimens. (D-E) Increased incidence of azoxymethane (AOM)/DSS induced colon cancer in BLT1−/− mice. The number of polyps in colon increased in BLT1−/− mice treated with AOM/DSS (D), and representative colon images of AOM-DSS treated BLT1−/− and BLT1+/+ mice are shown (E). The red arrows point to visible tumors. Statistical analysis was performed using the Mann-Whitney U test. Error bars, ± SEM. *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
Increased proliferation and decreased apoptosis in BLT1−/−ApcMin/+ tumors
Since colonic tumors appear earlier and progress more rapidly in BLT1−/−ApcMin/+ mice relative to ApcMin/+ mice, it is possible that molecular events required for the initiation and progression are accelerated in these mice. To explore these mechanisms, we determined the nuclear translocation of β-catenin, rates of proliferation and apoptosis in size matched tumors from BLT1−/−ApcMin/+ mice and ApcMin/+ mice. Colon tumors from BLT1−/−ApcMin/+ mice displayed an increase in β-catenin nuclear localization (), an increase in rate of proliferation (increase in BrdU positive cells) () as well as a decrease in apoptosis (decrease in TUNEL positive cells) compared with tumors from ApcMin/+ mice (). These results demonstrate that activities associated with rapid tumor development are increased in the tumors from BLT1−/−ApcMin/+ mice.
Enhanced colitis associated colon tumor development in BLT1−/− mice
Inflammatory bowel diseases (IBDs) such as Crohn's disease and ulcerative colitis are often associated with progression to colon cancer development in humans.Citation38 In mice, treatment with DSS induces colitis, presumably due to direct epithelial damage and ensuing inflammation. To examine the role of BLT1 in colitis, we subjected the BLT1−/− mice to the DSS model. Mice were given 3% DSS in drinking water and monitored for weight loss and survival over a 15 day period. The BLT1−/− mice are more susceptible to DSS-induced colitis and significant loss of body weight compared with WT mice (). The shortening of colon lengths is one of the hallmarks of colitis due to increased inflammation. The colon lengths of DSS-treated BLT1−/− mice were significantly reduced compared with DSS-treated WT mice (). The histopathological examination of colons (distal, medial, and proximal) showed significantly greater of loss of crypts in BLT1−/− mice compared with WT (Supplementary Figure 4). A portion of the colon, the mesenteric lymph nodes (MLN), spleen, and liver from the DSS-treated mice were analyzed for the total tissue bacterial content on tryptic soy agar (TSA) for aerobic and TSA Blood Agar for anaerobic bacteria. The total bacterial loads [colony-forming units (CFUs)] in various organs in DSS-treated mice indicated that BLT1−/− mice has increased systemic bacterial burden compared with WT mice (). DSS-induced colitis strongly promotes AOM-induced colon cancer in mice.Citation4 In AOM-DSS colon tumor model, the BLT1−/− mice showed a significant increase in colon tumor development ( and ). Overall, these studies suggest that absence of BLT1 significantly increased DSS-induced inflammation and promotes carcinogen induced colon tumors.
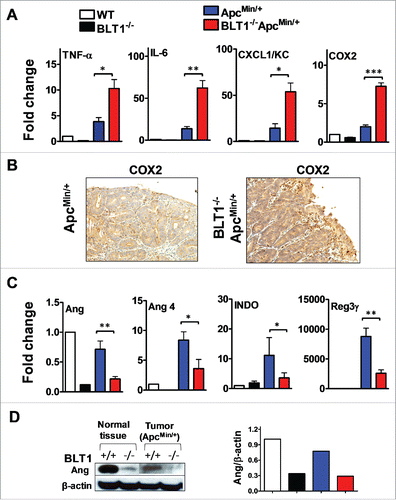
Figure 4. Increased inflammatory and decreased host defense markers in BLT1−/−ApcMin/+ tumors. (A-B). The mRNA levels were measured by qRT-PCR using SyBR green dye (Applied Biosystems) from size matched colon tumors of ApcMin/+ and BLT1−/−ApcMin/+ mice (105–110 d age old). Data are representative of tumors/tissues isolated from at least 5 different mice for each genotype. (A). mRNA levels of inflammatory mediators (TNF-α, IL-6, CXCL1 and COX2) are significantly increased in colonic tumors of BLT1−/−ApcMin/+ compared with ApcMin/+ mice. Statistical analysis was performed using the Mann-Whitney U test. Error bars, ± SEM. *, P < 0.05; **, P < 0.01; and ***, P < 0.001 (compared with ApcMin/+). (B). Immunohistochemical analysis indicates increased expression of COX2 in the colon tumors of BLT1−/−ApcMin/+ compared with ApcMin/+ mice. (C) The relative mRNA levels of host defense proteins (Ang, Ang 4, IDO and Reg3γ) were reduced in BLT1−/−ApcMin/+ compared with ApcMin/+ mice colon tumors. (D) Western blot analysis of angiogenin expression in colons and colon tumors of BLT1−/−ApcMin/+ compared with ApcMin/+.
Defective host-response in tumors from BLT1−/−ApcMin/+ mice
Increased inflammation promotes development of intestinal tumors.Citation1,4,39 Genes involved in promoting inflammation and tissue repair are upregulated in ApcMin/+ tumors in a MyD88 dependent manner.Citation22 Therefore, we measured the mRNA levels of inflammatory markers and positive regulators of tumorigenesis such as tumor necrosis factor-α (TNF-α), interleukin (IL)-6, CXCL1/KC and cyclooxygenase-2 (COX-2) Citation4,40 in size-matched tumors from the colon of BLT1−/−ApcMin/+ and ApcMin/+ (). The data shows that IL-6, CXCL1/KC, TNF-α and COX2 are elevated in tumors from BLT1−/−ApcMin/+ compared with ApcMin/+ mice. The increase in COX2 expression in BLT1−/−ApcMin/+ tumors relative to ApcMin/+ tumors was also observed by IHC ( and Supplementary Figure 5). Increased intestinal tumorigenesis in BLT1−/−ApcMin/+ mice may be directly related to the increased inflammation.
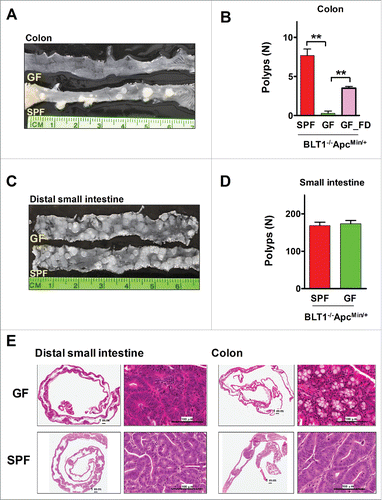
Figure 5. Germ-free BLT1−/−ApcMin/+mice were protected from colon cancer. (A). Gross appearance of longitudinally opened colons from BLT1−/−ApcMin/+mice raised and maintained in specific pathogen free (SPF) and germ free (GF) conditions. (B). BLT1−/−ApcMin/+ mice (n = 12) maintained in germ free facility are highly protected from developing colon tumors compared with SPF facility maintained BLT1−/−ApcMin/+ mice (n = 12). Oral gavage of fecal material from the SPF BLT1−/−ApcMin/+ mice to germ-free BLT1−/−ApcMin/+ mice (GF_FD; n = 8) resulted in colon tumor development in these mice. (C). Longitudinally opened distal small intestines of BLT1−/−ApcMin/+mice maintained specific pathogen free (SPF) or germ free (GF). (D). BLT1−/−ApcMin/+ mice maintained in germ free or SPF facility developed similar number of small intestinal tumors. E. The representative H and E stained swiss roll sections of small intestine (distal) (left panel), colon (right panels) of the BLT1−/−ApcMin/+ mice maintained at GF and SPF facilities. Images were captured using Aperio Image scope.
The increase in inflammation in tumors from BLT1−/−ApcMin/+ raises a critical question; why the loss of a pro-inflammatory mediator (LTB4-BLT1 axis) increased inflammation in this instance, whereas loss of BLT1 reduced inflammation in models of asthma, arthritis and atherosclerosis.Citation26-30 To explore the potential mechanisms, global changes in gene expression profiles in these tumors were determined using microarrays. In addition to increased expression of inflammatory/tumor promoting markers such as IL-6, KC, COX2, etc., the array analysis also revealed a novel pattern of genes that are downregulated in tumors from BLT1−/−ApcMin/+ mice relative to ApcMin/+ mice. In particular, the top 10 genes that were downregulated included angiogenins (Ang1 and Ang4), indoleamine-pyrrole 2, 3 dioxygenase (IDO), and regenerating islet-derived 3 γ (Reg IIIγ, REG3G) (Supplementary Fig. 6). Each of these proteins was shown to possess strong direct bactericidal activity.Citation41-46 Angs and Reg3 g display specific killing activity on gram-positive bacteria such as Listeria monocytogenes and Enterococcus faecalis. The relative expression of these genes determined in the individual tumor samples shows a direct contrast with the up regulation of inflammatory markers (). The angiogenin mRNA levels () were also consistent with the changes in protein levels determined using a common Ang-specific antibody by Western blotting ().
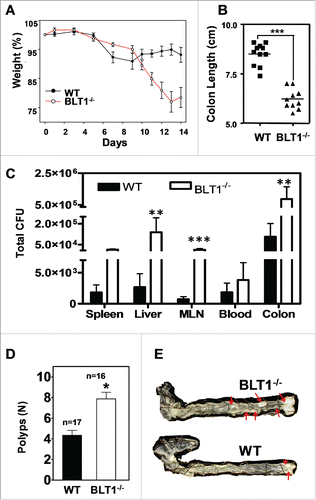
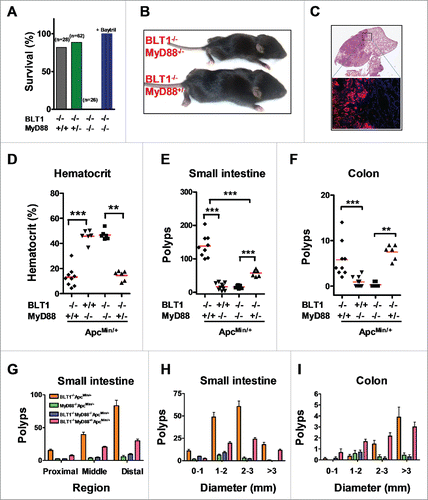
Figure 6. MyD88 acts downstream of BLT1 in promoting intestinal cancer in ApcMin/+ mice. (A). BLT1/MyD88 double knockout mice are susceptible to lethal neonatal infections. Percent survival of the offspring from BLT1−/−MyD88+/− cross (total of 15 litters from 8 different breeding pairs at day 15). Treatment of BLT1−/−MyD88+/− females during gestation and postpartum with a broad-spectrum antibiotic Baytril (enrofloxacin at 165 mg/L in drinking water that gave a daily dosage of ∼25 mg/kg) completely reversed the early lethality of BLT1−/−MyD88−/− pups. (B). Double deficient (BLT1−/−MyD88−/−) mice are smaller compared with the littermate heterozygous mice (BLT1−/−MyD88+/−). (C) H&E stained cross section images of lung from (BLT1−/−MyD88−/− mice (top panel). The bacterial infection was detected by FISH analysis (lower panel) with Eub338-Cy3 probe (red) and nuclei of lung parenchyma stained with DAPI (blue). (D-I). The BLT1−/−MyD88−/−ApcMin/+ and littermate control animals were generated and maintained on Baytril water. The hematocrits (D), number of small intestine polyps (E) and colon polyps (F) were evaluated at 110 d age-old mice. The frequency of small intestinal tumors (G) and size ranges of tumors in both small intestine (H) and colons (I) were analyzed. The overall tumor burden in mice lacking MyD88 significantly reduced in both BLT1+/+ and BLT−/−ApcMin/+ context indicating that MyD88 acts downstream of BLT1 in promoting intestinal cancer. Statistical analysis was performed using the Mann-Whitney U test. Error bars, ± SEM. **, P < 0.01; and ***, P < 0.001.
Germ-free BLT1−/−ApcMin/+ mice are free of colon cancer
LTB4-BLT1 axis has been implicated in protection against bacterial and viral infections by producing anti-microbial proteins and peptides.Citation47-50 To examine the role of BLT1 and microbiota mediated host response in colon tumor progression, germ free BLT1−/−ApcMin/+ mice were generated. The germ-free BLT1−/−ApcMin/+ mice showed complete absence of colon polyps (). Recolonizing these mice at the age of 50 d with fecal bacteria from specific pathogen free (SPF) BLT1−/−ApcMin/+mice led to the development of several colon polyps by 110 d indicating a critical role for fecal bacteria in promoting colon cancer (). The germ-free mice developed similar level of small intestinal polyps as mice housed under SPF condition (). Histopathological examination also confirms the complete absence of colon tumors and similar levels of small intestinal tumors in germ-free mice relative to mice maintained under SPF conditions ().
MyD88 is essential for promoting tumor development in BLT1−/−ApcMin/+ mice
MyD88 plays an important role in host defense and was shown to be a critical mediator of tumor promoting inflammation.Citation22,51 This is in contrast with the LTB4/BLT1 axis, which also appears to be important in host defense but seems to offer protection against tumor development. To examine the interplay of host defense and tumor promotion, we crossed MyD88−/− mice with BLT1−/− to generate double deficient mice. The 3 possible offspring from the BLT1−/−MyD88+/− crosses are born at the expected Mendelian ratios but all the double deficient (MyD88−/−BLT1−/−) mice died very early with few pups living up to 14 d (). The double deficient mouse which survived the longest was very sick and half the size of its heterozygous littermate (). Histopathological and microbiological examination showed infections of multiple organs including lungs and liver. The massive lung infections were delineated by Fluorescence In Situ Hybridization (FISH) analysis with bacterial probes (). Microbiological examination showed that the death of some of the double deficient mice could be attributed to Pasteurella pneumotropica infection that does not seem to affect the littermate heterozygous and WT animals. Treatment with a broad spectrum antibiotic, Baytril, completely protected the MyD88−/−BLT1−/− mice from neonatal lethality ().
Since the BLT1−/−MyD88−/− mice could be kept alive by treatment with broad spectrum antibiotics, we generated BLT1−/−MyD88−/−ApcMin/+ mice to examine the role of MyD88 mediated inflammation in tumor promotion in the BLT1−/−ApcMin/+ mice. As control, we also developed MyD88−/−ApcMin/+ mice. As reported earlier,Citation22 the MyD88−/−ApcMin/+ mice are highly protected with normal haematocrit () and greatly reduced tumor burden in both small intestines and colon (, ). Analysis of tumor development in BLT1−/−MyD88−/−ApcMin/+ mice showed that MyD88 dependent signaling is essential for the acceleration of tumor development seen in the BLT1−/−ApcMin/+ mice (). These results suggest that MyD88 acts downstream of BLT1 in tumor promotion and the absence of BLT1 likely results in enhanced activation of MyD88 dependent signaling.
Microbiota promotes colon tumorigenesis in BLT1−/−ApcMin/+ mice
Since germ-free mice were completely protected from colon tumor development, we examined the gut microbiota in Wild type, BLT1−/−, ApcMin/+, BLT1−/−ApcMin/+ along with MyD88−/− mice in the above backgrounds by 16 S rRNA gene sequencing. The fecal bacteria are majorly composed of Bacteroidetes and Firmicutes accounting up to ∼80% () total bacteria. The levels of Firmicutes and Verrucomicrobia significantly increased (26.8% vs 18%; 7.3% vs 2.1%) and levels of Bacteroidetes were significantly decreased (57.7% vs 52%) in BLT1−/−ApcMin/+ compared with ApcMin/+ ().
Figure 7. Modulation of gut microbiota and barrier dysfunction in BLT1−/−ApcMin/+. The gut microflora (fecal bacteria) was identified by sequencing 16 S rRNA gene using Roche 454 sequencer from indicated genotype (WT, n = 5; BLT1−/−, n = 5; ApcMin/+,n = 8; BLT1−/−ApcMin/+ n = 10; MyD88−/−ApcMin/+ (n = 5); BLT1−/−MyD8−/−ApcMin/+ (n = 5)). The 16 S rRNA gene (v1-v3 regions) sequences were uploaded on to QIIME pipe line (http://qiime.sourceforge.net/#). The distribution of phylum (A) and genus levels (B) among genotypes were identified. Genus level analysis suggested significant increase in Akkermansia muciniphila. (C). The relative amounts of Akkermansia muciniphila from the fecal contents of indicated mice was quantified using real time PCR as described in methods. Statistical analysis performed using unpaired t-test. ** P< 0.01, *P< 0.05. (D). The α diversity is measured using qiime pipeline. (E). Increased gut permeability and decreased tight junctional proteins in tumor bearing mice. The FITC-dextran (MWav = 4000; FD4) at 80 mg/mL (in 1X PBS) was administered by oral gavage (600 mg/kg bodyweight) to fasted mice. The fluorescence of FITC was measured in plasma after 2 hrs to estimate amount of FD4 leaked into blood and percent FD4 calculated using WT as base line. FD4 was significantly increased in BLT1−/−ApcMin/+ compared with ApcMin/+(*P< 0.05) and in ApcMin/+ compared with WT (*P< 0.05). (F). The mRNA levels of Cldn 5 and Jam C proteins was measured by RT-PCR using SyBr green. The mRNA levels were significantly reduced in tumors of ApcMin/+ and BLT1−/−ApcMin/+ mice.
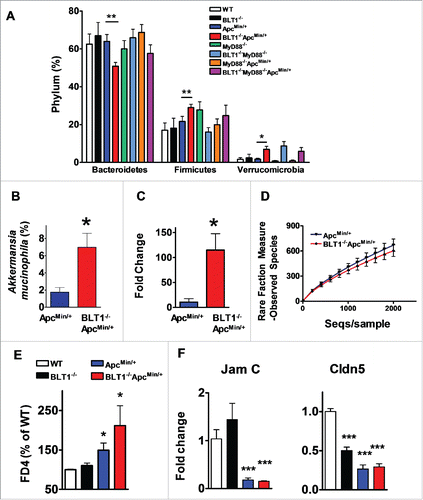
The analysis at genus levels indicated an overall significant increase in anaerobic bacteria, Akkermansia muciniphila in BLT1−/−ApcMin/+ compared with ApcMin/+ mice fecal samples (). Increased levels of Akkermansia muciniphila in BLT1−/−ApcMin/+ was further confirmed by independent assays based on real time PCR () and cloning of fecal 16 S rRNA gene amplicon and sequencing individual clones (data not shown). Interestingly, the analysis of MyD88−/−ApcMin/+ and BLT1−/−MyD88−/−ApcMin/+ microbiota also suggested that lack of BLT1 in ApcMin/+ background resulted in increase in Verricomicrobia (Akkermansia muciniphila) (6.84% vs 0.94%) and Firmicutes (24% vs 20%) and decrease in Bacteroidetes (67% vs 58%) indicating the role for BLT1 in MyD88 independent manner to modulate the gut flora in ApcMin/+ mice (). It is important to note that even in the absence of ApcMin/+ mutation in BLT1−/−MyD88−/− mice showed an increase in Akkermansia suggesting that the BLT1 and MyD88 synergistically control the Akkermansia. However, in the context of ApcMin/+ mutation, lack of BLT1 alone is sufficient to allow the outgrowth of Akkermansia. The α rare fraction analysis of observed species among ApcMin/+ and BLT1−/−ApcMin/+ shows distinct genotype associated differences between the groups ().
Adenoma induced barrier dysfunction is a major contributor to the progression of colon tumors in ApcMin/+ models.Citation52 Barrier dysfunction leading to intestinal permeability was measured in dextran-FITC fed mice. As shown in , the mice in ApcMin/+ background (both BLT1−/−ApcMin/+ and BLT1+/+ApcMin/+) showed increased intestinal leakage of dextran FITC into blood compared with non-cancerous mice suggesting damage of intestinal barrier in tumor bearing mice. The leakage of dextran-FITC is significantly more in BLT1−/−ApcMin/+ mice possibly due to increased tumor burden. The expression analysis of junctional proteins revealed a significant reduction in Cldn5 and Jam-C mRNA expression in colon tumors compared with normal colon (). These results suggest that adenoma induced barrier dysfunction likely forms the basis for host genotype dependent reshaping of microbiota ().
Discussion
Leukotriene B4 and its receptors have been a major focus in diverse inflammatory diseases over the past 2 decades. An emerging view from these studies is that inflammation promoted by the high affinity LTB4 receptor BLT1 is detrimental and is coincidental with the development and progression of asthma,Citation28 arthritis,Citation29,30 atherosclerosisCitation27 and lung cancers.Citation53 The results presented here outline a beneficial role for BLT1-mediated inflammation in host response to mucosal infections and control of spontaneous intestinal tumor development in ApcMin/+ mice.
In the ApcMin/+ mice most adenomas develop in the small intestine.Citation21 This is in contrast to Apc mutations in humans that invariably lead to colon tumor development. The BLT1−/−ApcMin/+ model described in this study, while retaining the small intestine specificity of the mouse ApcMin/+, develops very large multiple colonic adenomas reminiscent of human colon cancers. This phenotype retains the requirement for early mutations in the APC gene as the BLT1−/− mice do not develop any spontaneous tumors in colon. Previous studies have shown that regulators of the eicosanoid pathways such as cPLA2, COX2 and prostaglandin E2 receptor (EP2) to be tumor promoting in ApcMin/+ mice,Citation54-57 but the role of LTB4/BLT1 pathway was unknown. Use of dual 5-LOX/COX inhibitor, licofelone reduced the overall tumorigenesis in ApcMin/+ mouse model suggesting importance of leukotriene pathway in tumor development.Citation58 In support of this observation, 5-LO−/− in APCΔ468 background led to a dramatic reduction in the number and size of intestinal polyps.Citation59 It was suggested that haematopoietic expression (mast cells) of 5-LO is critical in recruitment of myeloid-derived suppressor cells (MDSCs) to promote tumorigenesis in this model.Citation59 We also generated 5LO−/−ApcMin/+ compound mice in our laboratory and evaluated the survival of mice. The deficiency of 5-LO increased the survival of ApcMin/+ mice as well as reduced their anemic status (Supplementary Figure 7A and B). Importantly, 5LO−/−ApcMin/+ displayed decreased polyps number (Supplementary Figure 7C) suggesting that 5-LO might play a role in tumor initiation corroborating with phenotype of 5-LO−/−APCΔ468. Thus, 5-LO derived products other than LTB4 could have a direct promotional activity on intestinal tumors. In this context, cystinyl leukotriene receptor 1 (CysLT1) deficient mice in the ApcMin/+background were shown to display reduced tumor burden.Citation60 Previously, Dreyling et al. showed that LTB4 levels are significantly increased in human gastrointestinal adenocarcinoma compared with normal colonic mucosa.Citation61 In ApcMin/+ mice, it was demonstrated that small intestinal tumors produced significantly higher levels of LTB4.Citation62 Mucosal mast cells might be the initial source of LTB4 that recruit additional LTB4 producing (granulocytes) cells involved in host response to infections.
The unexpected observations made in this study with BLT1−/− mice suggests that LTB4 pathway protects ApcMin/+ mice from developing intestinal tumors. Several lines of evidence point to an important role for BLT1 in host response to infection. The decrease in multiple antibacterial proteins at the mucosal surfaces and in intestinal tumors in BLT1−/−ApcMin/+ mice as well as decreased neutrophils influx and increased bacterial loads in response to peritoneal E. coli infection (Supplementary Figure 8) suggests that BLT1 is a mediator of host response to infection. These observations are consistent with the earlier findings that LTB4 directly activates production of β-defensinsCitation63,64 as well as decreased inflammatory cell influx in a model of septic peritonitis.Citation65 Addition of LTB4 was also shown to increase in bactericidal activity of alveolar macrophages from 5-lipoxygenase deficient mice.Citation66 The increased susceptibility to DSS induced colitis and increased bacterial load in BLT1−/− mice also support the notion that BLT1 is a critical mediator of host defense mechanisms. The protective role of BLT1 in colon tumor progression is also evident in the AOM-DSS induced colon cancer in mice ().
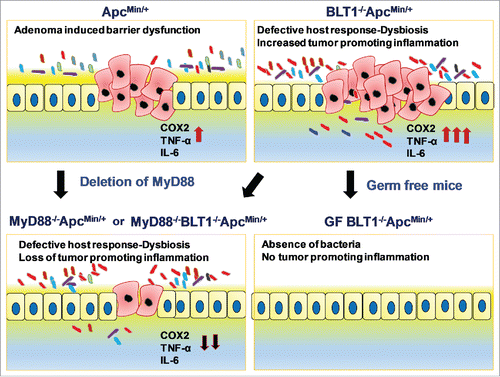
Figure 8. Interplay of BLT1 and MyD88 Signaling in Intestinal tumor development: Adenoma induced barrier dysfunction in ApcMin+ mice induces tumor promoting inflammation. Absence of BLT1 amplifies tumor initiated microbial dysbiosis leading to both bacteria (germ-free) and MyD88 dependent inflammation that promotes tumor progression.
The complete absence of colon tumors in germ-free BLT1−/−ApcMin/+ mice and their reappearance upon fecal transplantation from SPF BLT1−/−ApcMin/+ mice suggests that microbiota induced inflammation is a critical step in the development of spontaneous colon tumors. Interestingly, small intestinal tumor progression in BLT1−/−ApcMin/+ mice is bacteria independent as evident from unaltered tumor burden in germ free mice () that may be controlled by immune surveillance mechanisms. In this regard, we recently demonstrated using transplantable cervical cancer (TC1) and B16 melanoma models that expression of BLT1 on CD8+ T-cells is required for their migration into tumors to elicit effective anti-tumor immunity.Citation67,68 In these models, we showed accelerated tumor growth and decreased survival in BLT1−/− mice compared with wild type mice. T cell depletion and adoptive transfer of CTLs into Rag2−/− mice showed requirement for BLT1 on CD8+ T cells for effective immune surveillance. Numerous studies with human cancers showed a strong correlation between CD8+ T cell infiltration and long-term survival.Citation69 Our studies highlighted the critical nature of BLT1/LTB4 axis in the process of CTL recruitment to the sites of tumors for effective immune surveillance.
The current studies have uncovered a synergy between TLR- mediated inflammation and BLT1 mediated antibacterial response as seen by the lethal neonatal sepsis in BLT1/MyD88 double deficient mice. MyD88 was shown to directly influence the gut flora and these alterations have direct consequence to disease development.Citation70 The significant decrease in tumorigenesis in BLT1−/−MyD88−/−ApcMin/+ suggests that BLT1 acts upstream of MyD88 promoted inflammation in colon tumorigenesis. A single copy of MyD88 gene is sufficient to promote tumor growth in ApcMin/+ model () indicating the critical nature of MyD88 signaling cascade in tumor progression. Absence of MyD88 is highly protective in the small intestinal tumor despite lack of immune surveillance mechanisms mediated by BLT1 (BLT1−/−MyD88−/−ApcMin/+) suggesting a critical role for MyD88 in intestinal epithelial cell (IECs) proliferation apart from its classical role in host-defense mechanisms. In this regard, it was demonstrated that non-haematopoietic cells, such as IECs, required MyD88 for intestinal tumor growth.Citation71 It was suggested that TLR ligands derived from microbiota mediate the IEC tumor growth through ERK activation by stabilizing/increasing abundance of Myc protein, the product of oncogene c-Myc. Interestingly, our studies demonstrate that germ-free BLT1−/−ApcMin/+ mice did not alter the small intestinal tumor growth suggesting, bacterial independent, but MyD88 dependent tumor growth through yet unknown mechanisms. In contrast to spontaneous intestinal tumor model, deficiency of MyD88 in AOM-DSS induced colon tumor model significantly increased the colon tumor burden.Citation72 However, it is possible that extensive breach in the intestinal barrier caused by DSS could lead to leaky gut and bacteria, which will not be cleared in MyD88−/− mice leading to significantly increased inflammation associated colon tumors.
Microbiota is known to play a critical role in modulating the both innate and adaptive immune systems as well as regulate various disease conditions.Citation73-80 Our analysis of the 16 S rRNA gene sequences from fecal material of ApcMin/+ and BLT1−/−ApcMin/+ showed major and distinct differences with an increase in Akkermansia muciniphila in BLT1−/−ApcMin/+ compared with ApcMin/+ mice. A. muciniphila (belongs to Verrucomicrobia phylum) is known as a mucin degrading bacteria present in the human intestines.Citation81-83 A. muciniphila represented a relatively large percentage of sequences in human fecal microbiota of colon cancer patients,Citation84 ulcerative colitis-associated pouchitisCitation85 compared with healthy people. Mucin acts as a protective barrier between the intestinal contents and the mucosal wall and it is possible that enhanced degradation of mucin in BLT1−/−ApcMin/+ makes the intestines more permeable to pathogenic bacteria resulting in increased inflammation. Consistent with this notion, studies showed that a mucin deficient ApcMin/+ mice (Muc2−/−ApcMin/+) develop significant colon tumor burden.Citation86 Additional subtype taxonomy analysis (Supplementary Fig. 9) revealed that bacteria belonging to family S24–7 (Bacteroidetes phylum) are significantly downregulated in BLT1−/−ApcMin/+ mice. But absence of MyD88−/− in this context did not the alter the bacterial populations of S24–7. Most interestingly, Prevotella and bacteria belonging to Rikenellaceae are absent in mice lacking of MyD88 independent of ApcMin/+ mutation. The importance of these findings require additional studies. It is interesting to note that increase in A. muciniphila is BLT1 dependent (in context of either MyD88−/− or ApcMin/+ background), but requires MyD88 to promote colon tumorigenesis. The most significant observation of the current studies is the direct link between the altered microbiome in BLT1 deficient mice to increased MyD88 dependent inflammation in the intestinal tumors in the BLT1−/−ApcMin/+ mice.
Defective expression of tight junctional proteins (intestinal barrier proteins) such as JAM-A, JAM-B was an early event in colorectal cancer tumorigenesis followed by upregulation of IL-23 and IL-17 by microbial products to promote inflammation mediated colorectal cancer.Citation52 Our results also suggests that decreased expression of the junctional proteins (Cldn5 and JAM-C) and increased intestinal permeability in mice harbouring ApcMin/+ mutation. Defective host response in combination with increased intestinal permeability in BLT1−/−ApcMin/+ mice likely resulted in reshaping of the gut microbiota leading to increased inflammation and tumorigenesis. Further studies are needed to define the role of individual bacteria in promoting colon tumors.
These results suggest an intricate interplay of host response and inflammation in tumor development (). Adenoma induced barrier dysfunction in ApcMin+ mice initiates epithelial breach and induces inflammation. Absence of BLT1 in colon alters the host response reshaping the gut microbiota and sets up conditions for enhanced chronic inflammation and tumor promotion. The ensuing microbial dysbiosis amplifies bacteria induced colonic inflammation primarily through activation of a set of bacterial ‘sensors' exemplified by the typical TLR pathways that mobilize a group of ‘mediators' such as the IL-6 and inflammatory chemokines to clear these mucosal infections.Citation87 Under germ-free conditions or in the absence of MyD88, loss of bacteria induced inflammation prevents tumor progression. Our results highlight the importance of BLT1 mediated early inflammation in reducing dysbiosis, the underlying cause of gut inflammation and protecting from colon tumor development.
Author contributions
V.R.J, and P.M, contributed equally to this work; V.R.J., and B.H. designed research; P.M, E.K and V.R.J, S.R.B, M.W, S.M., K.S. performed research; A.B.J, M.L.P, E.R and R.N contributed analytic tools; P.M., V.R.J., and B.H. analyzed data; and V.R.J, S.R.B and B.H. wrote the paper.
downloadFromZipFile.pdf
Download PDF (1 MB)Acknowledgments
We thank Dr. Nejat Egilmez for critical reading of the manuscript. We thank Haritha Pallam and Michelle Smith for expert technical assistance. We thank Luke Ursell from Rob Knight's group in setting up the microbiota analysis. This work was supported by NIH grants CA-138623(BH) and James Graham Brown Cancer Center at U of L. Part of this work was performed with assistance of the U of L Microarray Facility, which is supported by NCRR COBRE P20RR018733, KY-INBRE NCRR P20RR016481 and the J. G. Brown Cancer Center at U of L. The authors declare no competing financial interests.
References
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860-867. doi:10.1038/nature01322
- Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823-835. doi:10.1016/j.cell.2006.02.016. PMID:16497591
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436-444. doi:10.1038/nature07205. PMID:18650914
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118, 285-296. doi:10.1016/j.cell.2004.07.013. PMID:15294155
- Dzutsev A, Goldszmid RS, Viaud S, Zitvogel L, Trinchieri G. The role of the microbiota in inflammation, carcinogenesis, and cancer therapy. European Journal of Immunology. 2015;45:17-31. doi:10.1002/eji.201444972. PMID:25328099
- Roy S, Trinchieri G. Microbiota: a key orchestrator of cancer therapy. Nature Reviews Cancer. 2017;17(5):271-285. doi:10.1038/nrc.2017.13. PMID:28303904
- Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology. 2009;136:65-80. doi:10.1053/j.gastro.2008.10.080. PMID:19026645
- Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, Corthier G, Tran Van Nhieu J, Furet JP, et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PloS One. 2011;6:e16393. doi:10.1371/journal.pone.0016393. PMID:21297998
- Schwabe RF, Jobin C. The microbiome and cancer. Nature Reviews Cancer. 2013;13:800-812. doi:10.1038/nrc3610. PMID:24132111
- Vipperla K, O'Keefe SJ. The microbiota and its metabolites in colonic mucosal health and cancer risk. Nutrition in Clinical Practice: Official Publication of the American Society for Parenteral and Enteral Nutrition. 2012;27:624-635. doi:10.1177/0884533612452012. PMID:22868282
- Waisberg J, Matheus Cde O, Pimenta J. Infectious endocarditis from Streptococcus bovis associated with colonic carcinoma: case report and literature review. Arq Gastroenterol. 2002;39:177-180. PMID:12778310
- Gold JS, Bayar S, Salem RR. Association of Streptococcus bovis bacteremia with colonic neoplasia and extracolonic malignancy. Arch Surg 2004;139:760-765. doi:10.1001/archsurg.139.7.760. PMID:15249410
- Klein RS, Recco RA, Catalano MT, Edberg SC, Casey JI, Steigbigel NH. Association of Streptococcus bovis with carcinoma of the colon. N Engl J Med. 1977;297:800-802. doi:10.1056/NEJM197710132971503. PMID:408687
- Zarkin BA, Lillemoe KD, Cameron JL, Effron PN, Magnuson TH, Pitt HA. The triad of Streptococcus bovis bacteremia, colonic pathology, and liver disease. Ann Surg. 1990;211:786-791; discussion 791-782. PMID:2357141
- Ruoff KL, Miller SI, Garner CV, Ferraro MJ, Calderwood SB. Bacteremia with Streptococcus bovis and Streptococcus salivarius: clinical correlates of more accurate identification of isolates. J Clin Microbiol 1989;27:305-308 PMID:2915024
- Tsuji S, Tsujii M, Murata H, Nishida T, Komori M, Yasumaru M, Ishii S, Sasayama Y, Kawano S, Hayashi N. Helicobacter pylori eradication to prevent gastric cancer: underlying molecular and cellular mechanisms. World J Gastroenterol. 2006;12:1671-1680. PMID:16586533
- Huang JQ, Zheng GF, Sumanac K, Irvine EJ, Hunt RH. Meta-analysis of the relationship between cagA seropositivity and gastric cancer. Gastroenterology. 2003;125:1636-1644. PMID:14724815
- Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM. Enhanced Escherichia coli adherence and invasion in Crohn's disease and colon cancer. Gastroenterology. 2004;127:80-93. PMID:15236175
- Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PloS One. 2012;7:e39743. doi:10.1371/journal.pone.0039743. PMID:22761885
- Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007;120:3327-3335. doi:10.1242/jcs.03485. PMID:17881494
- Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322-324. PMID:2296722
- Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317:124-127. doi:10.1126/science.1140488. PMID:17615359
- Toda A, Yokomizo T, Shimizu T. Leukotriene B4 receptors. Prostaglandins & Other Lipid Mediators. 2002;68-69:575-585.
- Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature. 1997;387:620-624. doi:10.1038/42506. PMID:9177352
- Yokomizo T, Kato K, Terawaki K, Izumi T, Shimizu T. A second leukotriene B(4) receptor, BLT2. A new therapeutic target in inflammation and immunological disorders. The Journal of Experimental Medicine. 2000;192:421-432. PMID:10934230
- Tager AM, Bromley SK, Medoff BD, Islam SA, Bercury SD, Friedrich EB, Carafone AD, Gerszten RE, Luster AD. Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nature Immunology. 2003;4:982-990. doi:10.1038/ni970. PMID:12949531
- Subbarao K, Jala VR, Mathis S, Suttles J, Zacharias W, Ahamed J, Ali H, Tseng MT, Haribabu B. Role of leukotriene B4 receptors in the development of atherosclerosis: potential mechanisms. Arterioscler Thromb Vasc Biol. 2004;24:369-375. doi:10.1161/01.ATV.0000110503.16605.15. PMID:14656734
- Miyahara N, Takeda K, Miyahara S, Matsubara S, Koya T, Joetham A, Krishnan E, Dakhama A, Haribabu B, Gelfand EW. Requirement for leukotriene B4 receptor 1 in allergen-induced airway hyperresponsiveness. American Journal Of Respiratory And Critical Care Medicine. 2005;172:161-167. doi:10.1164/rccm.200502-205OC. PMID:15849325
- Kim ND, Chou RC, Seung E, Tager AM, Luster AD. A unique requirement for the leukotriene B4 receptor BLT1 for neutrophil recruitment in inflammatory arthritis. The Journal of Experimental Medicine. 2006;203:829-835. doi:10.1084/jem.20052349. PMID:16567386
- Shao WH, Del Prete A, Bock CB, Haribabu B. Targeted disruption of leukotriene B4 receptors BLT1 and BLT2: a critical role for BLT1 in collagen-induced arthritis in mice. Journal of Immunology. 2006;176:6254-6261.
- Houthuijzen JM, Daenen LG, Roodhart JM, Oosterom I, van Jaarsveld MT, Govaert KM, Smith ME, Sadatmand SJ, Rosing H, Kruse F, et al. Lysophospholipids secreted by splenic macrophages induce chemotherapy resistance via interference with the DNA damage response. Nat Commun. 2014;5:5275. doi:10.1038/ncomms6275. PMID:25387467
- Haribabu B, Verghese MW, Steeber DA, Sellars DD, Bock CB, Snyderman R. Targeted disruption of the leukotriene B(4) receptor in mice reveals its role in inflammation and platelet-activating factor-induced anaphylaxis. J Exp Med. 2000;192:433-438. PMID:10934231
- Marr KA, Balajee SA, Hawn TR, Ozinsky A, Pham U, Akira S, Aderem A, Liles WC. Differential role of MyD88 in macrophage-mediated responses to opportunistic fungal pathogens. Infect Immun. 2003;71:5280-5286. PMID:12933875
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 2010;7:335-336. doi:10.1038/nmeth.f.303. PMID:20383131
- Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. The ISME Journal 2011;5:169-172. doi:10.1038/ismej.2010.133. PMID:20827291
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402-408. doi:10.1006/meth.2001.1262. PMID:11846609
- Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159-170. PMID:8861899
- Humphries A, Wright NA. Colonic crypt organization and tumorigenesis. Nat Rev Cancer. 2008;8:415-424. doi:10.1038/nrc2392. PMID:18480839
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749-759. doi:10.1038/nri1703. PMID:16175180
- Williams CS, et al. Elevated cyclooxygenase-2 levels in Min mouse adenomas. Gastroenterology. 1996;111:1134-1140. doi: S0016-5085(96)70083-5. PMID:8831610
- Carlin JM, Borden EC, Byrne GI. Interferon-induced indoleamine 2,3-dioxygenase activity inhibits Chlamydia psittaci replication in human macrophages. J Interferon Res 1989;9:329-337. PMID:2501398
- Thomas SM, Garrity LF, Brandt CR, Schobert CS, Feng GS, Taylor MW, Carlin JM, Byrne GI. IFN-gamma-mediated antimicrobial response. Indoleamine 2,3-dioxygenase-deficient mutant host cells no longer inhibit intracellular Chlamydia spp. or Toxoplasma growth. J Immunol. 1993;150:5529-5534 PMID:8515074
- Hooper LV, Stappenbeck TS, Hong CV, Gordon JI. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat Immunol. 2003;4:269-273. doi:10.1038/ni888ni888. PMID:12548285
- Boix E, Nogues MV. Mammalian antimicrobial proteins and peptides: overview on the RNase A superfamily members involved in innate host defence. Mol Biosyst. 2007;3:317-335. doi:10.1039/b617527a. PMID:17460791
- Rosenberg HF. RNase A ribonucleases and host defense: an evolving story. J Leukoc Biol. 2008;83:1079-1087. doi:10.1189/jlb.1107725. PMID:18211964
- Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313:1126-1130. doi:10.1126/science.1127119. PMID:16931762
- Soares EM, Mason KL, Rogers LM, Serezani CH, Faccioli LH, Aronoff DM. Leukotriene B4 enhances innate immune defense against the puerperal sepsis agent Streptococcus pyogenes. Journal of Immunology. 2013;190:1614-1622. doi:10.4049/jimmunol.1202932
- Sorgi CA, Secatto A, Fontanari C, Turato WM, Belangér C, de Medeiros AI, Kashima S, Marleau S, Covas DT, Bozza PT, et al. Histoplasma capsulatum cell wall {beta}-glucan induces lipid body formation through CD18, TLR2, and dectin-1 receptors: correlation with leukotriene B4 generation and role in HIV-1 infection. Journal of Immunology. 2009;182:4025-4035. doi:10.4049/jimmunol.0801795
- Gaudreault E, Gosselin J. Leukotriene B4-mediated release of antimicrobial peptides against cytomegalovirus is BLT1 dependent. Viral Immunology. 2007;20:407-420. doi:10.1089/vim.2006.0099. PMID:17931111
- Serezani CH, Perrela JH, Russo M, Peters-Golden M, Jancar S. Leukotrienes are essential for the control of Leishmania amazonensis infection and contribute to strain variation in susceptibility. Journal Of Immunology. 2006;177:3201-3208.
- Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121-124. doi:10.1126/science.1140485. PMID:17615358
- Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung K, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491:254-258. doi:10.1038/nature11465 PMID:23034650
- Satpathy SR, Jala VR, Bodduluri SR, Krishnan E, Hegde B, Hoyle GW, Fraig M, Luster AD, Haribabu B. Crystalline silica-induced leukotriene B4-dependent inflammation promotes lung tumour growth. Nat Commun. 2015;6:7064. doi:10.1038/ncomms8064. PMID:25923988
- Hong KH, Bonventre JC, O'Leary E, Bonventre JV, Lander ES. Deletion of cytosolic phospholipase A(2) suppresses Apc(Min)-induced tumorigenesis. Proc Natl Acad Sci U S A. 2001;98:3935-3939. doi:10.1073/pnas.051635898. PMID:11274413
- Kawai N, Tsujii M, Tsuji S. Cyclooxygenases and colon cancer. Prostaglandins & Other Lipid Mediators. 2002;68-69:187-196.
- Wang D, Wang H, Shi Q, Katkuri S, Walhi W, Desvergne B, Das SK, Dey SK, DuBois RN. Prostaglandin E(2) promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor delta. Cancer Cell. 2004;6:285-295. doi:10.1016/j.ccr.2004.08.011. PMID:15380519
- Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell. 1996;87:803-809. doi: S0092-8674(00)81988-1. PMID:8945508
- Mohammed A, Janakiram NB, Li Q, Choi CI, Zhang Y, Steele VE, Rao CV. Chemoprevention of colon and small intestinal tumorigenesis in APC(Min/+) mice by licofelone, a novel dual 5-LOX/COX inhibitor: potential implications for human colon cancer prevention. Cancer Prevention Research. 2011;4:2015-2026. doi:10.1158/1940-6207.CAPR-11-0233. PMID:21885812
- Cheon EC, Khazaie K, Khan MW, Strouch MJ, Krantz SB, Phillips J, Blatner NR, Hix LM, Zhang M, Dennis KL, et al. Mast cell 5-lipoxygenase activity promotes intestinal polyposis in APCDelta468 mice. Cancer Research. 2011;71:1627-1636. doi:10.1158/0008-5472.CAN-10-1923. PMID:21216893
- Mehdawi L, Osman J, Topi G, Sjolander A. High tumor mast cell density is associated with longer survival of colon cancer patients. Acta Oncol. 2016;55:1434-1442. doi:10.1080/0284186X.2016.1198493. PMID:27355473
- Dreyling KW, Hoppe U, Peskar BA, Morgenroth K, Kozuschek W, Peskar BM. Leukotriene synthesis by human gastrointestinal tissues. Biochim Biophys Acta. 1986;878:184-193. PMID:3019409
- Chiu CH, McEntee MF, Whelan J. Sulindac causes rapid regression of preexisting tumors in Min/+ mice independent of prostaglandin biosynthesis. Cancer Research. 1997;57:4267-4273. PMID:9331087
- Flamand L, Tremblay MJ, Borgeat P. Leukotriene B4 triggers the in vitro and in vivo release of potent antimicrobial agents. Journal of Immunology. 2007;178:8036-8045. doi: 178/12/8036
- Gaudreault E, Gosselin J. Leukotriene B4 induces release of antimicrobial peptides in lungs of virally infected mice. Journal of Immunology 2008;180:6211-6221. doi: 180/9/6211
- Scott MJ, Cheadle WG, Hoth JJ, Peyton JC, Subbarao K, Shao WH, Haribabu B. Leukotriene B4 receptor (BLT-1) modulates neutrophil influx into the peritoneum but not the lung and liver during surgically induced bacterial peritonitis in mice. Clinical and Diagnostic Laboratory Immunology. 2004;11:936-941. doi:10.1128/CDLI.11.5.936-941.2004. PMID:15358656
- Mancuso P, Standiford TJ, Marshall T, Peters-Golden M. 5-Lipoxygenase reaction products modulate alveolar macrophage phagocytosis of Klebsiella pneumoniae. Infect Immun. 1998;66:5140-5146. PMID:9784515
- Sharma RK, Chheda Z, Jala VR, Haribabu B. Expression of Leukotriene B4 Receptor-1 on CD8+ T Cells is required for their migration into tumors to elicit effective antitumor immunity. Journal of Immunology 2013;191:3462-3470. doi:10.4049/jimmunol.1300967
- Chheda ZS, Sharma RK, Jala VR, Luster AD, Haribabu B. Chemoattractant receptors BLT1 and CXCR3 regulate antitumor immunity by facilitating CD8+ T Cell migration into tumors. J Immunol. 2016;197:2016-2026. doi:10.4049/jimmunol.1502376. PMID:27465528
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782-795. doi:10.1016/j.immuni.2013.10.003. PMID:24138885
- Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109-1113. doi:10.1038/nature07336. PMID:18806780
- Lee SH, Hu LL, Gonzalez-Navajas J, Seo GS, Shen C, Brick J, Herdman S, Varki N, Corr M, Lee J, et al. ERK activation drives intestinal tumorigenesis in Apc(min/+) mice. Nature Medicine. 2010;16:665-670. doi:10.1038/nm.2143. PMID:20473309
- Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, Wang E, Ma W, Haines D, O'hUigin C, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. The Journal of Experimental Medicine. 2010;207:1625-1636. doi:10.1084/jem.20100199. PMID:20624890
- Kinross JM, Darzi AW, Nicholson JK. Gut microbiome-host interactions in health and disease. Genome Med. 2011;3:14. doi:10.1186/gm228. PMID:21392406
- Chung H, Kasper DL. Microbiota-stimulated immune mechanisms to maintain gut homeostasis. Curr Opin Immunol. 2010;22;455-460. doi:10.1016/j.coi.2010.06.008. PMID:20656465
- Lee YK, Mazmanian SK. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science. 2010;330:1768-1773. doi:10.1126/science.1195568. PMID:21205662
- Kuczynski J, Costello EK, Nemergut DR, Zaneveld J, Lauber CL, Knights D, Koren O, Fierer N, Kelley ST, Ley RE, et al. Direct sequencing of the human microbiome readily reveals community differences. Genome Biol. 2010;11:210. doi:10.1186/gb-2010-11-5-210. PMID:20441597
- Arthur JC, Jobin C. The struggle within: microbial influences on colorectal cancer. Inflamm Bowel Dis. 2011;17:396-409. doi:10.1002/ibd.21354. PMID:20848537
- Candela M, Guidotti M, Fabbri A, Brigidi P, Franceschi C, Fiorentini C. Human intestinal microbiota: cross-talk with the host and its potential role in colorectal cancer. Crit Rev Microbiol. 2011;37:1-14. doi:10.3109/1040841X.2010.501760. PMID:20874522
- Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, et al. Evolution of mammals and their gut microbes. Science. 2008;320:1647-1651. doi:10.1126/science.1155725. PMID:18497261
- Belizario JE, Napolitano M. Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front Microbiol. 2015;6:1050. doi:10.3389/fmicb.2015.01050. PMID:26500616
- Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. International Journal of Systematic and Evolutionary Microbiology. 2004;54:1469-1476. doi:10.1099/ijs.0.02873-0. PMID:15388697
- Collado MC, Derrien M, Isolauri E, de Vos WM, Salminen S. Intestinal integrity and Akkermansia muciniphila, a mucin-degrading member of the intestinal microbiota present in infants, adults, and the elderly. Applied and Environmental Microbiology. 2007;73:7767-7770. doi:10.1128/AEM.01477-07. PMID:17933936
- Derrien M, Collado MC, Ben-Amor K, Salminen S, de Vos WM. The Mucin degrader Akkermansia muciniphila is an abundant resident of the human intestinal tract. Applied and Environmental Microbiology. 2008;74:1646-1648. doi:10.1128/AEM.01226-07. PMID:18083887
- Weir TL, Manter DK, Sheflin AM, Barnett BA, Heuberger AL, Ryan EP. Stool Microbiome and Metabolome Differences between Colorectal Cancer Patients and Healthy Adults. PloS One. 2013;8:e70803. doi:10.1371/journal.pone.0070803. PMID:23940645
- Zella GC, Hait EJ, Glavan T, Gevers D, Ward DV, Kitts CL, Korzenik JR. Distinct microbiome in pouchitis compared to healthy pouches in ulcerative colitis and familial adenomatous polyposis. Inflammatory Bowel Diseases. 2011;17:1092-1100. doi:10.1002/ibd.21460. PMID:20845425
- Yang K, Popova NV, Yang WC, Lozonschi I, Tadesse S, Kent S, Bancroft L, Matise I, Cormier RT, Scherer SJ, et al. Interaction of Muc2 and Apc on Wnt signaling and in intestinal tumorigenesis: potential role of chronic inflammation. Cancer Res. 2008. 68:7313-7322. doi:10.1158/0008-5472.CAN-08-0598. PMID:18794118
- Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140:771-776. doi:10.1016/j.cell.2010.03.006. PMID:20303867