
ABSTRACT
Therapy of metastatic melanoma advanced recently with the clinical implementation of signalling pathway inhibitors, such as vemurafenib, specifically targeting mutant BRAFV600E. In general, patients experience remarkable clinical responses under BRAF inhibitor (BRAFi) treatment but eventually progress within 6–8 months due to resistance development. Responding metastases show an increased immune cell infiltrate, including also NK cells, that, however, is no longer detectable in BRAFi-resistant lesions, suggesting NK cell activity should be exploited to prevent disease progression. Here, we examined the effects of BRAFi on the expression of ligands targeting activating NK cells receptors immediately after treatment onset, prior to resistance development. We demonstrate that BRAFV600E mutant melanoma cells cultured in the presence of vemurafenib, strongly decreased surface expression of ligands for NK activating receptors including the NKG2D-ligand, MICA, and the DNAM-1 ligand, CD155, and became significantly less susceptible to NK cell attack. NKG2D-ligand protein downregulation was due to a significant decrease in mRNA levels, already detectable 24 h after drug treatment. Interestingly, vemurafenib-induced MICA downregulation could be counteracted by treatment of melanoma cells with the histone deacetylase (HDAC) inhibitor (HDACi) sodium butyrate, that also upregulated the DNAM1-ligand, Nectin-2. HDACi treatment enhanced surface expression of NKG2D-ligands in the presence of BRAFi, accompanied by recovery of NK cell recognition, but only upon simultaneous drug application. These results suggest that co-administration of BRAFi and HDAC inhibitors as well as having direct effects on melanoma cell survival, could also synergise to improve NK cell recognition and avoid tumour immune evasion.
Introduction
Recent advances in the molecular characterization of melanoma cells have led to the development of new therapies for this aggressive, devastating type of skin cancer. Until 2009, the average survival time with metastatic melanoma was 6–12 months with a 5-year survival rate under 10%.Citation1 Since then, survival was significantly improved with the clinical implementation of new treatments, including immune checkpoint inhibition and specific chemotherapies.Citation2,3
Nearly 50% of melanoma lesions have a valine to glutamic acid exchange at position 600 in the BRAF gene, known as BRAFV600E.Citation4 This mutation leads to constitutive activation of the mitogen-activated protein kinase (MAPK) pathway that regulates cell proliferation and survival and so became a target for new inhibitors and therapeutic approaches.Citation5 Treatment with BRAF inhibitors (BRAFi), such as vemurafenib (PLX4032)Citation6 and dabrafenib (GSK2118436), induces tumour regression in a high proportion of metastatic melanoma patients with tumours bearing the BRAFV600E mutation and improves overall survival.Citation7 However, an important limitation of this therapy is the emergence of drug resistance after several months.Citation8 To prevent resistance development, combinations of MAPK inhibitors with immunotherapy are now being tested in clinical trials. This is based on the observations that lesions responding to BRAFi show an enhanced infiltration of immune cells. Indeed, BRAFi treatment of melanoma leads to increased tumour infiltration by CD8+ T cells,Citation9,10 however it is also associated with increased expression in the tumour of markers such as TIM-3, PD1 and its ligand PDL1, that are associated with exhaustion and inhibition of effective immune responses. Interestingly, however, combination of BRAFi with checkpoint blockade produced only modest benefits when tested in murine modelsCitation11 and the combination of BRAFi with either anti-PD1Citation12 or anti-CTLA-4 immune checkpoint blocking antibodiesCitation13 was associated with significant, severe toxicities in human patients, suggesting that there may be important difficulties associated with the use of these combinations of targeted therapy with immune checkpoint blockade in clinical practice. Despite this, the induction of long-lasting clinical responses by immunotherapy argues for the development of strategies that enhance the sensitivity of melanoma cells to immune cells under BRAFi treatment. Interestingly, mouse experiments suggest that besides T cells, also NK cells contribute to the therapeutic efficacy of BRAFi in mouse melanoma models.Citation14,15 Since potentiation of cytotoxic T cell responses against malignant melanoma frequently faces the problem that melanoma cells readily acquire mutations that lead to evasion from T cell recognition,Citation16,17 the utilisation of Natural Killer (NK) cells as a component of immunotherapy for melanoma is of considerable interest.
NK cells respond to different targets depending on the balance of signals coming from a large array of receptor-ligand interactions.Citation18 It has been shown that NK cells can target melanoma cells for lysis via NKG2D, NKp46 and DNAM-1 and the absence of MHC Class I molecules only enhances this recognition.Citation19,20 NKG2D-ligands include, in human, two families of proteins, called MICA/B (MHC class I related Chain A/B) and ULBPs (UL16 Binding Proteins) that upon binding activate the NK cell cytotoxic machinery. DNAM1 participates in the adhesion between NK cells and their ligands which include CD155 (also called poliovirus receptor, PVR) and CD112 (Nectin-2) (Reviewed in.Citation21,22) These ligands also regulate invasion and migration of tumour cells.Citation23 NK cells are known to accumulate in lymph nodes invaded by melanoma and can kill these tumour cells after cytokine activation.Citation24,25 Indeed, in murine models, NK cells have been shown to play a critical role in the control of melanoma metastasis achieved by BRAF inhibition,Citation14 however the effects of BRAF inhibition on the recognition of melanoma cells by human NK cells have not been explored systematically.
Published data indicate that the immune infiltrate changes in response to MAPK inhibitors and, again, on tumour progression.Citation26 Moreover, in an early phase of treatment the immune infiltrate contains NK cells while at progression the NK cells have disappeared.
We investigated the early effects of the BRAFi, vemurafenib, on the recognition of melanoma cells by NK cells. We show that, in vitro, treatment of human melanoma cells with the BRAFi leads to downregulation of ligands for activating receptors including NKG2D and DNAM-1, as well as upregulation of MHC Class I, a ligand for NK inhibitory receptors. These observations suggest that NK ligand expression strongly depends on the signaling route activated by oncogenic B-RAF, as recently observed by Frazao et al.Citation27
Therefore, from a therapeutic perspective, although BRAFV600E inhibition attacks melanoma growth directly, our data indicate that this treatment also contributes to tumour evasion from NK cell responses which could contribute to the development of resistance to BRAFi. Here, we also go on to show that the combination of BRAFi with drugs such as HDAC inhibitors promotes tumour cell death directly and increases immunogenicity for NK cells demonstrating the validity of the concept that combination therapy with BRAFi and other chemotherapeutic agents could enhance tumour destruction and immunogenicity.
Results
Characterization of ligands affecting NK activation on metastatic melanoma cell lines
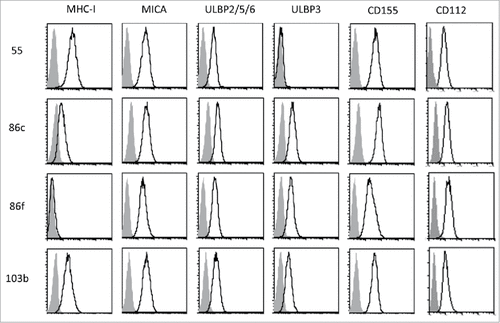
To define the impact of BRAFi treatment on the recognition of melanoma cells by NK cells we selected four cell lines established from metastatic patient lesions, three BRAF-V600E mutant cell lines (Ma-Mel-55, Ma-Mel-86c and Ma-Mel-86f) and one BRAF-WT cell line ().Citation17,28 The cell lines Ma-Mel-86c and Ma-Mel-86f were established from two consecutive patient metastases with Ma-Mel-86c cells showing an HLA class I haplotype loss and Ma-Mel-86f cells being HLA class I negative due to a lack of β-2 microglobulin expression. The cells were also genotyped for their MICA alleles. In agreement with the loss of one HLA class I haplotype, Ma-Mel-86c expressed only one of the two MICA alleles detected in Ma-Mel-86f cells. In contrast, the cell lines Ma-Mel-55 and Ma-Mel-103 showed normal HLA class I and MICA expression () (). Surface expression of the NKG2D-ligands (NKG2D-L), MICA and ULBPs, was also analysed by flow cytometry. All the melanoma cell lines studied expressed MICA and ULBP2/5/6; ULBP3 was expressed by three out of four, while none of them expressed ULBP1. MICB was not present at the surface of these melanoma cell lines as assessed with a monoclonal antibody specific for MICB (data not shown). Since DNAM-1/CD155 interactions have been described to mediate NK cell recognition and lysis of melanoma cells,Citation19 the surface expression of the two DNAM-1 ligands, CD155 and CD112, was also analysed in this panel of cells. Interestingly, the expression of these adhesion molecules necessary for the formation of the NK immune synapse varied considerably between the different cell lines, in particular CD155.
Table 1. Characteristics of cell lines established from metastatic melanoma.
Figure 1. Cell surface expression of MHC, NKG2D-ligands and DNAM1-ligands on melanoma cells. The metastatic melanoma cells Ma-Mel-55 (55), Ma-Mel-86c (86c), Ma-Mel-86f (86f) and Ma-Mel-103b (103b) were stained with the indicated antibodies and analysed by flow cytometry. Isotype control appears in grey, and signal for each antibody as a black line. The panel shows one representative experiment out of 3–5.
Vemurafenib modulates the expression of NKG2D-ligands and CD155 on BRAFV600E mutant melanoma cell lines
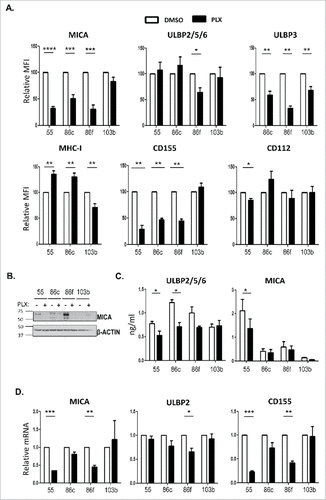
The effect of BRAF inhibition on the melanoma cells was analysed after 48 h of treatment with 1 μM vemurafenib. First, in MTT assays, the decrease in metabolic activity was confirmed to occur in BRAF mutant cells, but not in the non-mutant cell line; second, the decrease in pERK (Supplementary Figure 1A, B). Treatment with vemurafenib led to a decrease of surface MICA and ULBP3 on BRAFV600E mutant cells, however, in the case of ULBP2/5/6, only the decrease in Ma-Mel-86f was statistically significant, although the amount of this molecule was rather low initially (, Supplementary Figure 1C). In contrast, MHC expression increased in cells carrying the V600E mutation. Interestingly, CD155 expression followed a similar pattern to MICA and decreased in BRAFV600E expressing cell lines after vemurafenib treatment, while CD112 was not significantly affected in most cell lines. To investigate the mechanism of the decreased cell surface expression of NK ligands caused by vemurafenib, the amount of total MICA protein in treated cells was analysed by Western Blot, showing a similar pattern to that of surface expression (). Moreover, the release of NKG2D-L did not seem to be involved in this phenomenon, since vemurafenib treatment did not increase the amount of soluble protein detected by ELISA (). Thus, changes in mRNA content were studied (). The decrease of MICA and CD155 cell surface expression after vemurafenib treatment was accompanied by reduced levels of mRNA in two (out of three) cell lines carrying the BRAFV600E mutation, while mRNA levels remained unchanged in the non-mutant cell line. ULBP2 mRNA did not change significantly after vemurafenib treatment. These results show that treatment with BRAFi of melanoma cells bearing the V600E mutation in BRAF reduced cell surface expression of ligands for activating NK receptors at the same time that the expression of MHC molecules, ligands for inhibitory receptors, was increased, suggesting that the ability of NK cells to mediate cytotoxicity against BRAFi treated melanoma cells could also be influenced.
Figure 2. Effect of vemurafenib on NK ligand expression in melanoma cells. Melanoma cells Ma-Mel-55 (55), Ma-Mel-86c (86c), Ma-Mel-86f (86f) and Ma-Mel-103b (103b) were treated with 1 μM vemurafenib (PLX) for 48 h (A,B, D) or 24 h (C) (control cells were treated with the carrier DMSO) A. Flow cytometry. Melanoma cells were stained for detection of the indicated markers by flow cytometry. The plots represent the change in the mean fluorescence intensity (MFI) of each marker, as the percentage of the molecule present in control (DMSO) cells. Data are the mean and SEM. Isotype MFI was subtracted (n≥3, a representative experiment is shown in Suppl. ). B. Western blot showing the total amount of MICA in whole cell lysates, using antigen affinity-purified biotinylated goat polyclonal anti-MICA antibody BAF1300. Actin was used as loading control (n = 4). C. Soluble MICA and ULBP2/5/6 released to supernatants of vemurafenib -treated metastatic melanoma cells was analysed by ELISA at 24 h post-treatment. Plots represent the mean and SEM of protein concentration (ng/ml) (n = 3). D. mRNA detected using qPCR. Cells were recovered to extract RNA. cDNA was prepared and used as template in qRT-PCR experiments. Data are the mean and SEM, relative to control (DMSO) cells. RPLP0 mRNA levels were determined for normalization (#p < 0.05 ##p < 0.01, ### p < 0.001, #### p < 0.0001).
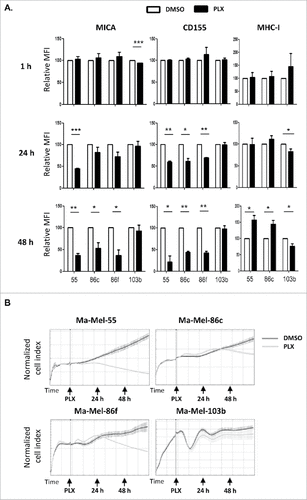
Since the data obtained above pointed to the possibility that NK recognition could be affected early after treatment with the BRAFi, the kinetics of the effect of vemurafenib on surface expression of NK ligands was analysed 1, 24 and 48 h after the addition of the inhibitor (). The decrease of MICA and CD155 was not detectable after 1 h of treatment with the inhibitor. Although the trend could be observed at 24 h, it was only statistically significant for the line Ma-Mel-55. So, we conclude that, at 48 h, the effect of vemurafenib was evident for BRAF mutant melanoma cell lines but not for non-mutant cells. Interestingly, monitoring of real-time proliferation of melanoma cells in the Xcelligence system, demonstrated that ligand expression was related to cell proliferation: while significant changes in surface MICA were observed after 24 h of treatment in Ma-Mel-55, a significant decrease was also observed at 48 h in Ma-Mel-86c and Ma-Mel-86f, and this parallels the differences in proliferation curves between DMSO and vemurafenib treated cells for Ma-Mel-55, Ma-Mel-86c and Ma-Mel-86f ().
Figure 3. Kinetics of the effect of vemurafenib on surface NK ligand expression and proliferation in melanoma cells. A. Effect of vemurafenib on NKG2D-ligand surface expression. Metastatic melanoma cells Ma-Mel-55 (55), Ma-Mel-86c (86c), Ma-Mel-86f (86f) and Ma-Mel-103b (103b) were treated with 1 μM vemurafenib for either 1 h, 24 h or 48 h (control cells with the carrier DMSO) and stained for detection of the indicated markers by flow cytometry. The plots represent the change in the mean fluorescence intensity (MFI) of each marker, as the percentage of the molecule present in control (DMSO) cells. Data show the mean and SEM. Isotype MFI was substracted (#p < 0.05 ##p < 0.01, ### p < 0.001) (n = 3). B. Proliferation assay. Metastatic melanoma cells were allowed to adhere to E-plates and then treated with 1 μM vemurafenib (control cells with the carrier DMSO). Real time electrical impedance was monitored to quantify cell proliferation for 48 h using x-CELLigence. Cell index normalized to the treatment time is represented. Arrows indicate the time of vemurafenib addition (PLX), 24 and 48 h post-treatment. The plots show quadruplicates of one representative of 3 independent experiments.
Vemurafenib affects the NK recognition of melanoma cells
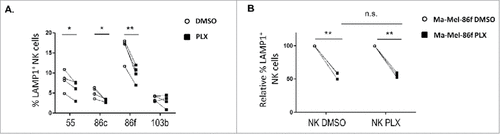
The recognition by autologous and allogenic NK cells of melanoma cell lines treated with BRAFi for 48 h was analysed next using either NK cells from unrelated healthy donors or autologous NK cells from patient Ma-Mel-86 activated in vitro. The response of allogeneic NK cells against melanoma cell lines, either untreated or treated with BRAFi, was evaluated in degranulation assays (). Treatment with vemurafenib led to a decrease in NK degranulation against the three metastatic melanoma cell lines with the V600E mutation in BRAF (). In these experiments, MHC class I was blocked to avoid donor-to-donor variation due to differences in inhibitory KIR expression of the donors. Even so, there was a decrease in NK recognition after treatment with vemurafenib which most likely is due to the decrease of activating ligands on melanoma cells. These data agree with reports showing that NK cells recognise melanoma and other tumour cells mainly through activating ligands such as NKG2D-L, NCR and DNAM-1Citation19.Citation20,29 Thus, enhanced expression of MHC class I by vemurafenib-treated melanoma cells is not the only factor that would facilitate NK evasion of vemurafenib-treated melanoma cells. Importantly, these data obtained in experiments with allogeneic NK cells could be confirmed in experiments using autologous NK cells from melanoma patient Ma-Mel-86, which showed that BRAF inhibition led to a reduction of around 40% in NK degranulation. In the autologous model, for comparison, NK cells were also incubated with vemurafenib. The decrease caused by vemurafenib on the recognition of autologous melanoma cells was similar for both BRAFi-treated and -untreated NK cells (), in agreement with prior data showing that NK cell activity was not affected by the use of vemurafenib.Citation14,30 Moreover, in the case of Ma-Mel-86f, no inhibitory signal could come from MHC-I because these cells showed a stable MHC-I-negative phenotype due to the loss β-2 microglobulin. Thus, the reduction in NK cell recognition of BRAFi-treated melanoma cells was most probably due to the decrease in ligands for activating NK receptors.
Figure 4. Effect of vemurafenib on NK cell recognition of melanoma cells. Metastatic melanoma cells Ma-Mel-55 (55), Ma-Mel-86c (86c), Ma-Mel-86f (86f) and Ma-Mel-103b (103b) were treated 48 h with either 1 μM vemurafenib or DMSO and used as targets in experiments of degranulation using NK cells. The plots represent the percentage of NK cells positive for surface CD107a (LAMP1) as a measure of degranulation. A. Healthy donor NK cells. Each symbol corresponds to an experiment with a different donor (n = 4). B. Autologous NK cells. Ma-Mel-86f were used as target cells. Autologous NK cell were also treated 48 h with DMSO or vemurafenib. Data represent the relative decrease as percentage of untreated cells. Three independent experiments are shown (n.s., non-significant; #p < 0.05 ## p < 0.01).
BRAF inhibitors in combination with HDACi could overcome NK resistance of melanoma cells
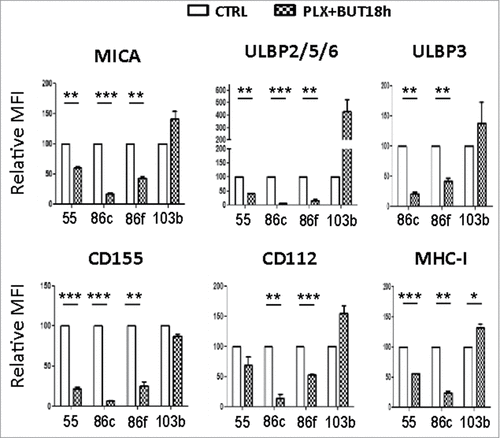
The above experiments show that treatment of malignant melanoma cells with BRAFi makes them less susceptible to NK cell recognition and attack. However, we and others, have shown that treatment with a number of other anti-cancer drugs can increase the immunogenicity of tumour cells by increasing expression of ligands for the NKG2D receptor.Citation31–35 Histone deacetylase inhibitors (HDACis) are currently under development for use as anticancer agents (reviewed inCitation36,37) and they are potent enhancers of NKG2D-L expression.Citation32 Initial experiments showed that the HDACi, sodium butyrate, consistently increased NKG2D-L surface expression in all the melanoma cell lines in a dose-dependent manner, the highest increase being observed with 5 mM and not affecting the viability of the V600E mutant melanoma cells (; Supplementary Figure 2). CD112 and MHC were affected with an 18 h exposure to the drug, but not CD155. Next, the effects on melanoma cells of treatment with the HDACi (sodium butyrate, 5 mM), in combination with vemurafenib (1 μM) were examined. MTT-based viability assays revealed that the combination of these drugs has a synergistic effect on the inhibition of the metabolic activity of the mutant melanoma cell lines (). In fact, cell numbers strongly decreased in the cultures after 48 h of incubation with the combination. Importantly, HDACi treatment on melanoma cells lines also resulted in increased NKG2D-L surface expression and, strikingly, the effect of this drug was dominant when combined with vemurafenib for 24 h, both in cell surface and mRNA levels of the NK ligands (, ). MHC-I and DNAM1-L surface expression was also checked in these treatments at 24 h. It is worth noting that the increase in surface NK ligands caused by either butyrate or the combination of drugs was higher for MICA and ULBP2/5/6 than for the rest of the molecules studied. The drug combination did not significantly increase CD155, however, it consistently increased the amount of CD112 at the cell surface of melanoma cells. In contrast, no clear pattern was observed for MHC with the combination of drugs. Thus, these data demonstrate that combined sodium butyrate and vemurafenib treatment not only increases melanoma susceptibility to be recognised by NK cells, but also causes a marked decrease in cell viability of all the melanoma cell lines tested. In order to separate toxicity from NK cell recognition, NK cell degranulation experiments were carried out after only 24 h of treatment with this drug combination (). Melanoma cells exposed to the butyrate+vemurafenib combination efficiently stimulated NK recognition and degranulation demonstrating that the diminished NK recognition of BRAFi-treated melanoma cells can be overcome by combining BRAF inhibition with the HDACi sodium butyrate. Antibody blocking confirmed a dominant role of NKG2D in NK recognition of melanoma and further demonstrated that the functional effect observed in the presence of the inhibitors was also mediated by the recognition of NKG2D ligands (). For consistency with the initial experiments, 48 h drug combination experiments were designed next but, to avoid the toxicity problem, melanoma cells were initially treated with vemurafenib only, then adding sodium butyrate for the last 18 h of the second day. Surprisingly, MICA downmodulation caused by vemurafenib was not rescued by a further incubation of melanoma cells with HDACi (), implying that the inhibition of BRAF leads to a persistent blockade of MICA expression. Moreover, this combination treatment also provokes the decrease of other NK ligands in mutant cells such as ULBP2/5/6, ULBP3, CD155, CD112 and MHC-I, indicating that timing is an important factor to consider when designing a new combination therapy.
Figure 5. Effect of the combination of HDACi and BRAFi on the immunophenotype and proliferation of melanoma cells. A. Titration of sodium butyrate and surface expression of NK ligands. Metastatic melanoma cells Ma-Mel-55 (55), Ma- Mel-86c (86c) and Ma-Mel-103b (103b) were treated with the indicated amounts of sodium butyrate or water for 18 h and stained with antibodies for detection of the indicated markers by flow cytometry. The plots represent the change in the mean fluorescence intensity (MFI) of each marker, as the percentage of the molecule present in control (water) cells. Data show the mean and SEM corresponding to 3 experiments. B. MTT assay of melanoma cells treated with the drug combination. Metastatic melanoma cells Ma-Mel-55 (55), Ma- Mel-86c (86c), Ma-Mel-86f (86f) and Ma-Mel-103b (103b) were treated with either 1 μM vemurafenib, 5 mM sodium butyrate or the combination of both for 24 h (control cells with the carrier, DMSO and water, CTRL). Cells were assayed for metabolic activity by incubation with MTT and OD570 was measured. Data represented are the mean as percentage of untreated cells and SEM of four replicates, from one experiment out of two. C. NK ligands on melanoma cells treated with the drug combination. Cells were stained for detection of the indicated markers by flow cytometry. The plots represent the mean fluorescence intensity (MFI) of each marker, isotype subtracted and the relative change as the percentage of the molecule present in control (DMSO and water, CTRL) cells. Data show the mean and SEM corresponding to 3 experiments. D. mRNA detected using qPCR. Cells were recovered to extract RNA. cDNA was prepared and used as template in qRT-PCR experiments. RPLP0 mRNA levels were determined for normalization (#p < 0.05 ##p < 0.01, ### p < 0.001). For simplicity, in B, C and D, only the p-values of each inhibitor related to the combination are shown.
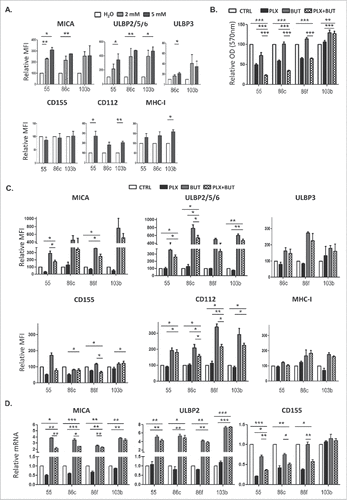
Figure 6. A. NK cell degranulation in the presence of HDACi- and BRAFi-treated melanoma cells. Melanoma cells Ma-Mel-55, Ma-Mel-86c, Ma-Mel-86f and Ma-Mel-103b cells treated with either 1 μM vemurafenib, 5 mM sodium butyrate or the combination of both for 24 h (control cells with the carrier, DMSO and water, CTRL) were used as targets in experiments of degranulation using NK cells from healthy donors PBMCs. The plots represent the percentage of NK cells positive for surface CD107a (LAMP1) as a measure of degranulation in 4 experiments (#p < 0.05 ##p < 0.01). For simplicity, only the p-values of each inhibitor related to the combination are shown. B. Effect of the blockade of activating NK receptors in degranulation experiments in the presence of HDACi- and BRAFi-treated melanoma cells. Melanoma cells Ma-Mel-86c treated with carrier (either DMSO or water, control) and the combination of 1 μM vemurafenib and 5 mM sodium butyrate for 24 h were used as targets in experiments of degranulation using NK cells from healthy donors PBMCs. NK receptors (NKG2D, DNAM-1, or both) were blocked, as indicated, incubating PBMCs with the corresponding antibodies, or isotype (ISO) as negative control. The plots represent the percentage of NK cells positive for surface CD107a (LAMP1) as a measure of degranulation in 3 experiments (#p < 0.05 ##p < 0.01###p < 0.001).
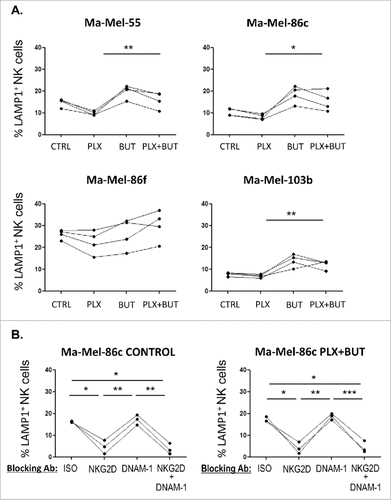
Figure 7. NK ligand expression on melanoma cells sequentially treated with vemurafenib (48 h) and sodium butyrate (last 18 h). Melanoma cells Ma-Mel-55 (55), Ma-Mel-86c (86c), Ma-Mel-86f (86f) and Ma-Mel-103b (103b) were treated with 1 μM vemurafenib (PLX) for 48 h, for the last 18 h of culture, a final concentration of 5 mM sodium butyrate was added (control cells with the carrier DMSO or water). Cells were stained for detection of the indicated markers by flow cytometry. The plots represent the mean fluorescence intensity (MFI) of each marker, isotype subtracted and the relative change as the percentage of the molecule present in control (DMSO and water, CTRL) cells. Data show the mean and SEM corresponding to 3 experiments. (#p < 0.05 ##p < 0.01, ### p < 0.001).
Discussion
In this paper, we explored whether treatment with BRAFi could influence the susceptibility of melanoma cells to NK cell recognition, in particular, via modulation of the expression of activating NK receptor ligands. NKG2D and its ligands constitute the best characterised system involved in NK surveillance of cancerCitation38 and melanoma cells are known to express various NKG2D-L affecting NK cell recognition.Citation20,Citation39,Citation40 Here, we demonstrate that exposure to the BRAFi, vemurafenib, modulates the surface expression of NKG2D-L, CD155 and MHC-I, all of which are important molecules regulating immune recognition by NK cells. The reduced cell surface expression of DNAM1-L and NKG2D-L were paralleled by decreases in the levels of DNAM1-L and NKG2D-L mRNA and were not due to an increase in the release of soluble proteins after BRAFV600E inhibition. This in turn indicates that expression of these ligands on melanoma cells is a consequence of downstream signaling via oncogenic BRAFV600E. Importantly, the overall result of the altered expression of ligands for activating and inhibitory NK receptors was a decrease in the susceptibility of the metastatic melanoma cells to NK cell attack. We further demonstrate that treatment of the tumour cells with a combination of BRAFi and HDACi, known to increase cell surface expression of NKG2D-L, counteracted the loss of activating ligand expression, favoring NK cell recognition of the melanoma cells. We propose that, in addition to trying to further block the MAPK route, the use of combination therapy with drugs that potentiate the susceptibility of the tumour cell to NK cells could be a viable approach to overcome the problems of tumour resistance to BRAFi that arise in melanoma patients.
Although most melanoma cells express at least one NKG2D-L,Citation29 the characterization of NK activating and inhibitory ligand expression by several metastatic melanoma cells shown here revealed heterogeneity, as previously reported for melanoma and other cancer cell lines.Citation20,41 This observation led to the hypothesis that, although melanoma is generally considered an immunogenic cancer, different melanoma cells from different metastatic lesions might show different susceptibilities to immune attack. As expected, treatment with the BRAFi vemurafenib led to a decrease in proliferation of BRAFV600E mutant melanoma cells, as well as an increase of MHC Class I expression which was previously reported using the BRAFi dabrafenib.Citation42 Surprisingly, however, BRAFi treatment of these melanoma cells also triggered a marked decrease in the levels of NKG2D-L, as recently described by Frazao,Citation27 and CD155 at the cell surface, sufficient to seriously impair NK cell recognition of these BRAFi-treated melanoma cells. Exposure to vemurafenib affected neither the proliferation of BRAFwt melanoma cells nor the expression of ligands for NK cell receptors, except for a decrease in ULBP3.
The mechanism underlying the reduced levels of cell surface ligands for the NKG2D and DNAM-1 receptors after vemurafenib treatment of BRAFV600E mutant melanoma appears to be a decrease in synthesis of these proteins due to significantly lower levels of specific mRNA. These data demonstrate a connection between the specific inhibition of proliferative signals from the BRAFV600E mutant kinase and a decrease in NKG2D-L and CD155 mRNA, suggesting either transcriptional control of the expression of these molecules coupled to this MAPK route in humans or a decrease in RNA stability. Interestingly, another oncogene present in many human tumours, H-RasV12, has previously been related with upregulation of ULBP and, in certain cells, MICA.Citation43 Ras activates a number of signalling pathways, including RAF-MAPK/MEK and PI3K, related with proliferation. Indeed, exposure to the tyrosine kinase inhibitor sorafenib and inhibitors of the PI3K and MEK pathways can also modulate MICA/B and other NKG2D-L surface expression.Citation43,44 Thus, it seems plausible to speculate that transcriptional/post-transcriptional regulation of MICA could be under control of the proliferation signals initiated by BRAF.
That NKG2D-L are expressed in immortalised cells and proliferative situations has been long knownCitation29 but, because proliferation not only occurs in tumour cells, the regulation of surface protein has to be very precise.Citation45 The finding of regulation by E2F transcription factors in the murine system provided the first molecular insight into how NKG2D-L regulation could be linked to proliferation.Citation46 However, NKG2D-L upregulation can also be ATM/ATR dependent in response to DNA-damage.Citation47 although this route does not seem to be involved in the expression of ligands in cells under other stresses. The effect of sequential treatment of melanoma cells with first vemurafenib and later sodium butyrate suggest that the regulation of expression is different for MICA and the other NKG2D-L as well as CD155 and CD112. The fact that, most probably, several pathways participate in driving the expression of NKG2D-L and other NK ligands makes biological sense because NKG2D-L have to be expressed only on target cells that suffer certain type of stress and should be eliminated.
The data presented here also support previous studies linking the biology of NKG2D-L with that of DNAM1-L, implying that there are likely common pathways in the regulation of these ligands for activating NK receptors, as previously proposed.Citation48 Since both MICA and CD155 decreased at the cell surface of melanoma cells treated with BRAFi, these data suggest that signals mediated by activation of the oncogenic BRAF route play some role in the regulation of both molecules. It is interesting to note that inhibition of BRAFV600E signaling decreased the expression of NKG2D and DNAM ligands that drive activation of NK cells, and increased the expression of MHC class I molecules, that are ligands for inhibitory NK receptors. Strikingly, however, while BRAFV600E modulates the expression of NK activating ligands transcriptionally (this paper), the kinase acts to decrease MHC-I expression at the cell surface by altering the intracellular trafficking of these molecules, so that they are rapidly internalised from the melanoma cell surface and are sequestered intracellularly.Citation42 Simultaneous increase in MHC-I expression and decrease in ligands for NK activating receptors would be expected to inhibit NK recognition and, indeed, BRAFi treatment decreased NK cell reactivity against BRAFV600E mutant melanoma cells ().
The most striking finding of this report was that the decrease of NKG2D-ligands that occurs in BRAFi-treated melanoma cell lines could be overcome by exposure to HDACi. Because HDACs have been suggested to contribute to tumorigenesis, due to the regulation of genes involved in DNA damage repair, and because this regulation has been co-related to patient prognosis, several FDA-approved HDACi are now used for the treatment of various types of cancer, the most successful being refractory cutaneous and peripheral T cell lymphoma.Citation37 However, the role of HDACi in cancer still has many unanswered questions and a possible contribution of immune system activation has been suggested only very recently.Citation36 For example, the use of HDACi has been shown to enhance T cell cytotoxicity towards BRAFi-treated melanoma which expressed more proapoptotic genes, although the involvement of NKG2D as co-receptor was not investigated in that system.Citation49 The expression of NKG2D in all human CD8+ T cells is important in this context, since these cells can be found infiltrating tumours (TILs) and could play a critical role in immunosurveillance. Independent experiments have also shown that exposure to HDACi causes an increase in the expression of several NKG2D-L in tumour cellsCitation32 and thus, we hypothesized that the decrease of NKG2D-L and NK cell recognition in BRAFi-treated cells could be reverted by the addition of HDACi or other drugs known to increase NKG2D-L expression. Here we show that treatment in vitro with the combination of both drugs, BRAFi and HDACi, can lead to the increase of NKG2D-L expression on melanoma cells. Blocking experiments confirmed that the expression of these ligands was the basis of the recovery of NK recognition. However, the addition of HDACi, after BRAFi-induced NKG2D-L downmodulation had already occurred, did not have the expected consequence of expression recovery. Instead, sodium butyrate needed to be incubated with vemurafenib simultaneously. Why NKG2D-L downmodulation by BRAFi could not be reverted when HDACi treatment was added later is not clear, but future experiments will investigate the hypothesis that increased phosphorylation of the translation initiation factor eIF2α (which would be expected to inhibit protein synthesis) induced by BRAFi pretreatment.Citation50 blocks the ability of HDACi to trigger increased NKG2D-L expression.
In any case our observation that simultaneous treatment with BRAFi and HDACi affected melanoma cell growth and immunogenicity more potently than treatment with either agent alone suggests the value of exploration of novel approaches of drug combinations aiming to potentiate distinct mechanisms of cancer elimination, i.e. direct drug cytotoxicity on the malignant cells and indirect increase of the susceptibility of the tumour to elimination by immune cells. Moreover, because different lesions from the same patient can either carry or not the BRAFV600E mutation, the effect of sodium butyrate on non-mutant cells could be of added value: the combination would avoid the undesirable decrease of NKG2D-L on BRAFV600E mutant cells, but also increase the activating NK ligands (including DNAM1-L) in non-mutant cells. In addition, in the BRAFV600E mutant lesions, the combination of HDACi and vemurafenib would synergise to decrease tumour cell viability. Our data demonstrate that combination of BRAFi and HDACi enhances NK cell recognition of melanoma cells. While the effect on T cell recognition still has to be addressed our data suggest that it would not be impaired because MHC class I expression is not significantly altered.
In aggregate, the data presented in this manuscript support the use of HDACi in combination with BRAFi to treat melanoma, based on the rescue of recognition by NK cells. The possibility of combining these two types of drugs has been suggested previously, however, in the context of their effect on cell death.Citation51 Although melanoma is generally considered very immunogenic, it is also quite heterogeneous at the level of genetic alterations that can lead to the lack of β2 m expression in one lesion of a patient and loss of heterozygosity in the MHC in a second one. Thus, it is very important to understand how the drugs directed to one type of lesion can affect other lesions in the same patient, as studied here in patient model Ma-Mel-86, and make sure that all of them can be controlled. Exploring the effect of chemotherapeutic drugs on tumor cell recognition by immune cells, as shown here for NK cells, may help proposing new avenues for intervention and facilitate the selection of drugs for combination in clinical trials for cancer therapy. Since it is not feasible to assess all the possible combinations in patients, proposing drug synergies in preclinical assays, as we show here for BRAFi and HDACi, may prove to be a useful strategy in the design of new therapeutic regimes.
Materials and methods
Reagents
Reagents were purchased from Sigma, unless otherwise stated. Vemurafenib was obtained from Selleckchem and used at a final concentration of 1 μM.
Cell lines and peripheral blood mononuclear cell culture
The metastatic melanoma cell lines Ma-Mel-55, Ma-Mel-86c, Ma-Mel-86f and Ma-Mel-103b were obtained as described previouslyCitation17,28 and typed for MICA subfamily by satellite analysisCitation52 and using Peak Scanner v1.0 software (Applied Biosystems). 721.221, RPMI-8866, K562 cells (used for functional assays) and the melanoma cell lines were grown in RPMI medium supplemented with 10% FCS, 1 mM glutamine, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 10 mM Hepes, 100 U/ml penicillin and 100 U/ml streptomycin (Biowest).
Peripheral blood mononuclear cells (PBMCs) were purified from healthy volunteer buffy coats (Regional Transfusion Centre, Madrid) or from donations from patient Ma-Mel-86 (University Hospital Essen). Approval from local ethical committees and informed consent from all participants were obtained and experiments have been conducted according to the principles expressed in the Declaration of Helsinki. Activated NK cells were prepared as described previously.Citation53 modified by the inclusion of IL-12 and IL-18 stimulation at the beginning of the culture. Briefly, PBMC were isolated by centrifugation on Ficoll-HyPaque. Adherent cells were eliminated by adherence to tissue culture plates and the remaining PBMCs were then cultured in the presence of irradiated feeder cells (721.221 and RPMI-8866 B cell lines) in RPMI-1640 medium (Lonza) supplemented with 10% FBS, 10% male AB human serum, 4 mM L-glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 U/ml streptomycin, 10 mM Hepes, 50 μM β-mercaptoethanol (Biowest), 10 U/ml IL-12(Peprotech) and 25ng/ml IL-18 (MBL). 5–6 d later, 50 U/ml of IL-2 (Peprotech) was added. After 7–8 d of culture, the proportion of NK cells was checked by flow cytometry using CD3 and CD56-specific mAbs, and used for functional experiments. If necessary, PBMCs were re-stimulated with IL-2.
Flow cytometry
Cells were incubated with the appropriate primary antibodies or isotype control. Monoclonal and PE-conjugated antibodies specific for ULBPs were purchased from R&D Systems (Abingdon, UK) (ULBP2/5/6, MAB1298 or FAB1298P; ULBP3, MAB1517); PE-conjugated antibody for MICA was purchased from R&D Systems (FAB1300P). 1H10 antibody specific for MICACitation54 and HP1F7 antibody recognizing MHC-ICitation55 were described before; monoclonal antibody anti-CD112 and anti-CD155 FITC were purchased from Santa Cruz Biotechnology, and anti-CD155 from Abcam followed by either PE- or FITC-labelled F(ab’)2 fragments of goat anti-mouse Ig (Dako). Cell death was evaluated by the analysis of 7-Aminoactinomycin D (7AAD) (Beckman Coulter) staining. Samples were analyzed using BD FACSCalibur (Becton Dickinson), Gallios Flow Cytometer or Cytomics FC 500 (Beckman Coulter). Analysis of the experiments was performed using Kaluza (Beckman Coulter) and FlowJo (Tree Star, Inc).
Viability/proliferation assays
Viability/Proliferation assays using MTT, were performed by plating 3000 cells per well in 96-well plates. After 16 h, either vehicle or inhibitor was added for 24 or 48 h, as indicated. Supernatants were then discarded, and MTT solution (20% complete medium, 70% PBS, 10% MTT stock solution 5 mg/ml in PBS) was added and incubated at 37°C for 4 h. MTT solution was discarded, and DMSO was added and OD at 554 nm was measured with TECAN Infinite M200 (Life Sciences). In combination treatments, the OD was measured at 570 nm with Thermo Scientific™ Multiskan™ FC Filter-based Microplate Photometer (Thermo Fisher Scientific). OD at 690 nm was used as reference measure. Four replicates per condition and cell line were done.
Proliferation assays using the xCELLigence® RTCA DP (Acea Bioscience) system were performed by plating 5000 cells in 16 well Eplates. Cells were allowed to adhere for 16 h and growth was followed for 2 d with the addition of vemurafenib or DMSO as control. Four replicates per condition and cell line were done. Blank was measured with complete RPMI medium. Data were analysed and normalized to time of treatment using the RTCA 2.0 Software.
Degranulation assays
PBMCs were co-cultured for 2 h with target cells at a final E:T ratio of 1 NK to 2 target cells. Surface expression of LAMP1 (CD107a) was analyzed by flow cytometry in NK cells CD3−CD56+ stained with directly conjugated antibodies (Biolegend). K562 cells were used as control for NK degranulation against a known NK target (internal experiment control). Basal degranulation was measured in PBMCs incubated alone (negative control). In the case of non-autologous PBMCs, target cells were pre-incubated 30 minutes at 25°C with 10 μg/ml of anti-MHC class I antibody HP1F7.Citation55 In experiments using autologous PBMCs, degranulation analyses were performed by incubating the effector cells for 3 h with monensin and anti-LAMP1 antibody. For blocking experiments PBMCs were incubated 30 minutes at RT with 5 μg/ml of the corresponding antibody (MOPC21, as isotype control, from Sigma; anti-NKG2D, MAB139 or anti-DNAM-1, MAB666, both from R&D Systems) before co-incubation with targets.
Quantitative PCR (qPCR)
After treatment, as indicated in the figure legends, melanoma cell lines were washed with PBS. Total mRNA was isolated from tumour cells using RNeasy Plus Kit (Qiagen, Hilden, Germany) and RNA was reverse transcribed into cDNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems), following the instructions from the manufacturers. Real-time quantitative RT-PCR was carried out with specific TaqMan Gene Expression assays. Relative mRNA expression was calculated by the 2−ΔΔCt method after normalizing expression levels of investigated mRNA to ribosomal protein lateral stalk subunit P0 (RPLP0) mRNA.
ELISA
NKG2D-L were detected using sandwich ELISAs on supernatants from cells cultured in the presence or absence of vemurafenib for 24 h. Plates were coated with purified mAb R&D for MICA (MAB13002) or goat polyclonal Ab for ULBP2/5/6 (AF1298) (R&D) in BBS (Borate Buffered saline). After incubation overnight at 4°C, the plates were blocked with 2% BSA-HBS (Hepes Buffered saline: 10 mM hepes pH 7.6, 150 mM NaCl) for 2 h at 37°C; then the samples were added and incubated overnight at 4°C. Bound proteins were detected using biotinylated goat anti-MICA (BAF1300) or biotinylated goat anti-ULBP2/5/6 (BAF1298), both from R&D, followed by streptavidin-HRP (Amersham) and developed using the peroxidase substrate system (ABTS; Roche). The absorbance was measured at 410 nm with a reference wavelength of 490 nm with Thermo Scientific™ Multiskan™ FC Filter-based Microplate Photometer (Thermo Fisher Scientific). Under these conditions, the cutoff for detection of recombinant soluble MICA (R&D Biosciences) (1300-MA) and ULBP2/5/6 (1298-UL) was around 100 pg/ml, and the ELISA absorbance values were directly proportional to the concentration of protein over the range of 90 pg/ml to 3 ng/ml.
Western blot
Cells were lysed in lysis buffer [50 mM Tris pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% NP-40 with protease inhibitors (leupeptin and pepstatin A)] and centrifuged to pellet nuclei. Samples were resolved on 10% SDS-PAGE and transferred to Immobilon-P (Millipore). The membrane was blocked using 5% nonfat-dry milk or BSA in PBS-0.1% Tween 20. MICA proteins were detected by incubating the membrane with biotinylated goat polyclonal anti-MICA antibody (R&D) (BAF1300). Mouse anti- β-actin (AC-15) was from Sigma, rabbit polyclonal anti-ERK, rabbit anti-p-ERK (20G11), rabbit anti-GAPDH (14C10) (Cell Signaling). Secondary antibodies [goat anti-mouse Ig-HRP (Dako), donkey anti-goat Ig-HRP (Santa Cruz Biotechnology), goat anti-rabbit Ig-HRP (Cell Signaling)] or streptavidin-HRP were used prior to developing using the ECL Plus system (GE Pharmaceuticals).
Stastistical analysis
Student's t-test was done for paired samples using GraphPad Prism 5. p-value is shown in each figure, when not indicated is because differences were non-significant.
Author contributions
SLC, AP, MV and HTR contributed to the conception and design of the study, acquisition, analysis and interpretation of data and wrote the manuscript; CCS and NP performed experiments; EGC provided data and/or reagents for the study; AP, MV and HTR provided material support.
2017ONCOIMM0454R1-f02-z-bw.pptx
Download MS Power Point (169.4 KB)Acknowledgments
SLC was the recipient of a mobility fellowship from the Spanish Education Ministry and a recipient of a FPU fellowship (MECD); EMG-C was a recipient of a PhD fellowship from “Fundación La Caixa”, CC-S was a recipient of a JAE-Intro fellowship (CSIC).
Disclosure statement
The authors declare no potential conflicts of interest.
Funding
This work was supported by the Madrid Regional Government under grant “Inmunothercan” S2010/BMD-2326; the Spanish Ministries of Economy and Health under grants SAF-2012-32293, SAF2015-69169-R (MINECO/FEDER) (MVG) and SAF2014-58752-R (HTR). AP received support from the “Hiege-Stiftung gegen Hautkrebs”. SLC and EMGC were recipients of Fellowships from Spanish Ministry of Education (FPU) and La Caixa, respectively. CC-S was a recipient of a JAE-Intro fellowship (CSIC).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- Agarwala SS. Current systemic therapy for metastatic melanoma. Expert Rev Anticancer Ther. 2009;9:587–95. doi:10.1586/era.09.25. PMID:19445576.
- Zhu Z, Liu W, Gotlieb V. The rapidly evolving therapies for advanced melanoma-Towards immunotherapy, molecular targeted therapy, and beyond. Crit Rev Oncol Hematol. 2016;99:91–9. doi:10.1016/j.critrevonc.2015.12.002.
- Schadendorf D, Fisher DE, Garbe C, Gershenwald JE, Grob JJ, Halpern A, Herlyn M, Marchetti MA, McArthur G, Ribas A, et al. Melanoma. Nature reviews Disease primers. 2015;1:15003. doi:10.1038/nrdp.2015.3. PMID:27188223.
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54. doi:10.1038/nature00766. PMID:12068308.
- Tran KA, Cheng MY, Mitra A, Ogawa H, Shi VY, Olney LP, Kloxin AM, Maverakis E. MEK inhibitors and their potential in the treatment of advanced melanoma: the advantages of combination therapy. Drug Des Devel Ther. 2016;10:43–52. PMID:26730180.
- Flaherty KT, Yasothan U, Kirkpatrick P. Vemurafenib. Nat Rev Drug Discov. 2011;10:811–2. doi:10.1038/nrd3579. PMID:22037033.
- Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. The New England journal of medicine. 2012;366:707–14. doi:10.1056/NEJMoa1112302. PMID:22356324.
- Fiskus W, Mitsiades N. B-Raf Inhibition in the Clinic: Present and Future. Annu Rev Med. 2016;67:29–43. doi:10.1146/annurev-med-090514-030732. PMID:26768236.
- Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, Kefford RF, Hersey P, Scolyer RA. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18:1386–94. doi:10.1158/1078-0432.CCR-11-2479. PMID:22156613.
- Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19:1225–31. doi:10.1158/1078-0432.CCR-12-1630. PMID:23307859.
- Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, Lo JA, Hodi FS, Freeman GJ, Bosenberg MW, et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol Res. 2014;2:643–54. doi:10.1158/2326-6066.CIR-13-0215. PMID:24903021.
- Johnson DB, Wallender EK, Cohen DN, Likhari SS, Zwerner JP, Powers JG, Shinn L, Kelley MC, Joseph RW, Sosman JA. Severe cutaneous and neurologic toxicity in melanoma patients during vemurafenib administration following anti-PD-1 therapy. Cancer Immunol Res. 2013;1:373–7. doi:10.1158/2326-6066.CIR-13-0092. PMID:24490176.
- Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. The New England journal of medicine. 2013;368:1365–6. doi:10.1056/NEJMc1302338. PMID:23550685.
- Ferrari de Andrade L, Ngiow SF, Stannard K, Rusakiewicz S, Kalimutho M, Khanna KK, Tey SK, Takeda K, Zitvogel L, Martinet L, et al. Natural killer cells are essential for the ability of BRAF inhibitors to control BRAFV600E-mutant metastatic melanoma. Cancer Res. 2014;74:7298–308. doi:10.1158/0008-5472.CAN-14-1339. PMID:25351955.
- Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, Haynes NM, Kinross K, Yagita H, Koya RC, et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J Clin Invest. 2013;123:1371–81. doi:10.1172/JCI66236. PMID:23454771.
- Mendez R, Aptsiauri N, Del Campo A, Maleno I, Cabrera T, Ruiz-Cabello F, Garrido F, Garcia-Lora A. HLA and melanoma: multiple alterations in HLA class I and II expression in human melanoma cell lines from ESTDAB cell bank. Cancer immunology, immunotherapy: CII. 2009;58:1507–15. doi:10.1007/s00262-009-0701-z. PMID:19340423.
- Zhao F, Sucker A, Horn S, Heeke C, Bielefeld N, Schrors B, Bicker A, Lindemann M, Roesch A, Gaudernack G, et al. Melanoma Lesions Independently Acquire T-cell Resistance during Metastatic Latency. Cancer Res. 2016;76:4347–58. doi:10.1158/0008-5472.CAN-16-0008. PMID:27261508.
- Pahl J, Cerwenka A. Tricking the balance: NK cells in anti-cancer immunity. Immunobiology 2017;222:11–20. doi:10.1016/j.imbio.2015.07.012. PMID:26264743.
- Lakshmikanth T, Burke S, Ali TH, Kimpfler S, Ursini F, Ruggeri L, Capanni M, Umansky V, Paschen A, Sucker A, et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J Clin Invest. 2009;119:1251–63. doi:10.1172/JCI36022. PMID:19349689.
- Casado JG, Pawelec G, Morgado S, Sanchez-Correa B, Delgado E, Gayoso I, Duran E, Solana R, Tarazona R. Expression of adhesion molecules and ligands for activating and costimulatory receptors involved in cell-mediated cytotoxicity in a large panel of human melanoma cell lines. Cancer immunology, immunotherapy: CII. 2009;58:1517–26. doi:10.1007/s00262-009-0682-y. PMID:19259667.
- Tarazona R, Duran E, Solana R. Natural Killer Cell Recognition of Melanoma: New Clues for a More Effective Immunotherapy. Frontiers in immunology. 2015;6:649. PMID:26779186.
- Molfetta R, Quatrini L, Santoni A, Paolini R. Regulation of NKG2D-Dependent NK Cell Functions: The Yin and the Yang of Receptor Endocytosis. Int J Mol Sci. 2017;18. doi:10.3390/ijms18081677. PMID:28767057.
- Gao J, Zheng Q, Xin N, Wang W, Zhao C. CD155, an onco-immunologic molecule in human tumors. Cancer science 2017. doi:10.1111/cas.13324.
- Messaoudene M, Fregni G, Fourmentraux-Neves E, Chanal J, Maubec E, Mazouz-Dorval S, Couturaud B, Girod A, Sastre-Garau X, Albert S, et al. Mature cytotoxic CD56(bright)/CD16(+) natural killer cells can infiltrate lymph nodes adjacent to metastatic melanoma. Cancer Res. 2014;74:81–92. doi:10.1158/0008-5472.CAN-13-1303. PMID:24225017.
- Ali TH, Pisanti S, Ciaglia E, Mortarini R, Anichini A, Garofalo C, Tallerico R, Santinami M, Gulletta E, Ietto C, et al. Enrichment of CD56(dim)KIR + CD57 + highly cytotoxic NK cells in tumour-infiltrated lymph nodes of melanoma patients. Nature communications. 2014;5:5639. doi:10.1038/ncomms6639. PMID:25472612.
- Cooper ZA, Reuben A, Spencer CN, Prieto PA, Austin-Breneman JL, Jiang H, Haymaker C, Gopalakrishnan V, Tetzlaff MT, Frederick DT, et al. Distinct clinical patterns and immune infiltrates are observed at time of progression on targeted therapy versus immune checkpoint blockade for melanoma. Oncoimmunology. 2016;5:e1136044. doi:10.1080/2162402X.2015.1136044. PMID:27141370.
- Frazao A, Colombo M, Fourmentraux-Neves E, Messaoudene M, Rusakiewicz S, Zitvogel L, Vivier E, Vély F, Faure F, Dréno B, et al. Shifting the Balance of Activating and Inhibitory Natural Killer Receptor Ligands on BRAFV600E Melanoma Lines with Vemurafenib. Cancer Immunol Res. 2017;5:582–93. doi:10.1158/2326-6066.CIR-16-0380. PMID:28576831.
- Ugurel S, Thirumaran RK, Bloethner S, Gast A, Sucker A, Mueller-Berghaus J, Rittgen W, Hemminki K, Becker JC, Kumar R, et al. B-RAF and N-RAS mutations are preserved during short time in vitro propagation and differentially impact prognosis. PloS one. 2007;2:e236. doi:10.1371/journal.pone.0000236. PMID:17311103.
- Pende D, Rivera P, Marcenaro S, Chang CC, Biassoni R, Conte R, Kubin M, Cosman D, Ferrone S, Moretta L, et al. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res. 2002;62:6178–86. PMID:12414645.
- Sottile R, Pangigadde PN, Tan T, Anichini A, Sabbatino F, Trecroci F, Favoino E, Orgiano L, Roberts J, Ferrone S, et al. HLA class I downregulation is associated with enhanced NK-cell killing of melanoma cells with acquired drug resistance to BRAF inhibitors. Eur J Immunol. 2016;46:409–19. doi:10.1002/eji.201445289. PMID:26564811.
- Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, Kaiser S, Jobst J, Smirnow I, Wagner A, et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005;65:6321–9. doi:10.1158/0008-5472.CAN-04-4252. PMID:16024634.
- Skov S, Pedersen MT, Andresen L, Straten PT, Woetmann A, Odum N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 2005;65:11136–45. doi:10.1158/0008-5472.CAN-05-0599. PMID:16322264.
- Vales-Gomez M. Selective induction of expression of a ligand for the NKG2D receptor by proteasome inhibitors. J Immunol. 2008;68:1546–54.
- Butler JE, Moore MB, Presnell SR, Chan HW, Chalupny NJ, Lutz CT. Proteasome regulation of ULBP1 transcription. J Immunol. 2009;182:6600–9. doi:10.4049/jimmunol.0801214.
- Chavez-Blanco A, De la Cruz-Hernandez E, Dominguez GI, Rodriguez-Cortez O, Alatorre B, Perez-Cardenas E, Chacón-Salinas R, Trejo-Becerril C, Taja-Chayeb L, Trujillo JE, et al. Upregulation of NKG2D ligands and enhanced natural killer cell cytotoxicity by hydralazine and valproate. Int J Oncol. 2011;39:1491–9. PMID:21805029.
- West AC, Smyth MJ, Johnstone RW. The anticancer effects of HDAC inhibitors require the immune system. Oncoimmunology. 2014;3:e27414. doi:10.4161/onci.27414. PMID:24701376.
- West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–9. doi:10.1172/JCI69738. PMID:24382387.
- Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. 2016;16:7–19. doi:10.1038/nrc.2015.5. PMID:26694935.
- Schwinn N, Vokhminova D, Sucker A, Textor S, Striegel S, Moll I, Nausch N, Tuettenberg J, Steinle A, Cerwenka A, et al. Interferon-gamma down-regulates NKG2D ligand expression and impairs the NKG2D-mediated cytolysis of MHC class I-deficient melanoma by natural killer cells. Int J Cancer. 2009;124:1594–604. doi:10.1002/ijc.24098. PMID:19089914.
- Fuertes MB, Girart MV, Molinero LL, Domaica CI, Rossi LE, Barrio MM, Mordoh J, Rabinovich GA, Zwirner NW. Intracellular retention of the NKG2D ligand MHC class I chain-related gene A in human melanomas confers immune privilege and prevents NK cell-mediated cytotoxicity. J Immunol. 2008;180:4606–14. doi:10.4049/jimmunol.180.7.4606.
- Garcia-Cuesta EM, Lopez-Cobo S, Alvarez-Maestro M, Esteso G, Romera-Cardenas G, Rey M, Cassady-Cain RL, Linares A, Valés-Gómez A, Reyburn HT, et al. NKG2D is a Key Receptor for Recognition of Bladder Cancer Cells by IL-2-Activated NK Cells and BCG Promotes NK Cell Activation. Frontiers in immunology. 2015;6:284. PMID:26106390.
- Bradley SD, Chen Z, Melendez B, Talukder A, Khalili JS, Rodriguez-Cruz T, Liu S, Whittington M, Deng W, Li F, et al. BRAFV600E Co-opts a Conserved MHC Class I Internalization Pathway to Diminish Antigen Presentation and CD8+ T-cell Recognition of Melanoma. Cancer Immunol Res. 2015;3:602–9. doi:10.1158/2326-6066.CIR-15-0030. PMID:25795007.
- Liu XV, Ho SS, Tan JJ, Kamran N, Gasser S. Ras activation induces expression of Raet1 family NK receptor ligands. J Immunol. 2012;189:1826–34. doi:10.4049/jimmunol.1200965.
- Romero AI, Chaput N, Poirier-Colame V, Rusakiewicz S, Jacquelot N, Chaba K, Mortier E, Jacques Y, Caillat-Zucman S, Flament C, et al. Regulation of CD4(+)NKG2D(+) Th1 cells in patients with metastatic melanoma treated with sorafenib: role of IL-15Ralpha and NKG2D triggering. Cancer Res. 2014;74:68–80. doi:10.1158/0008-5472.CAN-13-1186. PMID:24197135.
- Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. 2013;31:413–41. doi:10.1146/annurev-immunol-032712-095951. PMID:23298206.
- Jung H, Hsiung B, Pestal K, Procyk E, Raulet DH. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry. The Journal of experimental medicine. 2012;209:2409–22. doi:10.1084/jem.20120565. PMID:23166357.
- Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–90. doi:10.1038/nature03884. PMID:15995699.
- Cerboni C, Fionda C, Soriani A, Zingoni A, Doria M, Cippitelli M, Santoni A. The DNA Damage Response: A Common Pathway in the Regulation of NKG2D and DNAM-1 Ligand Expression in Normal, Infected, and Cancer Cells. Front Immunol. 2014;4:508. doi:10.3389/fimmu.2013.00508. PMID:24432022.
- Jazirehi AR, Nazarian R, Torres-Collado AX, Economou JS. Aberrant apoptotic machinery confers melanoma dual resistance to BRAF(V600E) inhibitor and immune effector cells: immunosensitization by a histone deacetylase inhibitor. American journal of clinical and experimental immunology. 2014;3:43–56. PMID:24660121.
- Beck D, Niessner H, Smalley KS, Flaherty K, Paraiso KH, Busch C, Sinnberg T, Vasseur S, Iovanna JL, Drießen S, et al. Vemurafenib potently induces endoplasmic reticulum stress-mediated apoptosis in BRAFV600E melanoma cells. Sci Signal. 2013;6:ra7. doi:10.1126/scisignal.2003057. PMID:23362240.
- Lai F, Jin L, Gallagher S, Mijatov B, Zhang XD, Hersey P. Histone deacetylases (HDACs) as mediators of resistance to apoptosis in melanoma and as targets for combination therapy with selective BRAF inhibitors. Adv Pharmacol 2012;65:27–43. doi:10.1016/B978-0-12-397927-8.00002-6. PMID:22959022.
- Zou Y, Han M, Wang Z, Stastny P. MICA allele-level typing by sequence-based typing with computerized assignment of polymorphic sites and short tandem repeats within the transmembrane region. Hum Immunol. 2006;67:145–51. doi:10.1016/j.humimm.2006.02.016. PMID:16698436.
- Mandelboim O, Reyburn HT, Vales-Gomez M, Pazmany L, Colonna M, Borsellino G, Strominger JL. Protection from lysis by natural killer cells of group 1 and 2 specificity is mediated by residue 80 in human histocompatibility leukocyte antigen C alleles and also occurs with empty major histocompatibility complex molecules. The Journal of experimental medicine. 1996;184:913–22. doi:10.1084/jem.184.3.913. PMID:9064351.
- Vales-Gomez M, Winterhalter A, Roda-Navarro P, Zimmermann A, Boyle L, Hengel H, Brooks A, Reyburn HT. The human cytomegalovirus glycoprotein UL16 traffics through the plasma membrane and the nuclear envelope. Cell Microbiol. 2006;8:581–90. doi:10.1111/j.1462-5822.2005.00645.x. PMID:16548884.
- Perez-Villar JJ, Melero I, Navarro F, Carretero M, Bellon T, Llano M, Colonna M, Geraghty DE, López-Botet M. The CD94/NKG2-A inhibitory receptor complex is involved in natural killer cell-mediated recognition of cells expressing HLA-G1. J Immunol. 1997;158:5736–43.