
ABSTRACT
Loss or mutation of TP53 has been linked to alterations in anti-tumor immunity as well as dysregulation of cell cycle and apoptosis. We explored immunologic effects and mechanisms following restoration of wild-type human TP53 cDNA in murine oral cancer cells using the therapeutic nanocomplex scL-53. We demonstrated scL-53 induces dose-dependent expression of TP53 and induction of apoptosis and immunogenic cell death. We further demonstrated both TP53-dependent and independent induction of tumor cell immunogenicity through the use of blocking mAbs, nanocomplex loaded with DNA plasmid with or without TP53 cDNA, empty nanocomplex and siRNA knockdown techniques. TP53-independent immune modulation was observed following treatment with nanocomplex loaded with DNA plasmid lacking TP53 cDNA and abrogated in STING-deficient tumor cells, supporting the presence of a cytoplasmic DNA sensing, STING-dependent type-I IFN response. Cooperatively, TP53- and STING-dependent alterations sensitized tumor cells to CTL-mediated lysis, which was further enhanced following reversal of adaptive immune resistance with PD-1 mAb. In vivo, combination scL-53 and PD-1 mAb resulted in growth control or rejection of established tumors that was abrogated in mice depleted of CD8+ cells or in STING deficient mice. Cumulatively, this work demonstrates 1) a direct anti-tumor effects of functional TP53; 2) non-redundant TP53- and STING-dependent induction of tumor cell immunogenicity following scL-53 treatment; and 3) that adaptive immune resistance following scL-53 treatment can be reversed with PD-based immune checkpoint blockade, resulting in the rejection or control of syngeneic murine tumors. These data strongly support the clinical combination of scL-53 and immune checkpoint blockade.
Introduction
Loss of functional TP53 has profound impacts on the cellular control of cell cycle progression and apoptosis and contributes to malignant conversion.Citation1 Emerging evidence also implicates TP53 in the induction of immunogenic cell death and regulation of expression of a number of genes involved in antigen processing and tumor cell immunogenicity.Citation2–Citation5 The majority of patients with head and neck squamous cell carcinoma (HNSCC) harbor genetic alterations in TP536, likely contributing to poor tumor immunogenicity and ultimately immune escape in these tumors. Reconstitution of functional TP53 into HNSCC cells could alter the tumor immune microenvironment and either directly enhance anti-tumor immunity or improve responses to immune-activating therapies.
Several therapeutic approaches to introduce functional wild-type TP53 (wtTP53) into carcinoma cells, including adenoviral delivery platforms, have been tested clinically.Citation7,Citation8 Impressive responses were observed in subsets of patients, particularly when wtTP53 was delivered via intratumoral adenovirus in combination with cytotoxic therapy.Citation9,Citation10 However, adenoviral approaches have not been adopted for widespread use due to a number of issues including low transduction efficiency and safety concerns.Citation11 scL-53 is a therapeutic nanocomplex made of a cationic liposome coated with an anti-transferrin receptor single-chain antibody fragment (scL) that delivers a wild-type human TP53 payload into target cells via transferrin receptor-mediated endocytosis. Intravenous delivery of scL-53 has demonstrated significant anti-tumor activity in a number of pre-clinical models.Citation12–Citation15 Two phase I clinical trials have demonstrated that scL-53 complex is well tolerated, selectively delivers wtTP53 cDNA to malignant but not normal tissues, and results in clinical anti-tumor activity in a subset of patients alone or in combination with docetaxel.Citation16,Citation17 However, the immunologic effects of reconstituting wtTP53 in HNSCC have not ben studied.
Pre-clinical evaluation of scL-53 has been performed primarily in xenograft models lacking adaptive immune responses.Citation12–Citation15 Here, we utilized a syngeneic murine model of oral cavity cancer to study how introduction of wild-type human TP53 into tumor cells alters anti-tumor immunity. We demonstrated that scL-53 targeting transferrin receptor CD71 expressed by cancer cells introduces a transcriptionally active transgene that not only directly promotes loss of cell viability, but also enhances tumor cell immunogenicity and induces immunogenic cell death in vitro. Mechanistically, enhanced expression of antigen-processing machinery and components of cell surface immunogenicity appeared to be due to both TP53-dependent gene activation as well as STING-dependent sensing of cytoplasmic DNA resulting in the induction of a type-I interferon (IFN) response. Using oral cancer cells engineered to express a model antigen, immune killing assays were used to demonstrate enhanced tumor cell susceptibility to T-lymphocyte killing after scL-53 treatment. In vitro, scL-53 induced expression of tumor cell PD-L1, and the addition of PD-1 mAb reversed adaptive immune resistance and enhanced T-lymphocyte killing of tumor cells. These alterations were validated in vivo as scL-53 treatment alone enhanced tumor cell immunogenicity and CD8 T-lymphocyte tumor infiltration. The combination of scL-53 treatment and PD-1 mAb in vivo significantly enhanced tumor growth control over either treatment alone and induced rejection of a subset of established tumors and immunologic memory. These results were largely abrogated following CD8+ cell depletion and in STING-deficient mice, validating a contribution of cytoplasmic DNA-sensing in both tumor and host cells to the induction of CD8-mediated anti-tumor immunity following scL-53 treatment. These data reveal a novel mechanism for induction of anti-tumor immunity following nucleic acid-based gene therapy and support the clinical investigation of scL-53 in combination with treatments that reverse adaptive immune resistance such as PD-based immune checkpoint blockade.
Results
MOC1 tumor cells express transferrin receptor in vitro and in vivo.
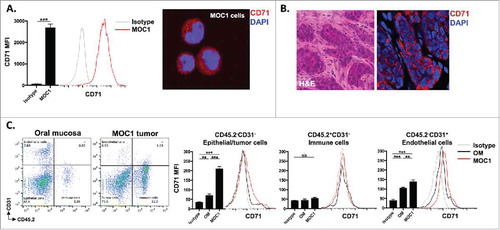
Most tumor cells express high levels of CD71 in response to increased metabolic demands for iron.Citation18 CD71 serves as the binding target for receptor-mediated endocytosis entry into cells via the anti-TfR single-chain antibody fragment that decorates the surface of the scL-53 nanocomplexes. To validate the expression of CD71 on MOC1 cells both in culture and in established tumors in syngeneic wild-type (WT) B6 mice, we used flow cytometry and immunofluorescence. demonstrates positive expression of CD71 on cultured MOC1 cells (A, flow on left, IF on right) and tumor but not stromal cells in established MOC1 tumors (B). To determine expression of CD71 in different cellular subsets within established MOC1 tumors and normal oral mucosa, we used flow cytometry to measure expression on tumor/epithelial, endothelial, and tissue infiltrating immune cells (). While CD71 expression was relatively lower on infiltrating CD45.2+ immune cells in both tumor and mucosa, expression was significant greater on MOC1 tumor cells compared to oral epithelial cells as well as on endothelial cells within MOC1 tumors compared to endothelial cells within oral mucosa. These data validate the expression of the target for cellular entry via the antibody fragment on scL-53 nanocomplexes.
Figure 1. CD71 is expressed on MOC1 cells and endothelial cells within the tumor microenvironment. A, MOC1 cells were assayed for CD71 expression by flow cytometry (clone R17217; left panel) or immunofluorescence (right panel). B, MOC1 tumors were harvested, sectioned, and assessed for CD71 expression via immunofluorescence (40x image, H&E on left). C, MOC1 tumor or oral mucosa was harvested, digested into a single cell suspension and assessed for CD71 expression on CD45.2−CD31− epithelial/tumor cells, CD45.2+CD31− immune cells or CD45.2−CD31+ endothelial cells via flow cytometry. Representative dot plots of the digested tissues on the left, with representative CD71 histograms and quantification (MFI, n = 5 mice) on the right for each cell type. **, p < 0.01; ***, P,0.001.
scL-53 treatment of MOC1 cells produces a functional TP53 transgene that induces loss of MOC1 cell viability and immunogenic cell death.
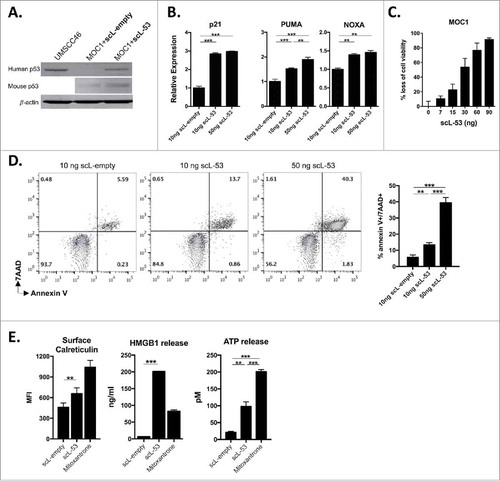
MOC1 tumor cells harbor a V170E non-synonymous Tp53 mutation and express low levels of TP53 protein and target gene expression.Citation19 Following exposure of MOC1 cells in culture to scL-53, western blot analysis was utilized to verify expression of human TP53 using a human-specific anti-TP53 antibody (). Murine TP53 expression was largely unaltered by scL-53 treatment. To validate expression of a functional TP53 transgene, qRT-PCR was used to demonstrate scL-53 dose-dependent induction of expression of TP53-target genes p21, PUMA and NOXA within treated MOC1 cells (). To assess whether introduction of wild-type human TP53 directly altered MOC1 cell survival, we performed XTT viability and apoptosis assays. demonstrates scL-53 dose-dependent loss of cell viability via XTT assay. Dose-dependent induction of dual 7AAD and annexin V positivity after scl-53 treatment () suggested that this loss of MOC1 cell viability was due at least in part to induction of apoptosis. Not all cellular stress or loss of cell viability induces cell surface expression and release of innate immune receptor ligands consistent with immunogenic cell death (ICD). demonstrates increased cell surface calreticulin expression, HMGB1 and ATP release following scL-53 treatment which, when combined with loss of cell viability, supports induction of ICD by defined criteriaCitation20 and is consistent with the result of others.Citation5
Figure 2. Treatment of MOC1 cells with scL-53 results in a functional TP53 protein that induces loss of cell viability, apoptosis and immunogenic cell death. A, MOC1 cells were exposed to 10 ng of scL-53 or scL-empty for 4 hours (volume equivalents), then incubated without treatment for 24 hours and assayed for human TP53 (clone DO-1) or mouse TP53 (clone 197643) expression by western blot. Lysates from human UMSCC46 cells served as a positive control for human TP53. B, expression of TP53 target genes p21, PUMA and NOXA was assessed via qRT-PCR following treatment with scL-empty or scL-53 as in A. C, cell viability was measured via XTT assay following treatment with increasing doses of scL-53 (exposed to scL-53 for 4 hours, then incubated without scL for 48 hours, indicated doses are per 1 × 104 cells). D, expression of the apoptotic marker annexin V and 7AAD uptake was measured via flow cytometry 48 hours after a 4 hour treatment with scL-empty or 10 ng or 50 ng scL-53. E, to assess for induction of immunogenic cell death, cell surface calreticulin (flow cytometry, clone EPR3924), HMGB1 release (ELISA) and ATP release (bioluminescence assay) was assessed in MOC1 cells treated as in A (scL-53 dose of 10 ng) . For experiments in C-E, all indicated doses are quantity of plasmid per 1 × 104 cells. All data are representative results from one of at least two independent experiments with similar results. **, p < 0.01; ***, P,0.001.
scL-53 treatment in vitro enhances tumor cell immunogenicity through TP53-dependent gene expression and induction of STING-dependent type-I IFN responses.
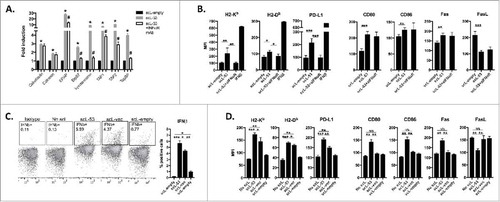
TP53 has been reported to regulate expression of a number of genes involved in antigen processing and presentation.Citation2-Citation4 Following treatment of MOC1 cells with scL-53, expression of antigen processing machinery (APM) genes (including calreticulin, calnexin, components of the β-immuno-proteasome, and TAP1/2) were significantly increased from 2- to 26-fold above untreated controls (). Similarly, scL-53 treatment of MOC1 cells increased cell surface expression of proteins involved in cellular immunogenicity such as MHC class I, CD80/86 and Fas as well as PD-L1 (). However, the presence of an IFNαR blocking mAb that blocks type I IFN (both IFNα and IFNβ) signaling either totally (Erp57, immunoproteasome, TapBP, H2-Kb, H2-Db, PD-L1) or partially (ERAP, TAP1, TAP2) abrogated the induced expression of APM and immunogenicity components observed following scL-53 treatment, suggesting a role for type I IFN signaling in this response. Hypothesizing that MOC1 cells could be producing type I interferon in response to cytoplasmic DNA introduced by scL-53, we treated MOC1 cells with scL nanocomplexes with variable payloads. MOC1 cells produced IFNβ following treatment with scL-53 or scL loaded with plasmid DNA vector lacking the TP53 cDNA insert (scL-vec) but not empty scL nanocomplex (scL-empty) (), suggesting the introduction of DNA into the target cells was inducing the production of IFNβ. We validated that induced expression of a subset of cell surface immunogenicity components occurred following treatment with scL-53 or scL-vec but not scL-empty ().
Figure 3. Treatment of MOC1 cells with scL-53 induces expression of type I IFN-dependent and independent immunogenicity. A, MOC1 cells were exposed to 10 ng scL-empty or scL-53 per 1 × 104 cells for 4 hours then incubated without scL for 24 hours in the presence or absence of an IFNαR mAb (clone MAR1-5A3), and expression of antigen processing machinery was assessed via qRT-PCR. B, similarly treated MOC1 cells were assessed for expression of cell surface components of immunogenicity via flow cytometry. For some targets known to be IFN-inducible, cells treated with IFNβ (500 units/ml for 24 hours) were used as a positive control. C, MOC1 cells were treated with scL-53 (10 ng), nanocomplex containing a noncoding DNA payload (scL-vec, 10 ng) or scL-empty as in A, then stained for intracellular IFNβ via flow cytometry. D, MOC1 cells treated as in C were assayed for expression of cell surface components of immunogenicity. *, p < 0.05; **, p < 0.01; ***, P,0.001.
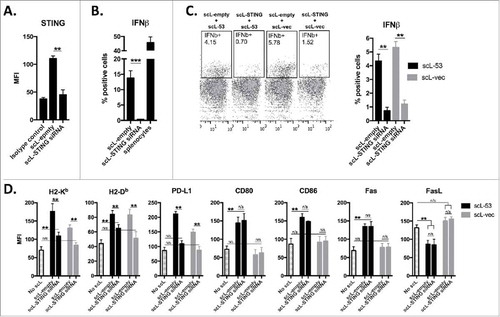
Through cGAS production of cGAMP, STING is known to serve as an innate immune receptor that can sense the presence of cytoplasmic DNA.Citation21,Citation22 To determine if STING was responsible for IFNβ production in treated MOC1 cells, we first used siRNA to knockdown expression of STING (). Functional loss of STING was validated with a lack of response to the STING agonist R,R-CDA (). We next treated MOC1 cells with scL-53 or scL-vec following siRNA knockdown of STING expression (). Abrogated STING expression largely reversed the ability of scL-53 or scL-vec to induce type I IFN expression. Treatment of MOC1 cells following STING knockdown with scL-53 or scL-vec revealed that induced PD-L1 expression appeared totally dependent on STING; that induced H2-Kb and H2-Db expression appeared to be dually dependent on STING and the presence of TP53-encoding DNA; and that increased CD80/86 and Fas expression and reduced FasL expression appeared to be totally dependent on the presence of TP53 encoding DNA (). Cumulatively, these data strongly suggest that while functional TP53 appears to be responsible for alterations in immunogenic components CD80/86, Fas and FasL, STING-dependent IFNβ production in response to cytoplasmic DNA solely or cooperatively drives alterations in the expression of H2-Kb, H2-Db and PD-L1.
Figure 4. Induction of IFN-dependent immunogenicity following scL-53 treatment is STING dependent. A, MOC1 cells were treated with scL loaded with TMEM173 siRNA (100 ng nucleic acid per 1 × 104 cells) or scL-empty (volume equivalent) for 4 hours and then incubated for 24 hours. TMEM173 protein expression was assessed via intracellular flow cytometry. B, following treatment as in A, MOC1 cells were treated with 20 μM R,R-CDA synthetic cyclic dinucleotide for 24 hours and IFNβ production was assayed by intracellular flow cytometry. C, following treatment as in A, MOC1 cells were treated with either scL-53 or scL-vec for 4 hours, then incubated for 24 hours. IFNβ production was assayed by intracellular flow cytometry. D, following treatment as in A, MOC1 cells were treated with either scL-53 or scL-vec for 4 hours, then incubated for 24 hours. Expression of cell surface components of immunogenicity was assayed via flow cytometry. **, p < 0.01; ***, P,0.001.
scL-53 treatment in vitro enhances tumor cell antigenicity and susceptibility to CTL-mediated killing
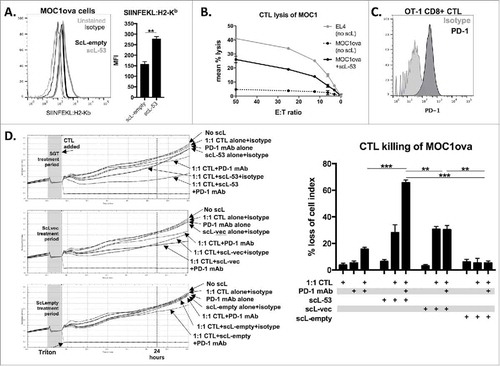
To investigate how treatment with different scL nanocomplexes functionally alters tumor cell sensitivity to CTL-mediated lysis, we utilized MOC1 cells engineered to express full-length ovalbumin as a model antigen (MOC1ova cells). Use of the 25-D1.16 antibody allows direct measurement of SIINFEKL presentation via H2-Kb on the surface of MOC1ova cells. Following scL-53 treatment, SIINFEKL expression was significantly enhanced (). To functionally validate this finding, we performed effector immune killing assays using SIINFEKL-specific cytotoxic T-lymphocytes (CTLs) generated from OT-1 splenocytes. In a standard Cr51-release assay, treatment of MOC1ova cells with scL-53 significantly enhanced MOC1ova sensitivity to CTL-mediated lysis (). We used flow cytometry to demonstrate that OT-1 CTLs express high levels of cell surface PD-1 (). Given the increased expression of tumor cells PD-L1 and the presence of PD-1 on the surface of OT-1 CTLs, we hypothesized that the addition of PD-1 mAb would enhance OT-1 CTL lysis of MOC1ova cells. CTL killing of MOC1ova cells treated with scL-53, scL-vec or scL-empty with or without PD-1 mAb was investigated using an impedance-based cytotoxicity assay allowing the real-time assessment of immune cell killing ( - individual growth curves on left, quantification on right). While the addition of PD-1 mAb did not significantly alter CTL lysis following scL-vec or scL-empty treatment, it significantly enhanced CTL lysis following treatment of MOC1ova cells with scL-53. These data support that scL-53 and scL-vec both functionally enhance susceptibility to CTL killing, but that only treatment with scl-53 results in adaptive immune resistance that is reversible with the addition of PD-1 mAb.
Figure 5. PD-1 mAb further enhances CTL lysis of engineered antigen-positive MOC1 cells following treatment with scL-53. A, to evaluate if scL-53 treatment enhances antigen presentation and antigen-specific CTL lysis, we performed experiments with MOC1 cells engineered to express full-length ovalbumin as a defined model antigen. SIINFEKL presentation via H2-Kb on the surface of MOC1ova cells was assessed via flow cytometry after treatment with scL-empty or scL-53 (10 ng per 1 × 104 cells for 4 hours then incubated for 24 hours; representative histograms on left, quantification on right). B, CTL-mediated lysis of MOC1ova cells following scL-53 treatment as in A was measured using a standard CR51-release assay, (SIINFEKL pulsed untreated EL-4 cells used as a positive control). C, PD-1 expression was assessed via flow cytometry on the surface of SIINFEKL-specific CTLs generated from OT-1 splenocytes. D, CTL-mediated lysis of MOC1ova cells following treatment with scL-53, scL-vec or scL-empty as in A with or without PD-1 mAb (clone RMP1-14, 1 μg/mL) or isotype control (rat IgG2 Ab) was measured using a real-time impedance-based cytotoxicity assay. OT-1 CTLs were added at the indicated vertical black line. scL treatment times indicated by grey shaded box. Conditional growth curves shown on left, quantification 24 hours after the addition of the CTLs on the right (vertical dashed line). Quantification of loss of cell index quantified on right. All data are representative results from one of at least three independent experiments. **, p < 0.01; ***, P,0.001.
scL-53 treatment in vivo enhances tumor cell immunogenicity and infiltration of effector immune cells
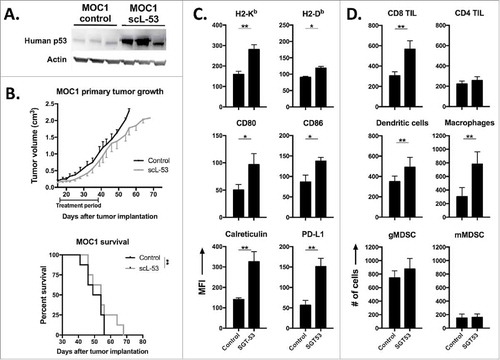
Mice bearing established MOC1 tumors were treated with single agent scL-53 and validated to express human TP53 within digested tumor tissue (). Evaluation of primary tumor growth and survival revealed a statistically significant but modest delay in primary tumor growth and enhancement of survival with scL-53 treatment alone (). However, flow cytometric analysis of digested tumor tissue revealed significantly enhanced immunogenicity of MOC1 tumor cells with increased expression of MHC class I, CD80/86, cell surface calreticulin and PD-L1 (), similar to our in vitro findings. This increased tumor cell immunogenicity was associated with enhanced tumor infiltration of CD8 but not CD4 TIL, dendritic cells and mature macrophages, without significant alteration in tumor infiltrating immunosuppressive myeloid derived suppressor cells (MDSC) (). While we did not assay specifically for regulatory T-cells in this dataset, total CD4 TIL were unaltered. These data suggested that while scL-53 monotherapy does not dramatically alter primary MOC1 tumor growth, it alters the tumor microenvironment in such a way to predict enhanced anti-tumor immunity and potentially adaptive immune resistance.
Figure 6. scL-53 treatment alone in vivo modestly alters MOC1 tumor growth but enhances tumor cell immunogenicity and effector immune cell tumor infiltration. A, established MOC1 tumors (WT B6 mice) were treated with scL-53 and digested whole tumors were assayed for the expression of human TP53 (clone DO-1) via western blot. B, treatment of established MOC1 tumors with single agent scL-53 (30 μg/injection, twice weekly x 3 weeks) resulted in a modest primary tumor growth delay (upper panel) and modest but statistically significant enhancement of survival (lower panel). Expression of MOC1 tumor cell (CD45.2−CD31−) surface components of immunogenicity (C, MFI shown) and tumor immune infiltration (D, absolute number of cells per 1 × 104 live cells collected) were assessed following treatment of established MOC1 tumors via flow cytometry. Identified CD45.2+ cells included CD8 TIL (CD3+CD8+), CD4 TIL (CD3+CD4+), dendritic cells (CD11c+CD11b+/−PDCA+/−), macrophages (CD11b+F4/80+), gMDSC (CD11b+Ly6G+Ly6Cint) and mMDSC (CD11b+Ly6G−Ly6Chi). Shown are representative results from one of two independent in vivo experiments. Control mice for these experiments received TVI of PBS alone. *, p < 0.05; **, p < 0.01.
Combination scL-53 and PD-1 mAb induces CD8+ cell and host STING-dependent rejection of a subset of established MOC1 tumors
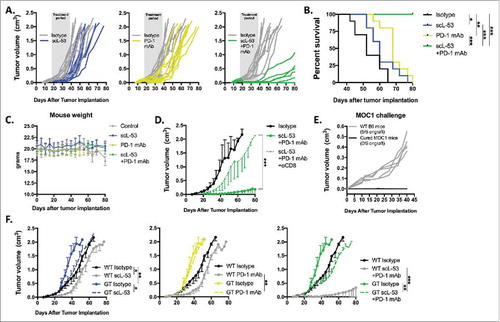
Given the above data, established MOC1 tumors were treated with scL-53 alone or in combination with PD-1 mAb. While neither treatment alone dramatically altered primary tumor growth, the combination resulted in rejection of 5/10 established MOC1 tumors and significantly delayed primary tumor growth in those tumors that did not reject (). This led to significantly better survival in mice treated with combination therapy (). Treated mice demonstrated no measureable toxicity with stable weight throughout treatment () and no labored breathing or behavioral change consistent with prior experiences with scL nanocomplexes.Citation12,Citation13 Tumor growth control after combination treatment was largely abrogated when combination treatment was preceded by systemic depletion of CD8+ cells (), and mice that rejected MOC1 tumors after combination treatment resisted engraftment with MOC1 cells indicating the presence of immunologic memory (). Given that CD71 is also expressed on stromal, endothelial and to a degree on hematopoietic cells, we hypothesized that STING expression within host cells would be important for the therapeutic effect of scL-53. Primary tumor growth inhibition following combination treatment was recapitulated when tumors were established in WT mice but subtotally reversed in STING-deficient mice (). Modest tumor growth inhibition was achieved with scL-53 treatment alone or in combination with PD-1 mAb in STING-deficient mice. These data demonstrate that primary tumor growth control or rejection following combination scL-53 plus PD-1 mAb treatment is dependent not only on CD8+ cells, but also on expression of STING in host cells.
Figure 7. Combination scL-53 and PD-1 mAb induces CD8 and host STING-dependent rejection of a subset of established MOC1 tumors. Individual growth curves (A) and survival (B) following treatment of established MOC1 tumors (n = 9–10 mice/condition) with scL-53 (30 μg/injection, twice weekly x 3 weeks) alone or in combination with PD-1 mAb (200 μg twice weekly 3 weeks). C, treated MOC1 tumor-bearing mouse weight was plotted over time. D, treatment with combination scL-53 plus PD-1 mAb as in A was repeated with or without CD8+ cell depletion (clone YTS 169.4, n = 5 mice/condition)). E, naïve WT B6 mice or WT B6 mice that rejected established MOC1 tumors after combination scL-53 and PD-1 mAb treatment were challenged with subcutaneous injection of 3 × 106 MOC1 cells and followed for tumor engraftment. F, MOC1 tumors were established in STING-deficient mice (GT; Tmem173gt, n = 5 mice/condition) and scL-53 treatment was repeated with or without PD-1 mAb as in A. Summary growth curves were analyzed via ANOVA of tumor volume at day 40 after tumor engraftment. For all in vivo experiments, shown are representative results from one of two independent experiments. Control mice for these experiments received TVI of PBS and IP injection of 200 μg of rat IgG2 Ab twice weekly. *, p < 0.05; **, p < 0.01; ***, P,0.001.
Discussion
Following demonstration of anti-tumor effects following restoration of functional TP53 over a decade agoCitation1, many therapeutic approaches aimed and introducing functional TP53 protein via gene therapy or activating endogenous TP53 have been attempted but failed to gain widespread use due to a number of clinical issues.Citation11,Citation23 Pre-clinical investigation of scL-53 gene therapy has primarily focused on direct anti-tumor cell activity. Here, using a syngeneic model system of oral cancer to study alterations in anti-tumor immunity, we report several in vitro and in vivo immunologic sequelae of introducing functional human TP53 into murine carcinoma cells. First, we identified induction of immunogenicity in tumor cells that appeared to be dependent on expression of functional TP53, STING-dependent type-I IFN production, or both. The observed cellular type-I IFN response appeared to be TP53-independent and due to the presence of cytoplasmic DNA as similar in vitro responses were observed following treatment with scL nanocomplex delivering a DNA plasmid with or without TP53 cDNA but not nanocomplexes lacking DNA. Second, scL-53 treatment of tumor cells appeared to induce adaptive immune resistance through increased STING-dependent PD-L1 expression. This resistance could be reversed with PD-1 mAb immune checkpoint blockade, leading to enhanced in vitro CTL killing of MOC1 tumor cells and significant primary tumor growth control or rejection in vivo following combination scL-53 and PD-1 mAb treatment. Finally, experiments using STING-deficient mice suggested the importance of both tumor cell and host cell STING expression in the observed therapeutic response with combination scL-53 and PD-1 mAb. While our experimental design did not allow us to specifically determine which host cell type was critical for this STING-dependent response, CD71 and STING expression profiles in endothelial and hematopoietic cells suggest a number of host as well as tumor cells could be playing a role.Citation24,Citation25
Our dissection of the relative roles of TP53 and cytoplasmic nucleic acid in induction of tumor cell immunogenicity provide some insight into the mechanisms that underlie the anti-tumor activity of scL-53. The scL-53 payload plasmid includes TP53 cDNA driven by a CMV promoter. The near total abrogation following STING knockdown in tumor cells treated with either scL-53 or scL-vec strongly supports a cytoplasmic DNA-sensing cGAS and STING-dependent mechanism of IFNβ production.Citation21,Citation22 It is also possible that introduction of functional TP53 into cancer cells lead to breakdown of nuclear stability and the introduction of endogenous nucleic acid into the cytoplasmic compartment. Tumor-derived DNA can lead to STING-dependent IFNβ production.Citation22 While we cannot rule out participation of tumor-derived DNA in the STING-dependent anti-tumor immunity that we see in mice, our in vitro observations in MOC1 cells treated with scL-vec suggest that scL-53 derived plasmid DNA is inducing the host STING pathway.
Our observations also indicated that scL-53 was superior to scL-vec in terms of sensitization to CTL-mediated lysis when combined with PD-1 mAb. One possible explanation is that several alterations is immunogenicity were observed with introduction of TP53 and not observed with scL-vec treatment, including increased CD80/86, increased Fas receptor, increased subsets of APM and decreased FasL expression. Many of these observations recapitulate the previous findings of others,Citation2-Citation4 and it is reasonable to hypothesize that despite similar levels of PD-L1 induction with scL-vec in cultured MOC1 cells, that expression of these other components of immunogenicity would be required to achieve efficient CTL-mediated lysis following reversal of adaptive immune resistance with PD-blockade. Thiery ET AL. also demonstrated that introduction of TP53 into target cells enhanced caspase 8 and BID-dependent induction of apoptosis following exposure to CTL-mediated cytotoxic insultsCitation4, suggesting that functional TP53 may reprogram intracellular responses downstream of TNF superfamily receptors. Additionally, TP53 has been reported to directly induce a type I IFN response following viral infection.Citation26 Our data demonstrates that while scL-53 did consistently induce higher expression of H2-Kb, H2-Db and PD-L1 in a largely STING-dependent fashion, scL-53 did not consistently induce significantly more IFNβ than scL-vec when measured directly. Cumulatively these data support several mechanisms to explain our observed enhanced CTL-sensitivity with scL-53 treatment plus PD blockade compared to scL-vec plus PD blockade.
Gene therapy approaches to increase activity of TP53 and subsequently induce tumor cell immunogenicity and anti-tumor immunity may be superior to therapeutic approaches that rely upon induction of endogenous TP53. While less than 10% of TP53 mutations in human cancers are nonsense mutations resulting in a truncated protein, the great majority of TP53 hotspot nonsynonymous missense mutations lead to functional loss due to defects in the DNA binding domain.Citation27 A recent report utilized nutlin-3a to induce activity of TP53, ICD, and anti-tumor immunity in mice bearing EL4 and B16 tumors. However, these cell lines harbor WT Tp535, 28 . It remains unclear if pharmacologic approaches to activate WT but under-expressed TP53 can be used with similar results in tumors harboring loss-of-function or nonsense TP53 mutations.
Contrary to recent results by Guo ET AL., scL-53 treatment in the MOC1 model did not appear to alter the tumor infiltration of immunosuppressive MDSC.Citation5 We did not assess ex vivo MDSC suppressive capacity, and it is possible that reduced suppressive potential of MDSC accounts for some of the increase in CD8 TIL and CD8+ cell-dependent anti-tumor immunity observed following scL-53 treatment in vivo. Others have reported that MDSC display high plasticity and can be converted into immunostimulatory antigen presenting cells, but our experience is that MDSC cell surface phenotypic markers tend to correlate well with immunosuppressive capacity in the MOC models.Citation29 Given no observed change in MDSC tumor infiltration, it is unlikely that a significant reduction in MDSC function accounts for the CD8+ cell- and STING-dependent growth control of MOC1 tumors.
To summarize, here we describe, in addition to direct TP53-induced loss of tumor cell viability, non-redundant mechanisms of enhanced tumor cell immunogenicity following scL-53 treatment through direct TP53-dependent activity and STING-dependent induction of type-I IFN responses. Conceptually, this suggests that scL-53 therapeutically can provide both the benefits of direct TP53 introduction and adjuvant immune activation through STING signaling. Further, suboptimal CTL lysis and in vivo immune responses following scL-53 therapy alone appeared to be restricted at least in part by PD-L1, and this adaptive immune resistance could be reversed through PD-based immune checkpoint blockade. These data provide a mechanistic rationale for combining scL-53 and PD-based immune checkpoint blockade in the clinical setting. HNSCC serves as a particularly attractive target for combination scL-53 and PD-1 mAb treatment given the high rate of TP53 genetic alterations and response rates of 15-20% to PD-based immune checkpoint blockade monotherapy in the recurrent/metastatic setting.Citation6,Citation30,Citation31
Materials and methods
Cell culture and murine experiments
MOC1 cells were obtained from Ravindra Uppaluri (Washington University in St. Louis) and maintained in culture as described.Citation32 Cells were used at low passage number and routinely verified to be negative for mycoplasma and murine associated pathogens. In some experiments, MOC1 cells stably transduced with full-length ovalbumin as a model antigen were used.Citation33 Cultured cells used for experiments were harvested with TrypLE-Select to minimize alteration of surface epitopes. All in vivo experiments were approved by the NIDCD animal care and use committee. Wild-type C57BL/6 (B6) mice purchased were from Taconic and TMEM176gt mice were purchased from Jackson Laboratories. Palate and oral tongue normal mucosa was harvested for analysis via standard surgical techniques. For tumor growth experiments, MOC1 tumors were established by subcutaneous flank injection of 5 × 106 cells. Treatments were started when tumor reached 0.1 cm3 volume, or approximately 14-18 days after injection. Tumors were measured three times weekly and tumor volume was calculated as: (tumor length x tumor widthCitation2)/2. For some experiments, CD8+ cells were depleted via intraperitoneal (IP) administration of 200 μg CD8 mAb clone YTX 169.4 twice weekly.Citation34 For other experiments, PD-1 mAb (clone RMP1-14) 200 μg administered IP twice weekly was used for immune checkpoint blockade.
Nanocomplex treatments
scL-53 (scL loaded with plasmid containing cDNA encoding TP53 under the control of a CMV promoter), scL-vec (scL loaded with plasmid without the TP53 cDNA insert) and scL-empty (empty scL) nanocomplex constructs were prepared as previously described.Citation12 For some experiments, scL constructs were loaded in commercially available STING siRNACitation35 (Thermo #507902). Lyophilized nanocomplexes were reconstituted into single elements in LAL water via sonication. For in vitro treatments, cells were treated with nanocomplex constructs in serum-free media for 4 hours before media with 2X serum was added for the remainder of the incubation. The quantity of nanocomplex construct added was standardized to a specified amount per 1 × 104 target cells and adjusted as needed for experimental size. For some in vitro experiments, IFNαR mAb (2 μg/mL; clone MAR1-5A3) was used to block type I IFN signaling or PD-1 mAb (clone RMP1-14, 2 μg/mL) was used to block PD-pathway signaling. Nanocomplex treatments in vivo were administered via tail vein injection (30 μg total plasmid content in 330 μL LAL water volume, LAL water alone used as a control).
Flow cytometry
Only fresh cultured cells or tissue prepared into single cell suspensions were analyzed. The harvest and digestion of MOC1 tumors or oral mucosa into single cell suspensions for analysis was performed as previously described.Citation25 Nonspecific staining was minimized by staining with CD16/32 (FcR) blocking antibodies for 10 minutes prior to the addition of primary antibodies. Anti-mouse CD71, CD45.2, CD31, PDGFR, H2-Kb, H2-Db, CD80, CD86, Fas, FasL, PD-L1, SIINFEKL:H2-Kb, CD3, CD8, CD4, CD11c, CD11b, PDCA, F4/80, Ly6G, and Ly6C antibodies were from Biolegend, anti-mouse calreticulin was from Abcam, and anti-mouse annexin V was from eBioscience. Primary antibodies were applied for 30-60 minutes at concentrations titrated for each antibody. Dead cells were excluded via 7AAD uptake and a “fluorescence-minus-one” technique was used to validate specific staining in all antibody combinations. For flow cytometric analysis of intracellular IFNβ (primary antibody ab24324; goat anti-rat IgG secondary), treated cells were exposed to brefeldin A for the final 4 hours of incubation. Fixation, permeation and staining were achieved with the intracellular fixation and permeabilization kit (eBioscience #88-8823-88) per manufacturer recommendations. All analyses were performed on a BD FACSCanto analyzer running FACSDiva software and interpreted using FlowJo (vX10.0.7r2).
Immunofluorescence
For immunocytofluorescence, cells were allowed to settle on charged slides before staining. For immunohistofluorescence, tumor tissue was embedded in optimum cutting temperature media. Sections (6 μm) were fixed with methanol, washed, and non-specific binding was blocked with a solution of 3% BSA and 0.1% Tween 20 in 1xPBS. PR-conjugated CD71 antibody (clone R17217) or isotype control antibody was incubated overnight at 4°C in a humidified chamber. Following washes, slides were counterstained with DAPI and mounted for analysis on a LSM 780 confocal microscope (Zeiss). Photomicrographs were analyzed using Zen 2012 SP1 software.
Western blot analysis
Tumor lysates were generated using the Tissue Lyser II per the manufacturer's recommendations. Whole-cell lysates were obtained using NP40 lysis buffer, mixed with NuPAGE LDS sample buffer and NuPAGE sample reducing agent (Life Technologies), heated at 95°C for 5 min and subjected to electrophoresis using 4 - 12% Bis-Tris precast gels (Life Technologies) at 150 V for 100 min. The Invitrogen iBlot Dry Blotting System was used to transfer proteins onto a PVDF membrane. Primary antibodies were diluted in 5% BSA prepared from Tween 20-TBS. Blots were incubated with Chemiluminescent HRP Antibody Detection Reagent (Denville Scientific Inc.) and imaged using Image Studio software (LI-COR Biosciences).
qRT-PCR
RNA was extracted from MOC1 cells using the RNEasy Mini Kit (Qiagen). cDNA was synthesized using high capacity reverse transcription. Gene expression was determined relative to GAPDH using the indicated commercial primers (Life Technologies) on a 7900HT Sequence Detection System (Applied Biosystems).
In vitro cell viability
Viability of MOC1 cells was measured via XTT (Trevigen) assay per manufacturer protocol.
HMGB1 ELISA
Used per manufacturer's (IBL International) recommendations.
ATP bioluminescence assay
Used per manufacturer's (CLS II; Sigma-Aldrich) recommendations.
Cytotoxic T-lymphocyte (CTL) killing assays
To generate SIINFEKL-specific CTLs, splenocytes from OT-1 mice were cultured in the presence of SIINFEKL (2 μg/ml) with daily 2:1 splitting. After 72 hours in culture, >80% of remaining cells are CD8+Vα2+ cells (data not shown).
Standard Cr51-release assay
OT-1 CTLs were combined with control or scL-53 treated MOC1ova cells or SIINFEKL pulsed and Cr51 labeled EL4 cells (positive control) at a various E:T ratios, and CTL activity was quantified via Cr51 release at 4 hours on a WIZARD2 Automatic Gamma Counter (PerkinElmer).
Real-time impedance assay
Following the addition of OT-1 CTL effectors to 1 × 104 target cells plated in a 96-well E-Plate (ACEA Biosciences), alteration of impedance was acquired using the xCELLigence Real-Time Cell Analysis (RTCA) platform per manufacturer recommendations. Triton X-100 (0.2%) was added to some wells to verify complete loss of cell index with total cell lysis, and CTLs alone were plated up to 1 × 106/well to verify that they do not contribute to gain of impedance (data not shown). Percent loss of cell index for a given time point was calculated as: 1-(experimental cell index/control cell index).
Statistics
Tests of significance between pairs of data are reported as p-values, derived using a Fisher's exact test or student's t-test with a two-tailed distribution and calculated at 95% confidence. Comparison of multiple sets of data was achieved with one or two-way analysis of variance (ANOVA). Kaplan-Meier curves were compared using the log-rank/Mantel Cox test. Differences in primary tumor summary growth curves were determined by one way ANOVA of tumor volumes at a specified day after treatment. Error bars reflect standard deviation from individual experiments. All analyses was performed using GraphPad Prism v6.
Disclosure of potential conflicts of interest
Dr. Chang is one of the inventors of the described nanodelivery technology, for which several patents owned by Georgetown University have been issued. The patents have been licensed to SynerGene Therapeutics Inc. for commercial development. Dr. Chang owns equity interests in SynerGene Therapeutics Inc. and serves as a non-paid scientific consultant to SynerGene Therapeutics Inc.
Financial support
This work was supported by the Intramural Research Program of the NIH, NIDCD, project number ZIA-DC000087 and DC000074.
Acknowledgments
We thank Drs. Ravindra Uppaluri and Nicole Schmitt for their critical review of this manuscript.
Funding
HHS | NIH | National Institute on Deafness and Other Communication Disorders (NIDCD), DC000074, HHS | NIH | National Institute on Deafness and Other Communication Disorders (NIDCD), DC000087
Related Research Data
References
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–5. doi:10.1038/nature05541. PMID:17251932.
- Wang B, Niu D, Lai L, Ren EC. p53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat Commun. 2013;4:2359. doi:10.1038/ncomms3359. PMID:23965983.
- Zhu K, Wang J, Zhu J, Jiang J, Shou J, Chen X. p53 induces TAP1 and enhances the transport of MHC class I peptides. Oncogene. 1999;18:7740–7. doi:10.1038/sj.onc.1203235. PMID:10618714.
- Thiery J, Abouzahr S, Dorothee G, Jalil A, Richon C, Vergnon I, Mami-Chouaib F, Chouaib S. p53 potentiation of tumor cell susceptibility to CTL involves Fas and mitochondrial pathways. J Immunol. 2005;174:871–8. doi:10.4049/jimmunol.174.2.871. PMID:15634909.
- Guo G, Yu M, Xiao W, Celis E, Cui Y. Local Activation of p53 in the Tumor Microenvironment Overcomes Immune Suppression and Enhances Antitumor Immunity. Cancer Res. 2017;77:2292–305. doi:10.1158/0008-5472.CAN-16-2832. PMID:28280037.
- Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–82. doi:10.1038/nature14129. PMID:25631445.
- Nemunaitis J, Nemunaitis J. Head and neck cancer: response to p53-based therapeutics. Head & neck. 2011;33:131–4. doi:10.1002/hed.21364.
- Nemunaitis J, Clayman G, Agarwala SS, Hrushesky W, Wells JR, Moore C, Hamm J, Yoo G, Baselga J, Murphy BA, ET AL. Biomarkers Predict p53 Gene Therapy Efficacy in Recurrent Squamous Cell Carcinoma of the Head and Neck. Clin Cancer Res : an official journal of the American Association for Cancer Research. 2009;15:7719–25. doi:10.1158/1078-0432.CCR-09-1044. PMID:19996201.
- Nemunaitis J, Swisher SG, Timmons T, Connors D, Mack M, Doerksen L, Weill D, Wait J, Lawrence DD, Kemp BL, ET AL. Adenovirus-mediated p53 gene transfer in sequence with cisplatin to tumors of patients with non-small-cell lung cancer. J Clin Oncol : official journal of the American Society of Clinical Oncology. 2000;18:609–22. doi:10.1200/JCO.2000.18.3.609. PMID:10653876.
- Swisher SG, Roth JA, Komaki R, Gu J, Lee JJ, Hicks M, Ro JY, Hong WK, Merritt JA, Ahrar K, ET AL. Induction of p53-regulated genes and tumor regression in lung cancer patients after intratumoral delivery of adenoviral p53 (INGN 201) and radiation therapy. Clin Cancer Res : an official journal of the American Association for Cancer Research. 2003;9:93–101. PMID:12538456.
- Chen GX, Zhang S, He XH, Liu SY, Ma C, Zou XP. Clinical utility of recombinant adenoviral human p53 gene therapy: current perspectives. Onco Targets Ther. 2014;7:1901–9. doi:10.2147/OTT.S50483. PMID:25364261.
- Xu L, Huang CC, Huang W, Tang WH, Rait A, Yin YZ, Cruz I, Xiang LM, Pirollo KF, Chang EH.. Systemic tumor-targeted gene delivery by anti-transferrin receptor scFv-immunoliposomes. Mol Cancer Ther. 2002;1:337–46. PMID:12489850.
- Xu L, Tang WH, Huang CC, Alexander W, Xiang LM, Pirollo KF, Rait A, Chang EH. Systemic p53 gene therapy of cancer with immunolipoplexes targeted by anti-transferrin receptor scFv. Mol Med. 2001;7:723–34. PMID:11713371.
- Camp ER, Wang C, Little EC, Watson PM, Pirollo KF, Rait A, Cole DJ, Chang EH, Watson DK.. Transferrin receptor targeting nanomedicine delivering wild-type p53 gene sensitizes pancreatic cancer to gemcitabine therapy. Cancer Gene Ther. 2013;20:222–8. doi:10.1038/cgt.2013.9. PMID:23470564.
- Kim SS, Rait A, Kim E, Pirollo KF, Chang EH. A tumor-targeting p53 nanodelivery system limits chemoresistance to temozolomide prolonging survival in a mouse model of glioblastoma multiforme. Nanomedicine. 2015;11:301–11. doi:10.1016/j.nano.2014.09.005. PMID:25240597.
- Senzer N, Nemunaitis J, Nemunaitis D, Bedell C, Edelman G, Barve M, Nunan R, Pirollo KF, Rait A, Chang EH. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol Ther : the journal of the American Society of Gene Therapy. 2013;21:1096–103. doi:10.1038/mt.2013.32. PMID:23609015.
- Pirollo KF, Nemunaitis J, Leung PK, Nunan R, Adams J, Chang EH. Safety and Efficacy in Advanced Solid Tumors of a Targeted Nanocomplex Carrying the p53 Gene Used in Combination with Docetaxel: A Phase 1b Study. Mol Ther : the journal of the American Society of Gene Therapy. 2016;24:1697–706. doi:10.1038/mt.2016.135. PMID:27357628.
- Miyamoto T, Tanaka N, Eishi Y, Amagasa T. Transferrin receptor in oral tumors. Int J Oral Maxillofac Surg. 1994;23:430–3. doi:10.1016/S0901-5027(05)80039-6. PMID:7890992.
- Onken MD, Winkler AE, Kanchi KL, Chalivendra V, Law JH, Rickert CG, Kallogjeri D, Judd NP, Dunn GP, Piccirillo JF, ET AL. A surprising cross-species conservation in the genomic landscape of mouse and human oral cancer identifies a transcriptional signature predicting metastatic disease. Clin Cancer Res. 2014;20:2873–84. doi:10.1158/1078-0432.CCR-14-0205. PMID:24668645.
- Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi:10.1146/annurev-immunol-032712-100008. PMID:23157435.
- Barber GN. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2014;35:88–93. doi:10.1016/j.it.2013.10.010. PMID:24309426.
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, ET AL. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–42. doi:10.1016/j.immuni.2014.10.017. PMID:25517615.
- Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical Overview of MDM2/X-Targeted Therapies. Front Oncol. 2016;6:7. doi:10.3389/fonc.2016.00007. PMID:26858935.
- Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, Gaide O, Michielin O, Hwu P, Petrova TV, ET AL. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A. 2015;112:15408–13. doi:10.1073/pnas.1512832112. PMID:26607445.
- Moore E, Clavijo PE, Davis R, Cash H, Van Waes C, Kim Y, Allen C. Established T Cell-Inflamed Tumors Rejected after Adaptive Resistance Was Reversed by Combination STING Activation and PD-1 Pathway Blockade. Cancer Immunol Res. 2016;4(12):1061–71. doi:10.1158/2326-6066.CIR-16-0104.
- Munoz-Fontela C, Macip S, Martinez-Sobrido L, Brown L, Ashour J, Garcia-Sastre A, Lee SW, Aaronson SA. Transcriptional role of p53 in interferon-mediated antiviral immunity. J Exp Med. 2008;205:1929–38. doi:10.1084/jem.20080383. PMID:18663127.
- Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi:10.1101/cshperspect.a001008. PMID:20182602.
- Melnikova VO, Bolshakov SV, Walker C, Ananthaswamy HN. Genomic alterations in spontaneous and carcinogen-induced murine melanoma cell lines. Oncogene. 2004;23:2347–56. doi:10.1038/sj.onc.1207405. PMID:14743208.
- Davis RJ, Moore EC, Clavijo PE, Friedman J, Cash H, Chen Z, Silvin C, Van Waes C, Allen C. Anti-PD-L1 Efficacy Can Be Enhanced by Inhibition of Myeloid-Derived Suppressor Cells with a Selective Inhibitor of PI3Kdelta/gamma. Cancer Res. 2017;77:2607–19. doi:10.1158/0008-5472.CAN-16-2534. PMID:28364000.
- Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, Heath K, McClanahan T, Lunceford J, Gause C, ET AL. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17:956–65. doi:10.1016/S1470-2045(16)30066-3. PMID:27247226.
- Ferris RL, Blumenschein G, Jr., Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, ET AL. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016;375:1856–67. doi:10.1056/NEJMoa1602252. PMID:27718784.
- Cash H, Shah S, Moore E, Caruso A, Uppaluri R, Van Waes C, Allen C. mTOR and MEK1/2 inhibition differentially modulate tumor growth and the immune microenvironment in syngeneic models of oral cavity cancer. Oncotarget. 2015;6:36400–17. PMID:26506415.
- Morisada M, Moore EC, Hodge R, Friedman J, Cash HA, Hodge JW, Mitchell JB, Allen CT. Dose-dependent enhancement of T-lymphocyte priming and CTL lysis following ionizing radiation in an engineered model of oral cancer. Oral Oncol. 2017;71:87–94. doi:10.1016/j.oraloncology.2017.06.005. PMID:28688697.
- Moore EC, Cash HA, Caruso AM, Uppaluri R, Hodge JW, Van Waes C, Allen CT. Enhanced Tumor Control with Combination mTOR and PD-L1 Inhibition in Syngeneic Oral Cavity Cancers. Cancer Immunol Res. 2016;4:611–20. doi:10.1158/2326-6066.CIR-15-0252. PMID:27076449.
- Pirollo KF, Zon G, Rait A, Zhou Q, Yu W, Hogrefe R, Chang EH. Tumor-targeting nanoimmunoliposome complex for short interfering RNA delivery. Hum Gene Ther. 2006;17:117–24. doi:10.1089/hum.2006.17.117. PMID:16409130.