
ABSTRACT
Using next-generation immunoglobulin (IGH) sequencing and flow cytometry, we characterized the composition, diversity and dynamics of non-malignant B cells in patients undergoing treatment with the Bruton tyrosine kinase (BTK) inhibitor ibrutinib or chemo-immunotherapy with fludarabine, cyclophosphamide, and rituximab (FCR). During ibrutinib therapy, non-malignant B cell numbers declined, but patients maintained stable IGH diversity and constant fractions of IGH-mutated B cells. This indicates partial preservation of antigen-experienced B cells during ibrutinib therapy, but impaired replenishment of the normal B cell pool with naïve B cells. In contrast, after FCR we noted a recovery of normal B cells with a marked predominance of B cells with unmutated IGH. This pattern is compatible with a deletion of pre-existing antigen-experienced B cells followed by repertoire renewal with antigen-naïve B cells. These opposite patterns in B cell dynamics may result in different responses towards neoantigens versus recall antigens, which need to be further defined.
Introduction
Successful therapy of patients with chronic lymphocytic leukemia (CLL) depends upon effective disease control, but, equally important, the preservation of immunocompetence, given that common and opportunistic infections remain a leading cause of morbidity and mortality.Citation1 Disease-related immunodeficiency in CLL affects the innate and adaptive immune systems, and includes T cell defects, hypogammaglobulinemia, impaired complement activity, and neutrophil as well as natural killer cell dysfunction.Citation2 Despite an expansion of T lymphocytes in untreated CLL patients, these T cells are functionally defective, with impaired immunological synapse formation being a characteristic feature.Citation3-5 Normal B cell function also is impaired, resulting in hypogammaglobulinemia, which is part of the natural history of the disease.Citation6,Citation7 Given these T and B cell deficiencies in CLL patients, antibody responses are often dampened, for example after prophylactic vaccination.Citation8,Citation9 These disease-inherent immunodeficiencies are further modulated by CLL-directed therapy. Chemoimmunotherapy (CIT) with FCR, a standard first-line therapy regimen for younger patients with (IGHV mutated) low-risk disease,Citation10,Citation11 and newer targeted agents, such as the BTK inhibitor ibrutinib, are highly effective treatments for CLL patients.Citation12-14 FCR, for example, effectively eliminates CLL cells in the majority of low-risk patients,Citation10,Citation11 but it also causes a profound and prolonged depletion of T cells.Citation15 Therefore, infections that are attributed to neutropenia and/or T lymphopenia are relatively common after CIT.Citation16 In contrast, ibrutinib generally is not toxic to normal hematopoiesis, and particularly not to T cells.Citation12,Citation17 Direct comparisons of infection rates between ibrutinib-based regimens and FCR are problematic due to lack of randomized data, but cross-trial comparisons suggest that the infectious risks in previously untreated CLL patients are lower with ibrutinib, at least during the first years on treatment.Citation10,Citation12,Citation18,Citation19 Lack of myelo- and T lymphotoxicity, along with a change from Th2-biased immunity due to inhibition of interleukin-2-inducible T cell kinase (ITK) by ibrutinib may account for relatively low rates of infection,Citation20 together with generally stable immunoglobulin levels that may even improve during therapy (IgA).Citation21 However, in previously treated patients, infections remain a more common problem with ibrutinib; about one third of patients experience grade ≥3 infections during the first 12 months of ibrutinib treatment which is more comparable to infection rates in patients undergoing other salvage regimens.Citation14,Citation22–24 In addition, atypical Pneumocystis jirovecii pneumonia can also occur during ibrutinib treatment.Citation25 Due to the importance of a normal B cell compartment for immune function, we analyzed the non-malignant B cell repertoire in patients undergoing treatment with ibrutinib or FCR with immunosequencing technology to dissect composition, dynamics and diversity of this immune compartment.
Material and methods
Patient and sample characteristics
The complete blood counts, immunoglobulin levels, and infectious complications history were recorded and analyzed for a total of 40 patients treated at MD Anderson Cancer Center with either CIT (FCR) or ibrutinib on protocols that were approved by The University of Texas MD Anderson Cancer Center institutional review board and registered at clinicaltrials.gov (NCT00759798, NCT02007044). The clinical trial NCT00759798 is a single center phase II study testing FCR treatment in untreated or rituximab pretreated patients.Citation26 FCR treated patients received up to 6 courses of fludarabine 25 mg/m2 given intravenously on days 2–4 of cycle 1 and days 1–3 of cycles 2 and beyond, cyclophosphamide 250 mg/m2 given intravenously on days 2–4 of cycle 1 and days 1–3 of cycles 2 and beyond, and rituximab 375 mg/m2 given intravenously on day 1 of course 1, and 500 mg/m2 given intravenously on day 1 of subsequent cycles. The clinical trial NCT02007044 is a single center phase II study primarily testing ibrutinib or ibrutinib and rituximab (only samples from the ibrutinib arm were analyzed) in pretreated patients or untreated patients with 17p deletion or TP53 mutation. Ibrutinib (420 mg daily by mouth) was given continuously until disease progression or toxicities or complications precluded further therapy. Comparison of the patient characteristics between samples from both studies (), revealed that ibrutinib treated patients were more heavily pretreated, had lower white blood cell counts (WBC), lower Rai stage, and less favorable genetic risk factors. FCR treated patients completed a median of 6 cycles (n = 20, range: 3–6) and all ibrutinb treated patients included were on treatment in the analyzed period (n = 20). The prophylactic use of antimicrobials was at the treating physician's discretion. Informed consent for collection of research samples was obtained in accordance with institutional guidelines and the Declaration of Helsinki. Peripheral blood mononuclear cells (PBMCs) for next-generation sequencing (NGS) and flow cytometry analysis were collected from 10 representative patients from each cohort during study visits before and after treatment initiation with FCR (after 24 months) or ibrutinib (after 12 and 24 months of continuous treatment). Late follow-up samples after 42 months were analyzed from 4 FCR treated and 2 ibrutinib treated patients. In addition, material from 9 age-matched healthy donors and 30 previously published control patients without a hematological malignancy were analyzed.Citation27
Table 1. Patient characteristicsFootnote#.
Multicolor flow cytometry for B cell phenotyping
As descriped previously,Citation27 within 2 hours of peripheral blood collection from control patients, erythrocyte lysis using a standard lysis buffer (ammonium chloride 8.29 g/l, EDTA 0.372 g/l, potassium hydrogen carbonate 1 g/l) was performed followed by flow cytometry using an 8-color flow cytometry panel (CD20-FITC, CD279(PD-1)-PE, CD38-ECD, IgM-PC-5.5, CD27-PC-7, CD19-APC – all purchased from Beckman Coulter). Measurements were performed on a Navios flow cytometer (Beckman Coulter, Krefeld, Germany). The gating strategy is shown in Figure S1.
Next-generation sequencing (NGS) of immunoglobulin heavy chain (IGH) immune repertoires
After isolation of genomic DNA from 5 × 106 – 3 × 107 PBMCs per sample using the GenElute Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich, Taufkirchen, Germany), the IGH gene locus containing the rearranged VH, DH and JH segments was amplified by multiplex PCR (Supplementary Figure S2) using previously published primers.Citation17,Citation28 Following purification with SPRIselect beads (Beckman Coulter, Krefeld, Germany), amplicon extension with Illumina adapter sequences and unique barcodes was achieved through a second PCR reaction. Primers were purchased from Metabion (Martinsried, Germany) and PCRs were performed using Phusion HS II (Thermo Fisher Scientific Inc., Darmstadt, Germany) according to the supplier's instructions. Finally, amplicons with the expected size were purified after agarose gel electrophoresis using the NucleoSpin® Gel and PCR Clean-up kit (Macherey-Nagel, Düren, Germany). After amplicon quantification and quality control with a Qubit (QIAGEN, Hilden, Germany) and an Agilent 2100 Bioanalyzer (Agilent technologies, Böblingen, Germany) sequencing was performed on an Illumina MiSeq platform.
Immunoglobulin isotype specific NGS from pre- and post-treatment RNA samples.
After isolation of RNA from 5 × 106 – 3 × 107 PBMCs per sample using the RNeasy Mini Kit (QIAGEN, Hilden, Germany), cDNA synthesis with the Mint-2 cDNA synthesis kit (Evrogen, Moscow, Russia) allowed subsequent immunoglobulin isotype specific amplification as described previously.Citation29 Afterwards, preparation for NGS including amplicon extension with Illumina adapter sequences and unique barcodes was performed as described above.
Determination of absolute sizes of malignant and non-malignant peripheral blood B cell compartments by integration of flow cytometry and NGS data
The CD19+ subpopulation of previously frozen PBMCs was determined by flow cytometry (antibodies CD19-APC, CD5-FITC purchased from Beckman Coulter), which was performed on a FACS Calibur cytometer (BD Biosciences) and analyzed using Flowing Software (http://flowingsoftware.btk.fi). As visualized in Figure S3, for every sample and time point the number of granulocytes was subtracted from the white blood cell count (WBC), which equaled the concentration of PBMCs in the blood. This was multiplied with the relative amount of CD19+ cells to calculate the CD19+ cell count. Based on the CDR3 amino acid sequences determined by NGS, non-malignant B cells and CLL clones were defined. The fraction of non-malignant B cells was calculated by subtraction of sequences belonging to the CLL clone from the total B cell sequences. Afterwards, the absolute count of non-malignant B cells was determined by multiplication of the fraction of non-malignant B cells with the CD19+ cell count.
Illumina NGS, data analysis, bioinformatics and statistics.
NGS was performed using an Illumina MiSeq sequencer [500 or 600 cycle single indexed, paired-end runs (V2 or V3 chemistry)] with adapter-ligated spiked-in PhiX library to increase diversity. The MiSeq reporter performed demultiplexing. Subsequently, FASTQ data analysis was computed in an analysis pipeline, which is based on the MiXCR software.Citation30 FASTQ data are available from the Sequence Read Archive, accession number PRJEB23571. Summarized NGS data is available as supplemental table S1. IGH hypermutation was defined as less then 97% identity between the VH-gene sequence and the corresponding germline gene. This definition was chosen based on correlation of the NGS results (pooled analysis of >360.000 aligned reads and >38.000 clonotypes) with flow cytometry measuring antigen-naïve and -experienced B cells in a cohort of 30 control patients (Figure S4 and S5). IGH clonotype diversity was quantified with the Shannon-Wiener and inverse Simpson diversity indices.Citation31,Citation32 Shannon-Wiener is a diversity index of first order disproportionately sensitive to the rare species/clonotypes, inverse Simpson of second order disproportionately sensitive to the most common species/clonotypes.Citation31 Indices were calculated for each sample and time point to be able to monitor changes in immune repertoire diversity before and after treatment initiation. Only when both diversity indices increased after treatment, IGH diversification was assumed. Student's t-test (paired samples) and one-way ANOVA followed by suitable posthoc tests (multiple samples) was used to calculate statistical significance as described in detail in the figure legends.
Results
Ibrutinib quantitatively suppresses peripheral blood non-malignant B cell repertoires
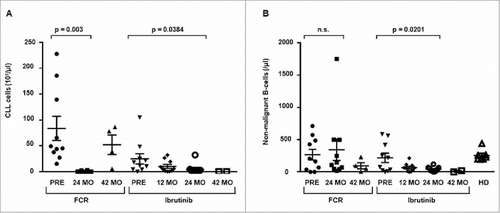
We quantitatively analyzed B cell counts in a subset of ten representative patients treated with ibrutinib versus ten patients treated with FCR by flow cytometry and NGS of the IGH locus. While many of the patients in the ibrutinib cohort were substantially pre-treated, all patients treated with FCR received this as first-line therapy (median number of 2 versus 0 prior treatments). In addition, ibrutinib treated patients had a less favorable genetic risk (5 versus 0 patients with del(17p)). Pre-treatment CLL cell counts were higher in the FCR cohort compared to the ibrutinib cohort and significantly reduced 24 months after initiation of treatment in both cohorts (). The post-treatment reduction of the CLL cell count was more pronounced in the FCR cohort and complete remission of the disease was achieved in 9 of 10 patients after 24 months. Responses to ibrutinib were mostly partial remissions (), which is in agreement with previous studies.Citation12,Citation14 Despite patients having received prior treatments in the ibrutinib cohort, the non-malignant B cell counts before treatment initiation were similar in both patient cohorts with relatively normal B cell counts (mean +/− SEM: FCR 268 +/− 83 cells/μl; ibrutinib 218 +/− 73 cells/μl). This is comparable to healthy elderly individuals in whom we measured B cell counts of 256 +/− 33 cells/μl (mean +/− SEM), which is in agreement with published values.Citation33 Ibrutinib treatment significantly decreased the non-malignant B cell count after 24 months of treatment (mean +/− SEM: 42 +/− 12 cells/μl, p = 0.0201, ), while B cell counts recovered back to baseline levels in the FCR cohort (mean +/− SEM: 344 +/− 166 cells/μl, ). After 42 months of ibrutinib treatment, B cell counts remained low in the two available follow-up samples (mean +/− SEM: 10 +/− 9 cells/μl, ). In four FCR treated patients with early recurrence of the disease, normal B cell counts were decreased nonsignificantly 42 months after treatment (mean +/− SEM: 96 +/− 48 cells/μl, ).
Figure 1. Global CLL and non-malignant B cell count in the peripheral blood. Changes in cell count were determined by flow cytometry and IGH next-generation sequencing prior, 24 months and 42 months after chemoimmuntherapy (FCR) or prior, 12 months, 24 months and 42 months after initiation of ibrutinib and from age-matched healthy donors (HD) A: Change in CLL cell count. B: Change in non-malignant B cell count. Horizontal lines show mean values and error bars show the SEM, n = 2-10 for each group. Statistical significance testing was performed using student's t-test.
Redistribution of IGH hypermutated, antigen-experienced non-malignant B cells over the course of treatment
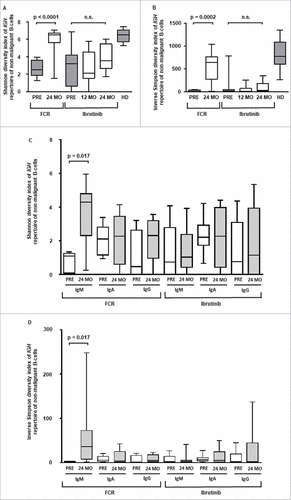
Next, we determined the dynamics of non-malignant B cell immune repertoire composition before and after FCR or during continuous ibrutinib treatment. Based on the mutational status of the VH gene, non-malignant B cells were classified as IGH hypermutated (<97% identity to the corresponding germline VH gene, corresponding to antigen-experienced B cells) or IGH unmutated (≥97% identity to the corresponding germline VH gene, corresponding to antigen-naïve B cells). This definition was experimentally determined in a control cohort of 30 individuals without hematological cancer through correlation of NGS immunosequencing with flow cytometry data from B cell immunophenotyping (Figure S4 and S5). Before treatment initiation, the mean percentage of antigen-experienced B cells was similar between the FCR cohort and age-matched healthy donors (HD) (mean +/− SEM of IGH mutated B cells: FCR 50 +/− 7%, HD 35 +/− 5%), while significantly less antigen-experienced B cells were measured in the ibrutinib cohort (mean +/− SEM of IGH mutated B cells: ibrutinib 22 +/− 6%, p = 0,0033). After 24 months, a significant decrease in antigen-experienced B cells was noted in the FCR cohort, while the ratio of antigen-experienced and antigen-naïve B cells remained unchanged in ibrutinib treated patients (mean +/− SEM of IGH mutated B cells: ibrutinib 34 +/− 7%, FCR 8 +/− 2%, p = 0.0001, ). Confirming the time point at 24 months, stable distributions were determined in available samples after 42 months (mean +/− SEM of IGH mutated B cells: ibrutinib 39 +/− 13%, FCR 12 +/− 1%, p = 0.0001, ) .Together, these data suggest a substantial deletion of the experienced B cell compartment, which is replaced with antigen-naïve B cells in the FCR cohort (exemplarily visualized in ). In contrast, ibrutinib treated patients had a preserved proportion of antigen-experienced B cells, while – quantitatively – the B cell repertoire was contracted (exemplarily visualized in ). We wished to confirm our results by IGH diversity analysis, assuming that the diversity of the regenerated FCR B cell repertoires should be highly increased, while ibrutinib B cell repertoires should show stable IGH diversity indices. Differences in IGH clonotype distributions were studied using the Shannon-Wiener diversity index, which is more sensitive to rare species/clonotypes, and the inverse Simpson diversity index, which is disproportionately sensitive to the most common species/clonotypes.Citation31 Indices were calculated for each sample and time point to be able to compare immune repertoire diversity before and after initiation of treatment. Before treatment initiation, the non-malignant IGH repertoire was composed of balanced numbers of antigen-experienced and antigen-naïve medium sized clones in both cohorts. In line with the IGH repertoire shift towards antigen-naïve B cells in FCR treated patients, the medium-sized clones disappeared after treatment, with large numbers of small-sized unmutated clones dominating after 24 months, resulting in increased repertoire diversity (, p < 0.0001 and 4B, p = 0.0002). In ibrutinib treated patients, the B cell repertoire showed an unchanged distribution of antigen-experienced and antigen-naïve B cells, resulting in stable repertoire diversity ( and ). Immunoglobulin isotype specific NGS from RNA samples before and 24 months after FCR treatment confirmed that the diversification was driven by IgM expressing naïve B cells, while IgA and IgG repertoire diversities remained stable ( and , p = 0.017). The IgM, IgA and IgG compartments from ibrutinib treated patients remained stable throughout treatment confirming our DNA-based sequencings ( and ).
Figure 2. Distribution of experienced and naïve non-malignant B cells. Hypermutated antigen-experienced non-malignant B cells determined through NGS. Samples were analyzed prior, 24 months and 42 months after FCR or prior, 12 months, 24 months and 42 months after initiation of ibrutinib treatment and from age-matched healthy donors (HD). Horizontal lines show mean values and error bars show the SEM, n = 2-10 for each group. Statistical significance testing was performed using student's t-test.
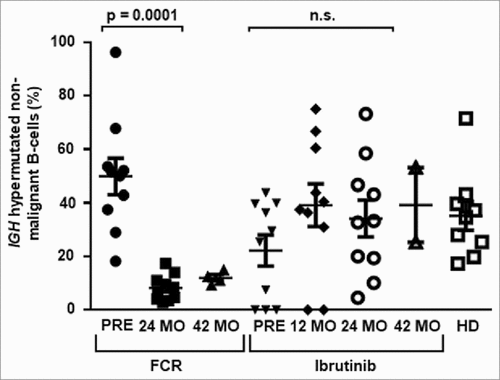
Figure 3. Exemplary IGH clonotype distribution of non-malignant B cell repertoires in the peripheral blood. A: Exemplary FCR treated patient. B: Exemplary ibrutinib treated patient. Coordinates of each dot are defined by the unique VH, DH and JH gene rearrangement. VH gene subgroups (V1-2 – V7-81) are shown from left to right, DH gene subgroups (D1-1 – D7-27) are shown from bottom to top. Dot size corresponds to the frequency. Blue color: unmutated VH gene sequence, red: hypermutated VH gene sequence.
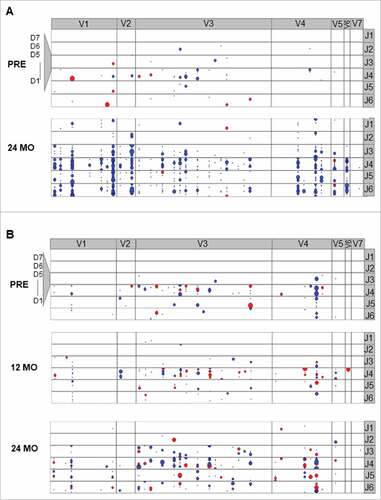
Figure 4. Diversity of the non-malignant B cell repertoire. A and B: Diversity of the non-malignant IGH repertoire based on genomic DNA with Shannon-Wiener and inverse Simpson diversity index. C and D: Isotype specific IGH repertoire diversity based on RNA sequencing with Shannon-Wiener and inverse Simpson diversity index. Horizontal lines show mean values and error bars show the SEM, n = 5-10 for each group. Statistical significance testing was performed using student's t-test.
Discussion
The adaptive immune system is severely impaired in CLL patients, with hypogammaglobulinemia and T cell dysfunction representing cardinal features.Citation2–4 This immune dysfunction is associated with ineffective immune responses towards the malignant B cells, autoimmune phenomena, and high susceptibility for infectious complications, which remain a leading cause for morbidity and mortality in CLL patients.Citation1 CIT with purine analogs, alkylating agents, and CD20 antibodies, such as FCR, remain a standard of care therapy option for the front-line treatment of younger low-risk patients, and are nowadays less commonly used for recurrent disease due to the advent of the kinase inhibitors targeting BCR signaling, such as ibrutinib, and the BCL-2 antagonist venetoclax.Citation18,Citation19,Citation34,Citation35 The myelo- and lymphotoxicity of CIT is associated with infectious complications in a significant proportion of patients.Citation15,Citation16,Citation36 In contrast, ibrutinib generally lacks myelotoxicity, neutropenia is rarely seen with ibrutinib, and – although it can modulate T cell function – it is thought to lack direct toxicity towards T cells. However, the consequences of long-term inhibition of BTK in CLL patients on the adaptive immune system and especially the B cell compartment remain incompletely defined. Children with XLA carrying functional null BTK mutations lack peripheral blood B cells and have markedly decreased or absent serum immunoglobulins of all isotypes.Citation37 This is due to failed development of B cell precursors in the bone marrow into mature B lymphocytes. In addition, BTK is also essential for germinal center reactions, as B cells from atypical XLA patients with mildly impaired BTK function show class switch defects and low frequencies of somatic hypermutation, while BTK overexpression increases germinal center formation.Citation38,Citation39 CLL patients treated with ibrutinib have relatively stable levels of immunoglobulins and do not exhibit an increasing incidence of infections during the first years of treatment, presumably because, in contrast to children with XLA, these patients have an antigen-experienced B cell compartment. Moreover, BTK is downregulated in plasma cells and therefore ibrutinib may not affect Ig production of preexisting plasma cells. However, the effect of ibrutinib on the normal B cell compartment and the composition of the B cell repertoire have not yet been studied.
Therefore, we examined the non-malignant B cell repertoire after or during treatment with CIT (FCR) or continuous BTK inhibition with ibrutinib using state-of-the-art next-generation immunosequencing. Interestingly, we noted strikingly different changes in the B cell compartment in both patient groups; FCR resulted in a major renewal of the B cell compartment with depletion of pre-existing B memory cells and emergence of large numbers of naïve antigen-inexperienced B cells. In contrast, ibrutinib treatment preserved, in part, the non-malignant pre-treatment B cell repertoire, but total B cell numbers declined and were not replenished by naïve B cells.
The declining numbers of B cells in ibrutinib treated patients, along with lack of replenishment by naïve B cells, are reminiscent of XLA patients, with the fundamental difference that CLL patients have a plasma cell pool to maintain Ig levels that is not affected by the treatment, which protects patients from frequent infections. However, our data also suggest that ibrutinib treated patients may have impaired responses towards neoantigens and consequently responses towards vaccines may be dampened. In XLA patients, this has been shown to be a characteristic feature that distinguishes XLA from other immunodeficiencies. In an elegant study, Ochs and colleagues explored responses to bacteriophage varphiX 174, and noted prolonged circulation of phage and no detectable antibody response as distinguishing characteristics of XLA.Citation40 In patients with CLL, vaccine studies might be complicated to conduct and interpret due to the pre-existing disease-inherent deficiencies in vaccine responses, but generally such studies are much-needed to refine our infection-preventive measures in this patient group.
Overall, comparisons between the two analyzed cohorts in this study need to be interpreted with caution, since patient characteristics differed, with ibrutinib treated patients having a less favorable genetic risk and being more heavily pretreated (median 2 versus 0 prior treatments). The effects of a drug on the immunocompetence through modulation of the B cell repertoire also needs to be seen in the context of baseline, disease-associated immunosuppression, as it is well established that not only more advanced CLL, but also a high number of pretreatments negatively impact immunocompetence.Citation22 Differences in the cohorts were also reflected by divergent levels of pretreatment ratios of naive versus antigen-experienced non-malignant B cells: While in the FCR cohort about equal numbers of naive and antigen-experienced B cells were seen, the percentage of naive B cells was higher in ibrutinib patients, who had received ibrutinib in a treatment line most often following CIT.
In summary, our study provides insight into the differential effects of CLL therapy on the normal B cell compartment. While FCR therapy depletes the experienced B cell compartment, followed by replenishment with naïve B cells, ibrutinib therapy over time results in a decline in normal B cell numbers and lack of replenishment with naïve B cells. This is consistent with the different mechanism of action of these types of therapy, acute lymphotoxicity in case of FCR and inhibition of BCR signaling-dependent growth and maturation of B cells in case of ibrutinib. In contrast, the data from the FCR treated cohort suggest that in these patients memory B cell responses may be more altered. Further studies about normal B cell development and function are warranted to address these questions.
Disclosure of potential conflicts of interest
A potential conflict of interest was declared: J.A.B. received research funding from Pharmacyclics. The remaining authors declare no potential conflicting interests.
Financial support
To J.A.B: Leukemia & Lymphoma Society Scholar Award in Clinical Research; MD Anderson's Moon Shot Program in CLL; and MD Anderson Cancer Center Support under Grant CA016672
To M.B.: Eppendorfer Krebs- und Leukämiehilfe e.V.; Annemarie Hilgemann Stiftung; Deutsche Krebshilfe under Grant 110906; and Hubertus Wald-Foundation.
Authorship contributions
M.B. designed the study, supervised the experiments, analyzed data and wrote the manuscript. J.B. designed the study, provided patient samples, analyzed data and wrote the manuscript. S.S. designed, performed and interpreted the experiments and wrote the manuscript. M.S and E.K. provided samples, analyzed clinical data and critically revised the manuscript. N.A. analyzed data and wrote the manuscript. L.W., B.T., C.F.-G., A.O. and D.S. analyzed data. T.T. and A.K.-G. provided the bioinformatics pipeline. C.B. interpreted data and critically revised the manuscript. N.J., Z.E., A.F., W.W. and M.K. provided samples and critically revised the manuscript. All authors reviewed the manuscript and approved the final version.
sup_data.zip
Download Zip (313.7 KB)Acknowledgments
We thank Christiane Horn, Katrin Kluge and Annette Schäfer for expert technical assistance with multicolour flow cytometry. We also thank the healthy donors.
Additional information
Funding
References
- Morrison V. Infectious complications in patients with chronic lymphocytic leukemia: pathogenesis, spectrum of infection, and approaches to prophylaxis. Clin Lymphoma Myeloma. 2009;9:365–70. doi:10.3816/CLM.2009.n.071. PMID:19858055.
- Forconi F, Moss P. Perturbation of the normal immune system in patients with CLL. Blood. 2015;126:573–81. doi:10.1182/blood-2015-03-567388. PMID:26084672.
- Rissiek A, Schulze C, Bacher U, Schieferdecker A, Thiele B, Jacholkowski A, Flammiger A, Horn C, Haag F, Tiegs G, et al. Multidimensional scaling analysis identifies pathological and prognostically relevant profiles of circulating T-cells in chronic lymphocytic leukemia. Int J Cancer. 2014;135:2370–9. doi:10.1002/ijc.28884. PMID:24723150.
- Ramsay AG, Johnson AJ, Lee AM, Gorgün G, Dieu R Le, Blum W, Byrd JC, Gribben JG. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest. 2008;118:2427–37. PMID:18551193.
- Christopoulos P, Pfeifer D, Bartholomé K, Follo M, Timmer J, Fisch P, Veelken H. Definition and characterization of the systemic T-cell dysregulation in untreated indolent B-cell lymphoma and very early CLL. Blood. 2011;117:3836–46. doi:10.1182/blood-2010-07-299321. PMID:21270444.
- Rozman C, Montserrat E, Viñolas N. Serum immunoglobulins in B-chronic lymphocytic leukemia. Natural history and prognostic significance. Cancer. 1988;61:279–83. doi:10.1002/1097-0142(19880115)61:2%3c279::AID-CNCR2820610215/3e3.0.CO;2-4. PMID:2446736.
- Boughton BJ, Jackson N, Lim S, Smith N. Randomized trial of intravenous immunoglobulin prophylaxis for patients with chronic lymphocytic leukaemia and secondary hypogammaglobulinaemia. Clin Lab Haematol. 1995;17:75–80. doi:10.1111/j.1365-2257.1995.tb00322.x. PMID:7621634.
- Hartkamp A, Mulder AH., Rijkers G, van Velzen-Blad H, Biesma D. Antibody responses to pneumococcal and haemophilus vaccinations in patients with B-cell chronic lymphocytic leukaemia. Vaccine. 2001;19:1671–7. doi:10.1016/S0264-410X(00)00409-6. PMID:11166890.
- Sinsalo M, Aittoniemi J, Käyhty H, Vilpo J. Haemophilus influenzae type b (Hib) antibody concentrations and vaccination responses in patients with chronic lymphocytic leukaemia: predicting factors for response. Leuk Lymphoma. 2002;43:1967–9. doi:10.1080/1042819021000015916. PMID:12481893.
- Keating MJ, O'Brien S, Albitar M, Lerner S, Plunkett W, Giles F, Andreeff M, Cortes J, Faderl S, Thomas D, et al. Early Results of a Chemoimmunotherapy Regimen of Fludarabine, Cyclophosphamide, and Rituximab As Initial Therapy for Chronic Lymphocytic Leukemia. J Clin Oncol. 2005;23:4079–88. doi:10.1200/JCO.2005.12.051. PMID:15767648.
- Hallek M, Fischer K, Fingerle-Rowson G, Fink A, Busch R, Mayer J, Hensel M, Hopfinger G, Hess G, von Grünhagen U, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376:1164–74. doi:10.1016/S0140-6736(10)61381-5. PMID:20888994.
- Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, Bairey O, Hillmen P, Bartlett NL, Li J, et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N Engl J Med. 2015;373:2425–37. doi:10.1056/NEJMoa1509388. PMID:26639149.
- O'Brien S, Furman RR, Coutre SE, Sharman JP, Burger JA, Blum KA, Grant B, Richards DA, Coleman M, Wierda WG, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open-label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014;15:48–58. doi:10.1016/S1470-2045(13)70513-8. PMID:24332241.
- Byrd JC, Brown JR, O'Brien S, Barrientos JC, Kay NE, Reddy NM, Coutre S, Tam CS, Mulligan SP, Jaeger U, et al. Ibrutinib versus Ofatumumab in Previously Treated Chronic Lymphoid Leukemia. N Engl J Med. 2014;371:213–23. doi:10.1056/NEJMoa1400376. PMID:24881631.
- Ysebaert L, Gross E, Kühlein E, Blanc A, Corre J, Fournié JJ, Laurent G, Quillet-Mary A. Immune recovery after fludarabine-cyclophosphamide-rituximab treatment in B-chronic lymphocytic leukemia: implication for maintenance immunotherapy. Leukemia. 2010;24:1310–6. doi:10.1038/leu.2010.89. PMID:20463751.
- Tam CS, O'Brien S, Wierda W, Kantarjian H, Wen S, Do K-A, Thomas DA, Cortes J, Lerner S, Keating MJ. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood. 2008;112:975–80. doi:10.1182/blood-2008-02-140582. PMID:18411418.
- Schliffke S, Akyüz N, Ford CT, Mährle T, Thenhausen T, Krohn-Grimberghe A, Knop S, Bokemeyer C, Binder M. Clinical response to ibrutinib is accompanied by normalization of the T-cell environment in CLL-related autoimmune cytopenia. Leukemia. 2016;30:2232–4. doi:10.1038/leu.2016.157. PMID:27220665.
- Hallek M. Chronic lymphocytic leukemia: 2015 Update on diagnosis, risk stratification, and treatment. Am J Hematol. 2015;90:446–60. doi:10.1002/ajh.23979. PMID:25908509.
- Eichhorst B, Fink A-M, Bahlo J, Busch R, Kovacs G, Maurer C, Lange E, Köppler H, Kiehl M, Sökler M, et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol. 2016;17:928–42. doi:10.1016/S1470-2045(16)30051-1. PMID:27216274.
- Dubovsky JA, Beckwith KA, Natarajan G, Woyach J, Jaglowski S, Zhong Y, Hessler JD, Liu T-M, Chang BY, Larkin KM, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122:2539–49. doi:10.1182/blood-2013-06-507947. PMID:23886836.
- Sun C, Tian X, Lee Y, Gunti S, Lipsky A, Herman S, Salem D, Stetler-Stevenson M, Yuan C, Kardava L, et al. Partial reconstitution of humoral immunity and fewer infections in patients with chronic lymphocytic leukemia treated with ibrutinib. Blood. 2015;126:2213–9. doi:10.1182/blood-2015-04-639203. PMID:26337493.
- Byrd JC, Furman RR, Coutre SE, Burger JA, Blum KA, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, et al. Three-year follow-up of treatment-naive and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125:2497–506. doi:10.1182/blood-2014-10-606038. PMID:25700432.
- Wierda WG, Kipps TJ, Mayer J, Stilgenbauer S, Williams CD, Hellmann A, Robak T, Furman RR, Hillmen P, Trneny M, et al. Ofatumumab As Single-Agent CD20 Immunotherapy in Fludarabine-Refractory Chronic Lymphocytic Leukemia. J Clin Oncol. 2010;28:1749–55. doi:10.1200/JCO.2009.25.3187. PMID:20194866.
- Perkins JG, Flynn JM, Howard RS, Byrd JC. Frequency and type of serious infections in fludarabine-refractory B-cell chronic lymphocytic leukemia and small lymphocytic lymphoma: implications for clinical trials in this patient population. Cancer. 2002;94:2033–9. doi:10.1002/cncr.0680. PMID:11932906.
- Ahn IE, Jerussi T, Farooqui M, Tian X, Wiestner A, Gea-Banacloche J. Atypical Pneumocystis jirovecii pneumonia in previously untreated patients with CLL on single-agent ibrutinib. Blood. 2016;128:1940–3. doi:10.1182/blood-2016-06-722991. PMID:27503501.
- Strati P, Keating MJ, O'Brien SM, Burger J, Ferrajoli A, Jain N, Tambaro FP, Estrov Z, Jorgensen J, Challagundla P, et al. Eradication of bone marrow minimal residual disease may prompt early treatment discontinuation in CLL. Blood. 2014;123:3727–32. doi:10.1182/blood-2013-11-538116. PMID:24705492.
- Akyüz N, Brandt A, Stein A, Schliffke S, Mährle T, Quidde J, Goekkurt E, Loges S, Haalck T, Ford CT, et al. T-cell diversification reflects antigen selection in the blood of patients on immune checkpoint inhibition and may be exploited as liquid biopsy biomarker. Int J Cancer. 2017;140:2535–44. doi:10.1002/ijc.30549. PMID:27925177.
- van Dongen JJM, Langerak AW, Brüggemann M, Evans PAS, Hummel M, Lavender FL, Delabesse E, Davi F, Schuuring E, García-Sanz R, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17:2257–317. doi:10.1038/sj.leu.2403202. PMID:14671650.
- Thiele B, Kloster M, Alawi M, Indenbirken D, Trepel M, Grundhoff A, Binder M. Next-generation sequencing of peripheral B-lineage cells pinpoints the circulating clonotypic cell pool in multiple myeloma. Blood. 2014;123:3618–21. doi:10.1182/blood-2014-02-556746. PMID:24753536.
- Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva E V, Chudakov DM. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods. 2015;12:380–1. doi:10.1038/nmeth.3364. PMID:25924071.
- Jost L. Partitioning diversity into independent alpha and beta components. Ecology. 2007;88:2427–39. doi:10.1890/06-1736.1. PMID:18027744.
- Kirsch I, Vignali M, Robins H. T-cell receptor profiling in cancer. Mol Oncol. 2015;9:2063–70. doi:10.1016/j.molonc.2015.09.003. PMID:26404496.
- Erkeller-Yuksel FM, Deneys V, Yuksel B, Hannet I, Hulstaert F, Hamilton C, Mackinnon H, Stokes LT, Munhyeshuli V, Vanlangendonck F. Age-related changes in human blood lymphocyte subpopulations. J Pediatr. 1992;120:216–22. doi:10.1016/S0022-3476(05)80430-5. PMID:1735817.
- Jain N, O'Brien S. Initial treatment of CLL: integrating biology and functional status. Blood. 2015;126:463–70. doi:10.1182/blood-2015-04-585067. PMID:26065656.
- Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016;374:311–22. doi:10.1056/NEJMoa1513257. PMID:26639348.
- García Muñoz R, Izquierdo-Gil A, Muñoz A, Roldan-Galiacho V, Rabasa P, Panizo C. Lymphocyte recovery is impaired in patients with chronic lymphocytic leukemia and indolent non-Hodgkin lymphomas treated with bendamustine plus rituximab. Ann Hematol. 2014;93:1879–87. doi:10.1007/s00277-014-2135-8. PMID:24951124.
- Ponader S, Burger JA. Bruton's tyrosine kinase: from X-linked agammaglobulinemia toward targeted therapy for B-cell malignancies. J Clin Oncol. 2014;32:1830–9. doi:10.1200/JCO.2013.53.1046. PMID:24778403.
- Mitsuiki N, Yang X, Bartol SJW, Grosserichter C. Mutations in Bruton’ s tyrosine kinase impair IgA responses. Int J Hematol. 2015;101:305–13. doi:10.1007/s12185-015-1732-1. PMID:25589397.
- Kil LP, de Bruijn MJW, van Nimwegen M, Corneth OBJ, van Hamburg JP, Dingjan GM, Thaiss F, Rimmelzwaan GF, Elewaut D, Delsing D, et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood. 2012;119:3744–56. doi:10.1182/blood-2011-12-397919. PMID:22383797.
- Ochs HD, Davis SD, Wedgwood RJ. Immunologic responses to bacteriophage φX 174 in immunodeficiency diseases. J Clin Invest. 1971;50:2559–68. doi:10.1172/JCI106756. PMID:5129308.