
ABSTRACT
Advancing chimeric antigen receptor (CAR)-engineered adoptive T cells for the treatment of solid cancers is a major focus in the field of immunotherapy, given impressive recent clinical responses in hematological malignancies. Prostate cancer may be amenable to T cell-based immunotherapy since several tumor antigens, including prostate stem-cell antigen (PSCA), are widely over-expressed in metastatic disease. While antigen selectivity of CARs for solid cancers is crucial, it is problematic due to the absence of truly restricted tumor antigen expression and potential safety concerns with “on-target off-tumor” activity. Here, we show that the intracellular co-stimulatory signaling domain can determine a CAR's sensitivity for tumor antigen expression. A 4-1BB intracellular co-stimulatory signaling domain in PSCA-CARs confers improved selectivity for higher tumor antigen density, reduced T cell exhaustion phenotype, and equivalent tumor killing ability compared to PSCA-CARs containing the CD28 co-stimulatory signaling domain. PSCA-CARs exhibit robust in vivo anti-tumor activity in patient-derived bone-metastatic prostate cancer xenograft models, and 4-1BB-containing CARs show superior T cell persistence and control of disease compared with CD28-containing CARs. Our study demonstrates the importance of co-stimulation in defining an optimal CAR T cell, and also highlights the significance of clinically relevant models in developing solid cancer CAR T cell therapies.
Introduction
Repairing defects in anti-tumor immunity has been a longstanding challenge in solid cancer therapy. In recent years, cellular immunotherapy has emerged as a promising approach for controlling advanced disease,Citation1,2 with impressive clinical efficacy seen in several hematological malignancies using chimeric antigen receptor (CAR)-engineered T cell therapy. CAR T cell-based approaches are now being actively investigated for the treatment of advanced solid cancers.Citation3,4 Prostate cancer may be targeted by CAR T cell therapy given that there are several overexpressed cell-surface tumor antigens, including prostate stem-cell antigen (PSCA) and prostate-specific membrane antigen (PSMA).Citation5-9 In the last decade, several major improvements to CAR constructs have allowed for more robust and persistent CAR T cells for use in the clinic, largely surrounding CD19-specific CAR T cells for B-cell malignancies.Citation10,11 However, moving beyond the restricted expression of CD19 as a tumor-associated antigen has highlighted potential “on-target off-tumor” safety concerns with CARs that must be addressed in developing this immunotherapy approach for solid cancers.
The design of CARs has evolved over the last several years, but the success of CAR T cell therapy for solid cancers is contingent upon the combination of robust therapeutic efficacy and enhanced tumor antigen selectivity, with minimal “on-target off-tumor” activity.Citation12-14 Recent studies indicate that both the affinity of the antigen-targeting domain and specificity for the epitope are important components in CAR activity.Citation15-18 Although fine-tuning scFv affinity may change the tumor antigen density requirement for CAR activity, it is not the only variable in defining CAR functionality. Major advancements in CAR design have been achieved by the addition of intracellular signaling domains (i.e. CD28, 4-1BB) to ‘first-generation’ CARs. These co-stimulatory domains have allowed for greater persistence of the so-called “second-generation” CAR T cells, with enhanced cytokine production, T cell proliferation, persistence and cytolytic activity.Citation19-21 Recent studies have also revealed that other components of CARs, namely the nonsignaling extracellular spacer can also impact CAR functional activity.Citation22-27 Given the importance of each of these variables in CAR design, empirical determination will likely be required to identify the optimal CAR T cell design for any given tumor target.
One of the biggest challenges for CAR T cell therapy is efficient trafficking and persistence of adoptively transferred T cells in the solid tumor microenvironment.Citation12,13,28 In metastatic castrate-resistant prostate cancer, a major site of metastasis is the bone, which is a hostile microenvironment that makes it problematic for effective adoptive T cell therapy.Citation3,4 Therefore, evaluation of CAR T cell immunotherapy in the context of clinically-relevant disease settings is imperative for preclinical development of CAR candidates. Here, we optimized CAR T cells targeting PSCA using both in vitro studies and orthotopic human xenograft models. By varying the intracellular co-stimulatory signaling domain of the CAR construct, we identified a CAR that selectively targets tumors with high antigen density and improves T cell persistence. These studies underscore the importance of CAR composition in optimizing activity and T cell persistence and highlight the importance of clinically-relevant models for the development of effective solid cancer CAR T cells.
Results
4-1BB-containing PSCA-CARs show more selectivity for high tumor antigen density compared to CD28-containing PSCA-CARs
Second-generation CARs containing intracellular co-stimulatory signaling domains have been shown to improve the overall functional activity and persistence of CAR T cells.Citation13,19 To investigate the impact of different intracellular co-stimulatory signaling domains in PSCA-specific CARs, we constructed CD28-containing (PSCA-28ζ) and 4-1BB-containing (PSCA-BBζ) PSCA-CARs. Both PSCA-CAR constructs contained the humanized PSCA scFv derived from 1G8 (A11 clone),Citation29 the ΔCH2 extracellular spacer, and the CD3ζ cytolytic domain; and both CAR constructs included a T2A ribosomal skip sequence followed by the truncated CD19 (CD19t), used as a marker of lentiviral transduction efficiency and cell tracking (). For the following studies, we utilized healthy donor peripheral blood mononuclear cells (PBMC) (Figure S1). Both PSCA-CARs were expressed on the surface of T cells as determined by flow cytometric detection of the scFv, albeit at lower levels for PSCA-BBζ compared to PSCA-28ζ (). The potential impact of differential CAR expression on the function of these T cells will be directly assessed in experiments detailed below. PSCA-28ζ and PSCA-BBζ CAR T cells exhibited comparable ex vivo T cell expansion kinetics () and similar cell surface T cell phenotypes ().
Figure 1. PSCA-CAR T cells containing CD28 or 4-1BB co-stimulatory domains. (a) Diagram of the lentiviral expression cassette with PSCA-CARs containing the humanized scFv (A11 clone) targeting PSCA, with a 129 amino acid modified human IgG4 Fc linker (void of the CH2 domain, ΔCH2), a CD28 or CD4 transmembrane domain, a cytoplasmic CD28 or 4-1BB costimulatory domain, and a cytolytic CD3ζ domain. A truncated non-signaling CD19 (CD19t), separated from the CAR with a T2A ribosomal skip sequence, was expressed for tracking CAR-expressing cells. (b) Mock (untransduced), PSCA-28ζ, or PSCA-BBζ CAR T cells were evaluated by flow cytometry for CD19t expression to detect lentiviral transduction of CARs (left) or Protein L to detect the scFv (right). (c) Ex vivo expansion kinetics for Mock and PSCA-CAR T cells over 25 days in culture. (d) Cell-surface expression of indicated cell-surface markers of PSCA-CAR T cells at end of ex vivo expansion as determined by flow cytometry. All data are representative of at least two independent experiments.
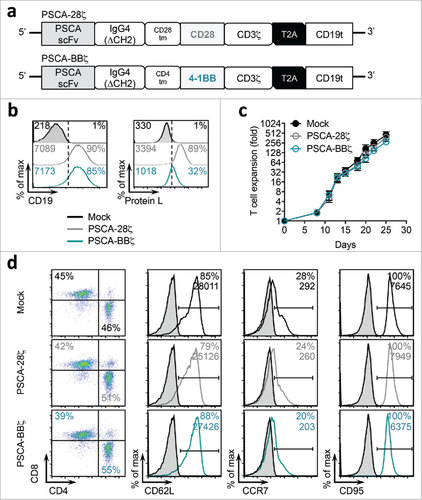
Next, we assessed the tumor targeting abilities of PSCA-28ζ and PSCA-BBζ CAR T cells by evaluating antigen-specific T cell activation. For these studies, we used several human prostate cancer cell lines that were engineered to stably express the human PSCA gene under the control of the EF1α promoter (). PC-3 tumor cells were also engineered with PSCA driven by a mutant PGK promoterCitation30 to derive a low antigen-density cell line (denoted “PGK100p”). LAPC-9 is a primary tumor xenograft derived from a patient with bone metastatic prostate cancerCitation31 that endogenously expresses PSCA. CAR T cell function was evaluated over 24 hours using CD137 (4-1BB) and CD69 as early markers of T cell activation, comparing the ability of the two CAR constructs to target tumors with varying tumor antigen densities. T cells induced little or no CD137 and CD69 expression at comparable levels as Mock (untransduced) T cells, when co-cultured with tumor cells for 1 hour (). At later time points (4 and 24 hours), PSCA-28ζ and PSCA-BBζ CAR T cells expressed similar peak levels of CD137 and CD69 against tumor cells with high levels of PSCA. However, PSCA-28ζ CAR T cells showed induction of CD137 and CD69 when co-cultured with tumor cells lacking cell surface expression of PSCA, and a slightly higher peak expression compared with PSCA-BBζ CAR T cells when co-cultured with PC-3-PGK100p cells. These early activation data suggest that while CARs with CD28- or 4-1BB-co-stimulation similarly target tumors with high antigen density, CD28 co-stimulation may also induce appreciable activation of CAR T cells against tumor cells lacking detectable cell surface antigen expression.
Figure 2. PSCA-BBζ CARs show antigen-dependent cytokine production in vitro. (a) Flow cytometric analysis of PSCA expression in human prostate cancer cell lines. DU145 and PC-3 cell lines were lentivirally transduced to over-express human PSCA under the control of the EF1α promoter (see materials and methods). PC-3-PGK100p cell line was generated by expressing human PSCA under the control of the indicated mutant PGK promoter (PGK100p). LAPC-9 cells endogenously express human PSCA. (b) Quantification of CD137 (top) and CD69 (bottom) expression on Mock, PSCA-28ζ, and PSCA-BBζ CAR T cells following a 1, 4, or 24 hour co-culture with the indicated tumor targets at a 1:2 effector:tumor (E:T) ratio. (c) IFNγ production quantified by ELISA in supernatants from PSCA-CAR T cells cultured overnight with DU145 or DU145-PSCA tumor cells. (d) Same as in (c) from PSCA-CAR T cells cultured overnight with PC-3, PGK100p, or PC-3-PSCA tumor cells. (e) IFNγ production quantified by ELISA in supernatants from PSCA-CAR T cells cultured overnight on plate-bound recombinant human PSCA at varying protein concentrations. (f) Representative FACS plots showing intracellular IFNγ and CD107a degranulation by PSCA-CAR T cells following a 4 – 6 hr co-culture with indicated tumor targets. (g) Quantification of intracellular IFNγ and CD107a degranulation by PSCA-CAR T cells from (f). Data are shown as n = 2 per group ± SD. All data are representative of at least two independent experiments.
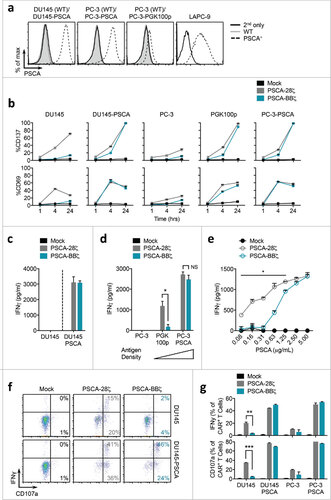
To confirm that functional differences between CD28- and 4-1BB-containing PSCA-CAR T cells were attributed to different co-stimulation and not a result of CAR expression differences,Citation32,33 we performed fluorescence-activated cell sorting (FACS) to enrich for both low CD19t-expressing PSCA-28ζ CAR T cells and high CD19t-expressing PSCA-BBζ CAR T cells. This was done to achieve similar cell surface CAR expression levels between the two groups as assessed by protein L to detect the CAR scFv (Figure S2 a). While sorted PSCA-28ζ and PSCA-BBζ CAR T cells showed comparable early activation against tumor cells with high PSCA expression, sorted PSCA-28ζ CAR T cells still induced CD137 and CD69 expression against PSCA-negative DU145 cells (Figure S2 b). It is noteworthy that sorted CARs did show some differences compared with unsorted cells (i.e. no activation with sorted CD28-containing PSCA-CARs was observed against wild-type PC-3 cells). Importantly, sorting for high CD19t-expressing PSCA-BBζ CAR T cells, and hence high CAR expression, did not increase the targeting of these CARs for tumor cells with little or no PSCA protein expression, as was observed with CD28-containing CARs. These data suggest that while expression of the CAR may play a role in tumor targeting, co-stimulation regulates PSCA-CAR sensitivity for tumor cells with varying antigen density.
We next sought to identify factors responsible for the functional activity of PSCA-28ζ CAR T cells against tumor cells with apparent lack of cell surface PSCA expression. We quantified PSCA at the mRNA level and found that while PSCA protein expression was undetectable by flow cytometry in wild-type DU145 and PC-3 cells, PSCA mRNA was detected in both these lines to varying degrees (Figure S3). Additionally, PSCA mRNA was expressed in other tumor cell lines that lacked detectable PSCA on their cell surface by flow cytometry, including A549, HT1080, and MDA-MB-468 (data not shown). The functional activity of PSCA-28ζ CAR T cells coincided with the low but detectable mRNA expression of PSCA in these tumor cell lines (Figure S4 a). SKBR3 tumor cells expressing higher levels of PSCA mRNA showed detectable cell surface PSCA (data not shown) and induced activation of both PSCA-28ζ and PSCA-BBζ CAR T cells. We also evaluated a series of normal human cell lines, and detected very low PSCA mRNA levels (Figure S5 a) with undetectable cell surface PSCA expression by flow cytometry (Figure S5 b). Low levels of activation of PSCA-28ζ CAR T cells, but not PSCA-BBζ CAR T cells, against these normal human cell lines were observed (Figure S5 c). We conclude that the activity observed by both PSCA-28ζ and PSCA-BBζ CAR T cells is antigen-specific at higher antigen density. However, the minimum antigen density required for activity is higher for 4-1BB co-stimulation than it is for CD28 co-stimulation.
We then evaluated cytokine production from CAR T cells as an additional measure of T cell activity. While both CAR T cells produced comparable IFNγ levels when co-cultured overnight with high-expressing DU145-PSCA and PC-3-PSCA cells, we observed higher IFNγ induction by PSCA-28ζ CAR T cells against low-expressing PC-3-PGK100p cells compared to PSCA-BBζ CAR T cells ( and d). To rule out potential tumor-specific effects on cytokine production, we performed similar IFNγ measurements by PSCA-CAR T cells against varying concentrations of plate-bound recombinant human PSCA protein. While CD28- and 4-1BB-containing CAR T cells produced similar IFNy against high antigen density, PSCA-BBζ CAR T cells showed an enhanced dose-responsiveness to decreasing concentrations of antigen (). Similar IFNγ trends were observed with FACS-sorted PSCA-28ζ and PSCA-BBζ CAR T cells with comparable CAR surface expression (Figure S2 c and d). We further confirmed these differences by measuring intracellular IFNγ and cell surface CD107a expression as an early measure of cytotoxicity ( and g).
4-1BB-containing PSCA-CARs demonstrate improved tumor killing, with reduced T cell exhaustion and greater expansion compared to CD28-containing PSCA-CARs
The development of second-generation CARs containing intracellular co-stimulatory signaling domains has shown improved anti-tumor potency and persistence of CAR T cells.Citation19,34 Current findings suggest that co-stimulation with 4-1BB yields a more persistent and durable therapy compared to CD28 in some CAR settings,Citation35,36 although the mechanisms driving this phenomenon are still being elucidated. To further investigate functional differences between CD28- and 4-1BB-containing PSCA-CARs, we performed in vitro tumor killing assays. PSCA-28ζ or PSCA-BBζ CAR T cells were co-cultured with various tumor targets and flow cytometry was used to quantify tumor cell killing. While both PSCA-28ζ and PSCA-BBζ CAR T cells killed high PSCA-expressing tumor cells with similar efficiency, PSCA-28ζ showed targeting of wild-type DU145 and PC-3 tumor cells to a greater extent than PSCA-BBζ ( and b). FACS-sorted PSCA-28ζ and PSCA-BBζ CAR T cells with comparable CAR surface expression showed similar in vitro trends in tumor cell killing (Figure S2 e). Across multiple tumor cell lines tested, CD28-containing PSCA-CAR T cells required a lower level of PSCA expression for tumor cell killing (Figure S4 b). In addition to more selective killing of tumor cells with high antigen density, PSCA-BBζ CAR T cells exhibited less evidence of exhaustion compared to PSCA-28ζ CAR T cells, as indicated by reduced expression of programmed death-1 (PD-1) ( and d), and other exhaustion markers including LAG3 and TIM3 (data not shown).
Figure 3. PSCA-CARs with 4-1BB co-stimulation demonstrate improved tumor killing, with reduced T cell exhaustion and greater antigen-specific expansion compared with CD28 co-stimulation in vitro. (a) Representative flow cytometry dot plots of a tumor killing assay comparing Mock, PSCA-28ζ, or PSCA-BBζ CAR T cells following a 3-day co-culture with DU145 or DU145-PSCA tumor cells at a 1:2 E:T ratio. (b) Quantification of tumor killing by PSCA-CAR T cells compared to Mock from (a). (c) Representative flow cytometry zebra plots of PD-1 expression in PSCA-CAR T cells following a 3-day co-culture with indicated tumor targets. (d) Quantification of PD-1 expression on CD8+ CAR+ T cells following a 3-day co-culture with indicated tumor targets at a 1:2 E:T ratio. (e) Quantification of tumor killing and CAR+ T cell count after 4 or 8 days of co-culture with PC-3-PSCA tumor cells at a 1:20 E:T ratio. (f) Quantification of CD137 and PD-1 expression on PSCA-CAR T cells from (e).
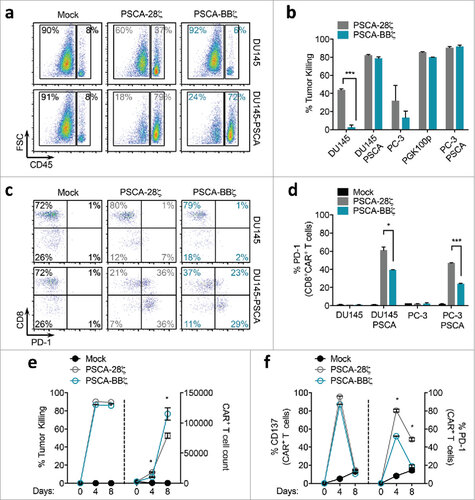
We next sought to determine if the differences in PD-1 expression could have an effect on antigen-dependent in vitro T cell expansion. Surprisingly, after 4 days of culture, PSCA-28ζ CAR T cells showed superior expansion compared with PSCA-BBζ CAR T cells against PSCA-expressing tumors despite markedly higher induction of PD-1. By day 8 of culture, however, PSCA-BBζ CAR T cells surpassed PSCA-28ζ CAR T cells in T cell numbers and maintained a lower exhaustion phenotype ( and f). Importantly, both PSCA-28ζ and PSCA-BBζ CAR T cells killed the tumor cells equivalently and showed comparable CD137 expression kinetics over the 8-day assay. T cells with similar cell surface CAR expression showed similar trends both in exhaustion phenotype and in antigen-dependent T cell expansion (Figure S2 f and g). Together, these data suggest that 4-1BB co-stimulation allows for potent and selective killing of high PSCA-expressing tumor cells while minimizing activity against low PSCA-expressing tumor cells. Additionally, PSCA-BBζ CAR T cells maintain lower levels of exhaustion and appear to have a superior ability to expand in vitro against prolonged antigen exposure compared to PSCA-28ζ CAR T cells.
PSCA-CAR T cells show robust therapeutic efficacy in subcutaneous prostate cancer models
As a first attempt to evaluate our PSCA-CAR T cells in xenograft models of prostate cancer, we treated mice bearing subcutaneous PC-3-PSCA tumors with a single intratumoral (i.t.) injection of 5 × 106 PSCA-BBζ CAR T cells. We observed complete tumor regression within two weeks following i.t. T cell injection (Figure S6 a). Although tumor regression was evident for over 30 days, tumors eventually recurred in the majority of animals with similar kinetics as the primary tumor, which we will further address below. To establish whether systemic therapy of CAR T cells was achievable in this solid tumor model, we intravenously (i.v.) delivered varying doses of PSCA-BBζ CAR T cells. While 5 × 106 PSCA-CAR T cells delivered i.v. showed complete regression of tumors, we observed similar yet delayed therapeutic efficacy with as little as 0.25 × 106 PSCA-CAR T cells (). Larger PC-3-PSCA tumors (∼500 mm3) also rapidly regressed with a single i.v. injection of 5 × 106 PSCA-BBζ CAR T cells (). We observed significant tumor infiltration of human T cells 11 days following CAR T cell i.v. infusion (, upper panel), which also expressed Granzyme B (, lower panel), a marker of T cell cytolytic activity. Tumors from Mock-treated mice showed few human T cells or Granzyme B expression at the same time point.
Figure 4. Robust therapeutic efficacy of PSCA-BBζ CAR T cells in subcutaneous and orthotopic bone metastatic human xenograft models of prostate cancer. (a) Tumor volume (mm3) in NSG mice bearing subcutaneous PC-3-PSCA (2.5 × 106) tumors on day 0, treated with Mock or PSCA-BBζ CAR T cells at the indicated doses by intratumoral (i.t.) injection on day 34. N = 4 mice per group. Data are representative of at least two independent experiments. (b) Mice with large tumors (approx. 500 mm3) treated with 5 × 106 Mock or CAR T cells by i.v. injection on day 51. N = 3 mice per group. Data are representative of at least two independent experiments. (c) Immunohistochemistry of PC-3-PSCA tumors, harvested 11 days post i.v. T cell treatment, stained with human CD3 (upper panels) and Granzyme B (lower panels). (d) Mice bearing intratibial tumors, with PC-3 (wild-type) cells (0.2 × 106) in the right hind leg, and PC-3-PSCA cells (0.2 × 106) in the left hind leg. On day 14, mice were treated with 5 × 106 firefly luciferase-positive (∼30%) Mock or PSCA-BBζ CAR T cells by i.v. injection. T cell trafficking was monitored at 4 hours, 1 day, 2 days, and 4 days by non-invasive optical imaging (Xenogen). Quantification of flux images, showing the ratio of T cells that trafficked to PC-3-PSCA vs. PC-3 (wild-type) tumors. N = 4 – 6 mice per group. (e) NSG mice bearing intratibial (left hind leg) PC-3-PSCA-eGFP-ffluc (0.2 × 106). Tumor growth kinetics were monitored by non-invasive optical imaging (Xenogen). On day 14, mice were i.v. injected with 5 × 106 Mock or varying doses of PSCA-BBζ CAR T cells. Representative flux images of mice on day 13 (pre-treatment) and day 33 are shown. (f) Quantification of flux images (with region of interest (ROI) at site of tumor injection) from tumor only, Mock T cells (5 × 106), and PSCA-BBζ CAR T cells (5 × 106, 2.5 × 106, 1 × 106, 0.5 × 106) groups. N ≥ 4 mice per group for CAR groups. Data are representative of at least two independent experiments.
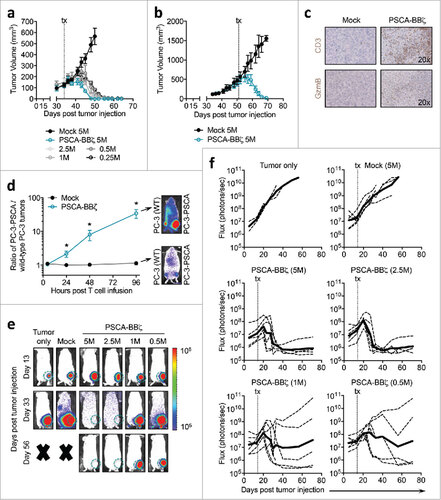
Recurrence following single antigen-specific CAR T cell therapy may be an expected phenomenon given the heterogeneous antigen profile of many solid tumors. To better understand the delayed tumor recurrences that were observed in , we performed immunohistochemistry to assess antigen expression in tumors from i.t. T cell-treated mice. Interestingly, while Mock T cell-treated tumors were highly positive for PSCA, tumors that recurred following PSCA-BBζ CAR T cell treatment were PSCA negative (Figure S6 b, upper panel). In the same recurring tumors, human T cells were present (Figure S6 b, middle panel), even though these tumors were harvested at least 2-months post-CAR T cell treatment. PC-3 cells also expressed HER2 in vitro (Figure S6 c), and we further confirmed that both Mock- and PSCA-BBζ CAR T cell-treated recurrent tumors expressed HER2 at equivalent levels in vivo (Figure S6 b, lower panel). To determine whether PSCA-negative tumors were still susceptible to CAR T cell therapy, we treated recurrent tumors by i.t. injection with either Mock, PSCA-directed- or HER2-directed-CAR T cells. Although recurrent PSCA-negative tumors were non-responsive to PSCA-CARs, they were susceptible to HER2-CAR T cells (Figure S6 d), suggesting that one of the mechanisms of CAR T cell evasion was antigen escape.
PSCA-CAR T cells traffic to bone and exhibit anti-tumor efficacy in bone metastatic disease
One of the major obstacles for cellular immunotherapy is the immunosuppressive microenvironment observed in solid cancers. This microenvironment is also evident in bone metastases, which can hamper effective trafficking and survival of T cells. To directly evaluate trafficking of CAR T cells to bone metastatic prostate tumors, we i.v. injected firefly luciferase-expressing Mock or PSCA-BBζ CAR T cells in mice bearing intratibial wild-type PC-3 (anatomical right tibia) and PC-3-PSCA (anatomical left tibia) tumors. Interestingly, while Mock and PSCA-CAR T cells showed equal early trafficking to both tumors (at 4 hours post T cell infusion), PSCA-CAR T cells were predominantly found in PSCA-expressing tumors at 1 day following T cell injection, which increased over the 4 days of imaging (), indicating antigen-dependent trafficking and/or CAR T cell expansion in PSCA-positive tumors. We next performed a therapeutic study by injecting PC-3-PSCA tumor cells into the intratibial space, and on day 14 post tumor engraftment, we i.v. treated these mice with varying doses of PSCA-BBζ CAR T cells (0.5 × 106 to 5 × 106) (). The majority of mice treated with either 5 × 106 or 2.5 × 106 CAR T cells showed complete tumor regression whereas mice treated with either 1 × 106 or 0.5 × 106 CAR T cells had a more heterogeneous therapeutic response (). Importantly, when compared to the s.c. tumor model (), it appears that higher doses of i.v. administered CAR T cells are required for efficacy with this orthotopic intratibial tumor model. These data highlight potential differences in the solid tumor microenvironment and their impact on CAR T cell efficacy.
4-1BB-containing PSCA-CARs provide superior persistence and durable anti-tumor responses in a clinically relevant bone metastatic prostate cancer model
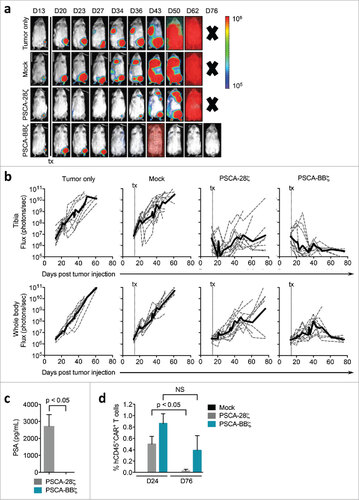
We extended these orthotopic studies using the endogenous PSCA-expressing bone metastatic prostate cancer patient-derived xenograft cell line, LAPC-9. On day 14 post tumor engraftment, mice treated with a single i.v. injection of 5 × 106 PSCA-BBζ CAR T cells showed near complete regression of tumors at the intratibial tumor site (). Although intratibial tumors were effectively targeted, LAPC-9 tumors disseminated to other sites in the body, which were found to be particularly evident in lymphoid tissue (axillary and inguinal) and the thymus as confirmed by immunohistochemistry (data not shown). While these tumors seemingly grew for several weeks after initial regression in the bone, they were ultimately eradicated by PSCA-BBζ CAR T cells. Based on the requirement of persistent T cells for complete anti-tumor activity of PSCA-CARs, we decided to compare PSCA-CARs containing either CD28 or 4-1BB co-stimulatory signaling domains. Due to limitations in the quantity of FACS-sorted T cells to yield CD28- and 4-1BB-containing PSCA-CARs with similar CAR surface expression and the overall comparability in our in vitro findings with these cells whether sorted or not, the following in vivo studies were carried out using unsorted PSCA-CAR T cells. While both PSCA-CAR T cells showed dramatic regression of intratibial tumors, the majority of mice treated with PSCA-BBζ CAR T cells demonstrated curative responses (8 of 11 mice), while PSCA-28ζ CAR T cell-treated mice had tumor recurrences in primary (7 of 11) and disseminated tumors (8 of 11) ( and b). We confirmed tumor recurrence in PSCA-28ζ CAR T cell-treated mice by quantifying PSA levels in the blood at Day 76 post CAR T cell treatment (). We also quantified CAR T cells in the blood of treated animals, and while CAR T cells were observed in both groups at Day 24 post tumor injection, PSCA-BBζ CAR T cells were more abundant than PSCA-28ζ CAR T cells at Day 76, indicating greater T cell persistence (). Further, tumor recurrences in mice treated with PSCA-28ζ CAR T cells retained PSCA expression (Figure S7). Overall, these studies demonstrate potent and durable anti-tumor efficacy with PSCA-BBζ CAR T cells in multiple tumor systems, including orthotopic bone metastatic patient-derived xenograft models of prostate cancer.
Figure 5. Durable anti-tumor efficacy of PSCA-BBζ CAR T cells compared with PSCA-28ζ CAR T cells in a prostate cancer patient-derived bone metastatic xenograft model. (a) NSG mice bearing intratibial (left hind leg) LAPC-9-eGFP-ffluc (0.15 × 106). Tumor growth kinetics were monitored by non-invasive optical imaging (Xenogen). On day 14, mice were i.v. injected with 5 × 106 Mock, PSCA-28ζ or PSCA-BBζ CAR T cells. Representative flux images of mice on indicated days are shown. (b) Quantification of flux images, with ROI at the tibia (upper panels) or from whole body (lower panels) from each treatment group. (c) PSA levels determined by ELISA from serum harvested from treated mice (n = 2 – 3 per group) at day 76 post tumor injection. (d) Flow cytometric analysis of peripheral blood of mice 24 and 76 days post tumor injection (n = 2 – 3 per group). Data are compiled from two independent in vivo experiments.
Discussion
In this study, we identified a PSCA-specific CAR with enhanced selectivity for tumor cells with high expression of cell-surface PSCA by modifying the co-stimulatory signaling domain. While PSCA represents a promising target for solid cancer immunotherapy, it is expressed at lower levels on some normal “noncancerous” tissue.Citation6,7 The importance of CAR selectivity for high tumor antigen density was underscored in a recent case report where toxicity of CAR T cells was observed, and likely attributed to targeting normal tissue with low levels of antigen expression.Citation37 Prior preclinical studies have tested PSCA-CAR T cells, however, the extent to which the CAR T cells targeted non- or low-PSCA expressing tumor cells was not thoroughly investigated. The first example of a PSCA-CAR was a first-generation CAR based on the humanized 7F5 scFv, with efficient in vitro cytolytic activity against PSCA-positive bladder cancer cells.Citation38 More recently, a third-generation PSCA-CAR (containing both OX40 and CD28 co-stimulatory domains) was evaluated using melanoma cells engineered to express PSCA.Citation39 While these CARs utilized the same humanized A11 scFv we used in the current study, they did not evaluate additional CAR designs for improved functionality. PSCA-CAR T cells based on a novel, fully human Ha1-4.117 scFv were recently described, demonstrating significant delay and even regression of some pancreatic tumors treated with either second-generation (containing CD28) or third-generation (containing 4-1BB and CD28) CAR T cells.Citation40 In that study, although third-generation 4-1BB-CD28-containing PSCA-CARs conferred a survival advantage over second-generation CD28-containing CARs, superior anti-tumor efficacy of second-generation CARs was demonstrated with the addition of three injections of recombinant human IL-2. In contrast, our studies described here show that 4-1BB-containing PSCA-CAR T cells eradicated significant tumor burden with a low T cell dose without the requirement for exogenous IL-2, which was also recently validated in subcutaneous xenograft models of prostate cancer.Citation41
Our data reveal co-stimulatory signaling to be a critical modular component that requires empirical determination in the specific disease and target setting to achieve a fully optimized CAR. Our current studies with PSCA-CARs support recent findings by Cherkassky et al. that 4-1BB-containing Mesothelin-specific CAR T cells provided more durable anti-tumor therapy compared with CARs with CD28 co-stimulation.Citation42 Our study also identified differences between CD28- and 4-1BB-containing CAR T cells in targeting tumor cells with varying antigen density. 4-1BB-containing PSCA-CAR T cells more selectively targeted tumor cells with higher antigen density, which could potentially result in reduced CAR activity in normal tissues expressing low levels of PSCA. While this may hold true against normal cells with low but detectable cell surface PSCA expression, our in vitro studies using five normal cell lines lacking PSCA protein expression induced very low activity of PSCA-28ζ CARs. In an attempt to identify CARs that selectively target tumor tissue over normal tissue with low antigen density, other preclinical studies have focused on modifying the antigen-binding domain. A recent study optimizing HER2-specific CAR T cells indicated that generating scFvs with reduced affinity for the tumor antigen achieved CARs with greater selectivity for tumors with high tumor antigen density.Citation18 In some scenarios, the extracellular spacer domain is decisive for CAR anti-tumor activity, and future studies may elucidate roles for this domain in generating CARs with similar high tumor antigen density requirements. Additional studies are warranted to determine whether PSCA-BBζ CARs target normal tissues that express PSCA (e.g., esophagus, stomach, and colon). This phenomenon is particularly important for non-HLA-restricted solid tumor antigens, where targeting only higher tumor antigen-expressing tumor cells is likely imperative to minimize the “on-target off-tumor” activity of CAR T cells.
Our data using clinically relevant bone metastatic prostate cancer models highlights the need for better preclinical models to evaluate different CAR designs in vivo. Our PSCA-CARs effectively homed to bone metastases, as shown by imaging of T cell trafficking. Compared with subcutaneous models of prostate cancer, where complete responses were achieved using T cell doses as low as 0.25 × 106, we also showed that nearly a 10-fold higher CAR T cell dose (2.5 × 106) was required to obtain similar responses in orthotopic bone metastasis models. We are, to our knowledge, the first to evaluate PSCA-CARs using an endogenous PSCA-expressing patient-derived prostate cancer xenograft model. Although testing CARs in orthotopic tumors more closely recapitulates the clinical setting than do subcutaneous tumors, an important next step would be the use of syngeneic mouse models to better interrogate the contributions of the immune-stromal tumor microenvironment and cytokine milieu to an overall CAR T cell therapy response. Collectively, these studies show that modification of CAR components and the use of improved preclinical models are required to create an optimal CAR T cell for solid cancer clinical applications.
Materials and methods
Cell lines
Human metastatic prostate cancer cell lines DU145 (ATCC HTB-81) and PC-3 (ATCC CRL-1435) were cultured in RPMI-1640 (Lonza) containing 10% fetal bovine serum (FBS, Hyclone), and 1X antibiotic-antimycotic (AA, Gibco) (complete RPMI). DU145 and PC-3 cells were authenticated by STR Profiling and verified mycoplasma negative (DDC Medical, OH). The human fibrosarcoma cell line, HT1080 (ATCC CCL-121), the human lung epithelial cancer cell line, A549 (ATCC CCL-185), the human breast cancer cell line MDA-MB-468 (ATCC HTB-132), and the human embryonic kidney cell line, 293T (ATCC CRL-3216), were cultured in Dulbecco's Modified Eagles Medium (DMEM, Life Technologies) containing 10% FBS, 1X AA, 25 mM HEPES (Irvine Scientific), and 2 mM L-Glutamine (Fisher Scientific) (complete DMEM). The human pancreatic cancer cell line, HPAC (ATCC CRL-2119), and the human breast cancer cell line, SKBR3 (ATCC HTB-30) were cultured in Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F12, Life Technologies) containing 10% FBS and 1X AA. The human pancreatic cancer cell line, Capan-1 (ATCC HTB-79) was cultured in Iscove's Modified Dulbecco's Medium (IMDM, Irvine Scientific) containing 20% FBS and 1X AA (complete IMDM). Human cardiac myocytes (PromoCell) and human skin fibroblastsCitation43 (a kind gift from Dr. Timothy O'Connor) were cultured in complete DMEM. Human bronchial epithelial cellsCitation44 (a kind gift from Drs. Timothy O'Connor and Jerry Shay) were cultured in Keratinocyte-SFM media (Life Technologies). Human pulmonary artery smooth muscle cells (PromoCell) were cultured in Smooth Muscle Cell Medium (PromoCell). Human renal proximal tubule epithelial cells (ATCC) were cultured in Renal Epithelial Cell Basal medium with supplements (ATCC).
The human prostate cancer-derived xenograft LAPC-9 (a kind gift from Dr. Robert Reiter, UCLA) was cultured in complete IMDM. LAPC-9 cells were serially passaged in male NOD.Cg-Prkdcscid IL2rgtm1Wjl/ SzJ (NSG) mice, and single-cell suspensions were prepared as previously described.Citation31 Briefly, tumor tissue was harvested, minced in a petri dish, and digested with 1% Pronase E (Roche). Following a wash with complete IMDM, single-cell suspensions were filtered through a 40 µm cell strainer (Falcon), washed again, and frozen immediately.
DNA constructs and lentivirus production
DU145 and PC-3 tumor cells were engineered to express PSCA by transduction with epHIV7 lentivirus carrying the human PSCA gene (Accession #: NM_005672.4) under the control of the EF1α promoter. PSCA+ cells were stained with the mouse anti-human PSCA antibody (1G8) as described below (see ‘Intracellular/Extracellular Staining and Flow Cytometry’ section), and then underwent fluorescence-activated cell sorting (FACS) using the BD FACSAria™ Special Order Research Product (SORP) cell sorter. For generation of tumor cells with low PSCA expression, the PSCA gene was placed under the control of mutated versions of the PGK promoter as previously described.Citation30 The A11 scFvCitation29 sequence was kindly provided by Drs. Anna Wu and Robert Reiter (UCLA). CAR constructs with a truncated CD19 gene (CD19t) separated by a T2A ribosomal skip sequence were cloned in an epHIV7 lentiviral backbone. The extracellular spacer domain included the 129-amino acid middle-length CH2-deleted version (ΔCH2) of the IgG4 Fc spacer.Citation22 The intracellular co-stimulatory signaling domain contained that of either CD28 with a CD28 transmembrane domain, or 4-1BB with a CD4 transmembrane domain. The CD3ζ cytolytic domain was previously described.Citation45 The scFv for the HER2-BBζ CAR construct was based on Herceptin.
Lentivirus was generated by plating 293T cells in T-225 tissue culture flasks 1-day prior to transfection with packaging plasmids and desired CAR lentiviral backbone plasmid. Supernatants were collected after 3 to 4 days, filtered and centrifuged to remove cell debris, and incubated with 2mM magnesium and 25U/mL Benzonase® endonuclease (EMD Millipore) to remove contaminating nucleic acids. Supernatants were combined and concentrated via high-speed centrifugation (6080g) overnight at 4°C. Lentiviral pellets were then resuspended in phosphate-buffered saline (PBS)-lactose solution (4g lactose per 100 mL PBS), aliquoted and stored at −80°C for later use. Lentiviral titers, as determined by CD19t expression, were quantified using HT1080 cells.
T cell isolation, lentiviral transduction, and ex vivo expansion
Leukapheresis products were obtained from consented research participants (healthy donors) under protocols approved by the City of Hope (COH) Internal Review Board (IRB). On the day of leukapheresis, peripheral blood mononuclear cells (PBMC) were isolated by density gradient centrifugation over Ficoll-Paque (GE Healthcare) followed by multiple washes in PBS/EDTA (Miltenyi Biotec). Cells were rested overnight at room temperature (RT) on a rotator, and subsequently washed and resuspended in X-VIVO-15 (Lonza) with 10% FBS (complete X-VIVO). PBMCs were immediately frozen in CryoStor® CS5 cryopreservation media (BioLife Solutions) until further processing.
Freshly thawed PBMC were washed once and cultured in complete X-VIVO containing 100 U/mL recombinant human IL-2 (rhIL-2, Novartis Oncology) and 0.5 ng/mL recombinant human IL-15 (rhIL-15, CellGenix). For CAR lentiviral transduction, T cells were cultured with CD3/CD28 Dynabeads® (Life Technologies), protamine sulfate (APP Pharmaceuticals), cytokine mixture (as stated above) and desired lentivirus at varying MOI either the day of, or the day following, bead stimulation. Spinoculation was performed by centrifugation at 2000 rpm for 30 min at 32°C with no brake. Cells were then cultured in and replenished with fresh complete X-VIVO containing cytokines every 2–3 days. After 7–9 days, beads were magnetically removed, and cells were further expanded in complete X-VIVO containing cytokines to achieve desired cell yield. CAR T cells were positively selected for CD19t using the EasySep™ CD19 Positive Enrichment Kit I or II (StemCell Technologies) according to the manufacturer's protocol. Following further expansion, cells were frozen prior to in vitro functional assays and in vivo tumor models. Purity and phenotype of CAR T cells were verified by flow cytometry.
Intracellular/extracellular staining and flow cytometry
For flow cytometric analysis, cells were resuspended in FACS buffer (Hank's balanced salt solution without Ca2+, Mg2+, or phenol red (HBSS−/−, Life Technologies) containing 2% FBS and 1x AA). For PSCA staining, the mouse anti-human PSCA antibody (1G8) was kindly provided by Dr. Robert Reiter, UCLA. For detecting CAR scFv, biotinylated Protein-L (GenScript USA) was used as previously described.Citation46 Cells were incubated with primary antibodies for 30 minutes at 4°C in the dark before proceeding to secondary staining. For extracellular and secondary staining, cells were washed twice prior to 30 min incubation at 4°C in the dark with fluorescein isothiocyanate (FITC), phycoerythrin (PE), peridinin chlorophyll protein complex (PerCP), PerCP-Cy5.5, PE-Cy7, allophycocyanin (APC), and APC-Cy7 (or APC-eFluor780)-conjugated antibodies. CD3 (BD Biosciences, Clone: SK7), CD4 (BD Biosciences, Clone: SK3), CD8 (BD Biosciences, Clone: SK1), CD14 (BD Biosciences, Clone: MΦP9), CD19 (BD Biosciences, Clone: SJ25C1), CD25 (BD Biosciences, Clone: 2A3), mouse CD45 (BioLegend, Clone: 30-F11), human CD45 (BD Biosciences, Clone: 2D1), CD45RA (BD Biosciences, Clone: HI100), CD45RO (BD Biosciences, Clone: UCHL1), CD62L (BD Biosciences, Clone: DREG-56), CD69 (BD Biosciences, Clone: L78), CD95 (BD Biosciences, Clone: DX2), CD107a (BD Biosciences, Clone: H4A3), CD137 (BD Biosciences, Clone: 4B4-1), LAG3 (CD223) (eBiosciences, Clone: 3DS223H), PD-1 (CD279) (eBiosciences, Clone: J105), TIM3 (CD366) (eBiosciences, Clone: F38-2E2), CCR7 (BD Biosciences, Clone: 3D12), IFNγ (BioLegend, Clone: MD-1), Goat Anti-Mouse Ig (BD Biosciences), and streptavidin (BD Biosciences) were used. Cell viability was determined using 4′, 6-diamidino-2-phenylindole (DAPI, Sigma). For intracellular staining, cells were fixed, permeabilized, and processed according to the PE Active-Caspase-3 Apoptosis kit (BD Biosciences) manufacturer's protocol. Cells were then incubated with fluorophore-conjugated antibodies for 30 minutes at 4°C in the dark, and washed twice prior to resuspension in FACS buffer and acquisition on the MACSQuant Analyzer 10 (Miltenyi Biotec). Data were analyzed with FlowJo software (v10, TreeStar). Where indicated, CAR T cells were FACS sorted using the BD FACSAria™ SORP cell sorter for cells expressing high or low levels of CD19t (termed CD19thi or CD19tlo).
In vitro T cell functional assays
For degranulation and intracellular cytokine assays, CAR T cells and tumor targets were co-cultured at varying effector:target (E:T) ratios in complete X-VIVO in the absence of exogenous cytokines in round-bottom 96-well tissue culture-treated plates (Corning). FITC-CD107a was added to cultures and after incubating for 4 – 6 hrs at 37°C, cells were fixed and permeabilized before analysis by flow cytometry as described above. For tumor killing assays, CAR T cells and tumor targets were co-cultured at varying E:T ratios in complete X-VIVO in the absence of exogenous cytokines in 96-well plates for 1 – 5 days and analyzed by flow cytometry as described above. For extended tumor killing assays, CAR T cells and tumor targets were co-cultured at an E:T ratio of 1:20 in complete X-VIVO in the absence of exogenous cytokines in 96-well plates for 4 – 8 days (with media replenishment every 2 – 4 days) and analyzed by flow cytometry. Tumor killing by CAR T cells was calculated by comparing CD45-negative cell counts relative to that observed by Mock (untransduced) T cells. For T cell activation assays, CAR T cells and tumor targets were co-cultured at an E:T ratio of 1:2 in complete X-VIVO in the absence of exogenous cytokines in 96-well plates for the indicated time points and analyzed by flow cytometry for specific markers of T cell activation.
ELISA and multiplex cytokine assays
Varying concentrations of recombinant human PSCA protein (amino acids 23–95; Abnova) was coated overnight in 1X PBS at 4°C on high-affinity 96-well flat bottom plates (Corning). Wells were washed twice with 1X PBS, blocked with 10% FBS for 1 hr, and washed again. CAR T cells (5 × 103 in 200 μL) were added to coated wells. Where specified, tumor targets (5 × 103) were incubated with T cells in non-coated wells (final volume of 200 μL). Following an overnight incubation at 37°C, supernatants were harvested and processed according to the Human IFNγ ELISA Ready-SET-GO!® (eBioscience) manufacturer's protocol. Plates were read at 450 nm using the Wallac Victor3 1420 Multilabel Counter (Perkin-Elmer) and Wallac 1420 Workstation software. Alternatively, supernatants were analyzed for multiple cytokines using the Multiplex Bead Immunoassay Kit (Invitrogen) according to the manufacturer's protocol. Mouse serum was analyzed for prostate-specific antigen (PSA) using the human PSA/KLK3 ELISA (Abcam) according to the manufacturer's protocol.
Quantitative PCR
Tumor cells (plated at 0.5 × 106/mL) were cultured for one day prior to RNA isolation. RNA was extracted using RNeasy® Mini Kit column purification (Qiagen). cDNA was prepared using SuperScript™ IV First-Strand Synthesis System (Invitrogen). RNA primers were generated using TaqMan® Gene Expression Assays specific to either PSCA (Hs04166224_g1, Life Technologies) or GAPDH (Hs02758991_g1, Life Technologies). qPCR was performed on a ViiA™ 7 Real-Time PCR System (Thermo Fisher). Primer sets were validated using a standard curve across a specified dynamic range with a single melting curve peak. Expression of target genes was normalized to GAPDH.
In vivo tumor studies
All animal experiments were performed under protocols approved by the City of Hope Institutional Animal Care and Use Committee. For subcutaneous tumor studies, PC-3 and DU145 cells (2.5 × 106) were prepared in HBSS−/− and injected subcutaneously in the left depilated belly of male NSG mice. Tumor growth was monitored 3 times per week via caliper measurement. Once tumor volumes reached 50 – 500 mm3, CAR T cells were prepared in PBS and injected either intratumorally (i.t.) or intravenously (i.v.). Once tumors reached 15 mm in diameter, mice were euthanized and tumors were harvested and processed for immunohistochemistry as described below. When subcutaneous tumors recurred, mice were treated by i.t. injection with either PSCA-CARs or HER2-CARs. Peripheral blood was collected from isoflurane-anesthetized mice by retro-orbital (RO) bleed through heparinized capillary tubes (Chase Scientific) and into polystyrene tubes containing a heparin/PBS solution (1000 units/mL, Sagent Pharmaceuticals). Approximately 150 µL of blood was collected per mouse. Blood was lysed with 1X Red Cell Lysis Buffer (Sigma) according to the manufacturer's protocol, and then washed, stained and analyzed by flow cytometry as described above.
For orthotopic intratibial tumor studies, LAPC-9 and PC-3-PSCA were transduced with lentivirus carrying enhanced green fluorescent protein (eGFP)/firefly luciferase (ffluc) to allow for non-invasive optical imaging (Xenogen) once implanted into mice (resulting lines named LAPC-9-eGFP-ffluc and PC-3-PSCA-eGFP-ffluc). Briefly, these lines were incubated with polybrene (4 mg/mL, Sigma) and the eGFP-ffluc lentivirus (see above), followed by cell sorting for GFP+ cells using the BD FACSAria™ SORP cell sorter. Freshly sorted LAPC-9-eGFP-ffluc cells were serially passaged in NSG mice as described above. PC-3-PSCA-eGFP-ffluc cells (2 × 105) or LAPC-9-eGFP-ffluc cells (1.5 × 105) were prepared as in subcutaneous models. Mice were anesthetized by intraperitoneal (i.p.) injection of ketamine/xylazine and gaseous isoflurane prior to tumor injection. Tumor cells (in 30 µL HBSS−/−) were injected in the intratibial space of the mouse hind leg. After 14 days, mice were i.v. injected with CAR T cells. Tumor growth was monitored via biweekly optical imaging (IVIS, Xenogen) and flux signals were analyzed with Living Image software (Xenogen). For imaging, mice were injected i.p. with 150 µL D-luciferin potassium salt (Perkin Elmer) suspended in PBS at 4.29 mg/mouse.
For T cell trafficking studies, mice were implanted in the right intratibial space with wild-type (non-firefly luciferase expressing) PC-3 cells (2 × 105) and in the left intratibial space with PC-3-PSCA cells (2 × 105). After 14 days, mice were i.v. injected with 5 × 106 Mock or PSCA-BBζ CAR T cells that had been co-transduced with eGFP-ffluc lentivirus. T cells were CAR enriched using CD19 enrichment kits as described above, and determined to be approximately 30% eGFP+ by flow cytometry. T cell trafficking was monitored by non-invasive optical imaging (Xenogen) at 4 hr, 1 day, 2 days, and 4 days post T cell infusion. Flux signals were analyzed as described above.
Immunohistochemistry
Tumor tissue was fixed for up to 3 days in 4% paraformaldehyde (Boston BioProducts) and stored in 70% ethanol until further processing. Histology was performed by the Pathology Core at City of Hope. Briefly, paraffin-embedded sections (10-µm) were stained with hematoxylin & eosin (H&E, Sigma-Aldrich), mouse anti-human CD3 (DAKO), mouse anti-human PSCA (Abcam), rat anti-human HER2 (DAKO), and rat anti-human Granzyme-B (eBioscience). Images were obtained using the Nanozoomer 2.0HT digital slide scanner and the associated NDP.view2 software (Hamamatzu).
Statistical analysis
Data are presented as mean ± SEM, unless otherwise stated. Statistical comparisons between groups were performed using the unpaired two-tailed Student's t test to calculate p value. #p < 0.05, ##p < 0.01, ###p < 0.001; ns, not significant.
Conflict of interest
The authors declare no potential conflicts of interest. Patents associated with this work have been licensed by Mustang Bio, Inc., for which S.J.P., C.E.B., S.J.F. receive royalties.
Author contributions
Study concept and design: S.J.P., E.A.G., D.T., K.T.K. and S.J.F. Experimental methodology: S.J.P., E.A.G., D.T., K.T.K., J.P.M., A.K.P., B.J., Y.Y., X.Y., J.P.M., R.U., L.W., W-C.C., S.W. Data analysis and interpretation of results: S.J.P., E.A. G., D.T., K.T.K. Writing and review of manuscript: S.J.P., E.A.G., K.T.K., S.P., R.E.R., A.M.W., C.E.B., and S.J.F.
supplemental_material.pdf
Download PDF (7.4 MB)Acknowledgments
We thank staff members of the Flow Cytometry Core, the Animal Facility Core, and the Pathology Core in the Beckman Research Institute at the City of Hope Comprehensive Cancer Center for excellent technical assistance. Research reported in this publication was supported by the Movember-Prostate Cancer Foundation (PCF) Challenge Award 2013 (S. Forman), the PCF Young Investigator Award 2015 (S. Priceman), the PCF Special Challenge Award 2016 (S. Priceman, S. Pal, S. Forman), and the National Comprehensive Cancer Network Young Investigator Award 2016 (S. Priceman). We thank Drs. Sandra Thomas and Julie Ostberg for manuscript editing and scientific feedback. We also thank Drs. Robert Reiter and Anna Wu for input on PSCA scFv development for the aforementioned CAR constructs.
References
- Schweizer MT, Drake CG. Immunotherapy for prostate cancer: Recent developments and future challenges. Cancer Metastasis Rev. 2014;33(2–3):641-55. doi:10.1007/s10555-013-9479-8. PMID:24477411
- Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480-9. doi:10.1038/nature10673. PMID:22193102
- Kakarla S, Gottschalk S. CAR T cells for solid tumors: Armed and ready to go? Cancer J. 2014;20(2):151-5. doi:10.1097/PPO.0000000000000032. PMID:24667962
- Hillerdal V, Essand M. Chimeric antigen receptor-engineered T cells for the treatment of metastatic prostate cancer. BioDrugs. 2015;29(2):75-89. doi:10.1007/s40259-015-0122-9. PMID:25859858
- Saeki N, Gu J, Yoshida T, Wu X. Prostate stem cell antigen: A Jekyll and Hyde molecule? Clin Cancer Res. 2010;16(14):3533-8. doi:10.1158/1078-0432.CCR-09-3169. PMID:20501618
- Reiter RE, Gu Z, Watabe T, Thomas G, Szigeti K, Davis E, Wahl M, Nisitani S, Yamashiro J, Le Beau MM, et al. Prostate stem cell antigen: A cell surface marker overexpressed in prostate cancer. Proc Natl Acad Sci U S A. 1998;95(4):1735-40. doi:10.1073/pnas.95.4.1735. PMID:9465086
- Gu Z, Thomas G, Yamashiro J, Shintaku IP, Dorey F, Raitano A, Witte ON, Said JW, Loda M, Reiter RE. Prostate stem cell antigen (PSCA) expression increases with high gleason score, advanced stage and bone metastasis in prostate cancer. Oncogene. 2000;19(10):1288-96. doi:10.1038/sj.onc.1203426. PMID:10713670
- Fong L, Small EJ. Immunotherapy for prostate cancer. Semin Oncol. 2003;30(5):649-58. doi:10.1016/S0093-7754(03)00350-6. PMID:14571412
- Ristau BT, O'Keefe DS, Bacich DJ. The prostate-specific membrane antigen: Lessons and current clinical implications from 20 years of research. Urol Oncol. 2014;32(3):272-9. doi:10.1016/j.urolonc.2013.09.003. PMID:24321253
- Kochenderfer JN, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol. 2013;10(5):267-76. doi:10.1038/nrclinonc.2013.46. PMID:23546520
- Sadelain M. CAR therapy: The CD19 paradigm. J Clin Invest. 2015;125(9):3392-400. doi:10.1172/JCI80010. PMID:26325036
- Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388-98. doi:10.1158/2159-8290.CD-12-0548. PMID:23550147
- Priceman SJ, Forman SJ, Brown CE. Smart CARs engineered for cancer immunotherapy. Curr Opin Oncol. 2015;27(6):466-74. doi:10.1097/CCO.0000000000000232. PMID:26352543
- Abate-Daga D, Davila ML. CAR models: Next-generation CAR modifications for enhanced T-cell function. Mol Ther Oncolytics. 2016;3:16014. doi:10.1038/mto.2016.14. PMID:27231717
- Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, Mi T, Switzer K, Singh H, Huls H, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 2015;75(17):3505-18. doi:10.1158/0008-5472.CAN-15-0139. PMID:26330164
- Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, Dimitrov DS, Morgan RA, FitzGerald DJ, Barrett DM, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121(7):1165-74. doi:10.1182/blood-2012-06-438002. PMID:23243285
- James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, Raubitschek AA, Forman SJ, Press OW. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol. 2008;180(10):7028-38. doi:10.4049/jimmunol.180.10.7028. PMID:18453625
- Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, Cogdill AP, Li N, Ramones M, Granda B, et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 2015;75(17):3596-607. doi:10.1158/0008-5472.CAN-15-0159. PMID:26330166
- van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14(7):499-509. doi:10.1038/nrd4597. PMID:26129802
- Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106(9):3360-5. doi:10.1073/pnas.0813101106. PMID:19211796
- Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453-64. doi:10.1038/mt.2009.83. PMID:19384291
- Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, Chang WC, Bretzlaff W, Starr R, Priceman S, et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther. 2015;23(4):757-68. doi:10.1038/mt.2014.208. PMID:25366031
- Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, Jensen MC, Riddell SR. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3(2):125-35. doi:10.1158/2326-6066.CIR-14-0127. PMID:25212991
- Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, Riddell SR. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 2013;19(12):3153-64. doi:10.1158/1078-0432.CCR-13-0330. PMID:23620405
- Kunkele A, Johnson AJ, Rolczynski LS, Chang CA, Hoglund V, Kelly-Spratt KS, Jensen MC. Functional tuning of CARs reveals signaling threshold above which CD8+ CTL antitumor potency is attenuated due to cell Fas-FasL-dependent AICD. Cancer Immunol Res. 2015;3(4):368-79. doi:10.1158/2326-6066.CIR-14-0200. PMID:25576337
- Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O'Neill A, Irlam J, Chester KA, Kemshead JT, Shaw DM, et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: Evaluation of four different scFvs and antigens. J Immunother. 2005;28(3):203-11. doi:10.1097/01.cji.0000161397.96582.59. PMID:15838376
- Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J Immunol. 2010;184(12):6938-49. doi:10.4049/jimmunol.0901766. PMID:20483753
- Beatty GL, Moon EK. Chimeric antigen receptor T cells are vulnerable to immunosuppressive mechanisms present within the tumor microenvironment. Oncoimmunology. 2014;3(11):e970027. doi:10.4161/21624011.2014.970027. PMID:25941599
- Lepin EJ, Leyton JV, Zhou Y, Olafsen T, Salazar FB, McCabe KE, Hahm S, Marks JD, Reiter RE, Wu AM. An affinity matured minibody for PET imaging of prostate stem cell antigen (PSCA)-expressing tumors. Eur J Nucl Med Mol Imaging. 2010;37(8):1529-38. doi:10.1007/s00259-010-1433-1. PMID:20354850
- Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, Kawalekar OU, Guedan S, McGettigan SE, Posey AD, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res. 2015;3(4):356-67. doi:10.1158/2326-6066.CIR-14-0186. PMID:25600436
- Craft N, Chhor C, Tran C, Belldegrun A, DeKernion J, Witte ON, Said J, Reiter RE, Sawyers CL. Evidence for clonal outgrowth of androgen-independent prostate cancer cells from androgen-dependent tumors through a two-step process. Cancer Res. 1999;59(19):5030-6. PMID:10519419
- James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Budde LE, Till BG, Raubitschek AA, Forman SJ, Press OW. Mathematical modeling of chimeric TCR triggering predicts the magnitude of target lysis and its impairment by TCR downmodulation. J Immunol. 2010;184(8):4284-94. doi:10.4049/jimmunol.0903701. PMID:20220093
- Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, Odak A, Gönen M, Sadelain M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113-7. doi:10.1038/nature21405. PMID:28225754
- Zhao Z, Condomines M, van der Stegen SJ, Perna F, Kloss CC, Gunset G, Plotkin J, Sadelain M. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28(4):415-28. doi:10.1016/j.ccell.2015.09.004. PMID:26461090
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507-17. doi:10.1056/NEJMoa1407222. PMID:25317870
- Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581-90. doi:10.1038/nm.3838. PMID:25939063
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843-51. doi:10.1038/mt.2010.24. PMID:20179677
- Morgenroth A, Cartellieri M, Schmitz M, Gunes S, Weigle B, Bachmann M, Abken H, Rieber EP, Temme A. Targeting of tumor cells expressing the prostate stem cell antigen (PSCA) using genetically engineered T-cells. Prostate. 2007;67(10):1121-31. doi:10.1002/pros.20608. PMID:17492652
- Hillerdal V, Ramachandran M, Leja J, Essand M. Systemic treatment with CAR-engineered T cells against PSCA delays subcutaneous tumor growth and prolongs survival of mice. BMC Cancer. 2014;14:30. doi:10.1186/1471-2407-14-30. PMID:24438073
- Abate-Daga D, Lagisetty KH, Tran E, Zheng Z, Gattinoni L, Yu Z, Burns WR, Miermont AM, Teper Y, Rudloff U, et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum Gene Ther. 2014;25(12):1003-12. doi:10.1089/hum.2013.209. PMID:24694017
- Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, Newick K, Lo A, June CH, Zhao Y, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016;76(6):1578-90. doi:10.1158/0008-5472.CAN-15-2524. PMID:26979791
- Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126(8):3130-44. doi:10.1172/JCI83092. PMID:27454297
- Bates SE, Zhou NY, Federico LE, Xia L, O'Connor TR. Repair of cyclobutane pyrimidine dimers or dimethylsulfate damage in DNA is identical in normal or telomerase-immortalized human skin fibroblasts. Nucleic Acids Res. 2005;33(8):2475-85. doi:10.1093/nar/gki542. PMID:15863724
- Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, Peyton M, Zou Y, Kurie JM, Dimaio JM, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64(24):9027-34. doi:10.1158/0008-5472.CAN-04-3703. PMID:15604268
- Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A, Wright C, Popplewell L, Raubitschek A, Forman SJ, et al. T-cell clones can be rendered specific for CD19: Toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101(4):1637-44. doi:10.1182/blood-2002-07-1989. PMID:12393484
- Zheng Z, Chinnasamy N, Morgan RA. Protein L: A novel reagent for the detection of chimeric antigen receptor (CAR) expression by flow cytometry. J Transl Med. 2012;10:29. doi:10.1186/1479-5876-10-29. PMID:22330761