
ABSTRACT
Triple-negative breast cancer (TNBC) is an aggressive and molecularly diverse breast cancer subtype typified by the presence of p53 mutations (∼80%), elevated immune gene signatures and neoantigen expression, as well as the presence of tumor infiltrating lymphocytes (TILs). As these factors are hypothesized to be strong immunologic prerequisites for the use of immune checkpoint blockade (ICB) antibodies, multiple clinical trials testing single ICBs have advanced to Phase III, with early indications of heterogeneous response rates of <20% to anti-PD1 and anti-PDL1 ICB. While promising, these modest response rates highlight the need for mechanistic studies to understand how different ICBs function, how their combination impacts functionality and efficacy, as well as what immunologic parameters predict efficacy to different ICBs regimens in TNBC. To address these issues, we tested anti-PD1 and anti-CTLA4 in multiple models of TNBC and found that their combination profoundly enhanced the efficacy of either treatment alone. We demonstrate that this efficacy is due to anti-CTLA4-driven expansion of an individually unique T-cell receptor (TCR) repertoire whose functionality is enhanced by both intratumoral Treg suppression and anti-PD1 blockade of tumor expressed PDL1. Notably, the individuality of the TCR repertoire was observed regardless of whether the tumor cells expressed a nonself antigen (ovalbumin) or if tumor-specific transgenic T-cells were transferred prior to sequencing. However, responsiveness was strongly correlated with systemic measures of tumor-specific T-cell and B-cell responses, which along with systemic assessment of TCR expansion, may serve as the most useful predictors for clinical responsiveness in future clinical trials of TNBC utilizing anti-PD1/anti-CTLA4 ICB.
Introduction
The importance of the tumor microenvironment, especially tumor infiltrating lymphocytes (TILs), on the immunologic control of cancers has long been recognized; however, multiple layers of immune suppression have prevented immune targeted therapies from being broadly successful.Citation1 One such layer is the balance between inhibitory and stimulatory signals, also known as immune checkpoints, which normally maintain self-tolerance and immune homeostasis.Citation2,Citation3 In naïve T-cells, these inhibitory checkpoint molecules are upregulated in parallel with T-cell activation and are now recognized as powerful pathways by which tumors can suppress T-cells.Citation4,Citation5 Checkpoint molecules are also highly expressed on T regulatory cells (Tregs) and can mediate direct suppression of T-cell responses.Citation6,Citation7
Of the many immune checkpoint proteins described, the two best validated therapeutically are cytotoxic T lymphocyte antigen 4 (CTLA4, targeted by ipilimumab and tremelimumab) and programmed cell death protein 1 (PD1, targeted by nivolumab and pembrolizumab). Antibodies against these proteins are approved by the FDA for treatment of six types of cancer, including melanoma, lung, renal cell, bladder, types of colorectal, and head and neck cancer, with numerous clinical trials underway for other types of cancer.Citation8-11 As clinical studies have progressed, several observations have been made: 1) treatment is more effective for certain types of cancer than others, 2) clinical efficacy is heterogeneous within responsive cancers, with only a fraction of individuals responding to treatments, and 3) many responsive patients experience durable clinical responses lasting more than 3 years, presumably mediated by effective tumor-specific immunity.Citation12 While effective antitumor responses are hypothesized to depend on the presence of tumor infiltrating lymphocytes (TILs), emergence of mutated neoantigens, and expression of immune checkpoint molecules, it is unknown exactly what parameters can best predict the emergence of effective immune responses at an individual level.Citation13-15
These points are illustrated in early trials of estrogen receptor (ER)-/progesterone receptor (PR)-/human epidermal growth factor receptor 2 (HER2)- (triple-negative) breast cancer (TNBC), an aggressive breast cancer subtype with only chemotherapeutic treatment options, for which anti-PD1/PDL1 therapy has resulted in tumor regression in a modest 12–20% of patients across multiple clinical trials to date.Citation16-20 This response rate is comparable to those demonstrated in other responsive cancers and occurs despite widespread infiltration of TILs and expression of PDL1 in the majority of TNBC patient samples, with tumoral PDL1 expression being highest in TNBC compared to other breast cancer subtypes.Citation21-27 Additionally, ∼80% of TNBC possess p53 mutations and are characterized by a high degree of genomic dysregulation that results in a comparatively high mutational rate and subsequent elevated neoepitope burden.Citation28 Over 40 clinical trials are ongoing or will soon begin enrollment to evaluate the efficacy of these checkpoint inhibitors in TNBC, most of which combine single ICB therapies with other interventions. However, few studies have investigated the mechanisms that underlie responsiveness of these antibodies in isolation making it difficult to propose rational therapeutic combinations.Citation8,Citation29,Citation30
Currently the only approved combination of ICB antibodies in oncology is anti-PD1 and anti-CTLA4. While this combination has demonstrated elevated toxicities in early studies of melanoma (>50% Grade III/IV), recent studies employing reduced doses and sequential use have achieved more clinically acceptable toxicity profiles, which along with the use of corticosteroids, has controlled toxicities in most patients.Citation31 Thus, while toxicity remains a critical concern, these approaches have led to more widespread use of the anti-CTLA4/anti-PD1 combination in Phase III trials for NSCLC, renal cell, head and neck, gastric, lung, mesothelioma and glioblastoma. Early indications suggest that this combination will be more effective than either monotherapy, but it remains unknown how it will synergize in TNBC. More importantly, it is unknown which specific patients of a particular type of cancer may benefit from these combinations. Thus, while there is considerable promise in the use of these approved ICB antibodies in cancers like TNBC, there remain critical gaps in a complete mechanistic understanding of their function in specific tumor types, how these ICB antibodies interact at an individual level, as well as how important certain factors are in influencing local immunotherapeutic outcomes (i.e. presence of neoantigens, TIL repertoire, PDL1 expression, FoxP3+ Treg infiltration, etc.).
To address these gaps in knowledge and guide the future clinical use of approved ICBs in TNBC, we established a mouse model of TNBC and tested the efficacy of anti-PD1 and anti-CTLA4 checkpoint blockade, both alone and in combination. We demonstrated that while treatment with anti-PD1 or anti-CTLA4 alone had a moderate anti-tumor effect, complete tumor regression was only observed in animals treated with both antibodies. We further confirmed this finding in several other models of TNBC. We analyzed the impact of these therapies on the TCR repertoire of tumor bearing mice and identified significant changes following treatment with anti-CTLA4 that were not altered by the addition of anti-PD1. We went on to investigate these therapies using a model of TNBC expressing a non-self gene, ovalbumin (OVA), with multiple possible neoantigenic epitopes. These studies confirmed the individual role of anti-PD1 in suppressing immune responses against TNBC and anti-CTLA4 in expanding immunodominant T-cell clones and inducing epitope spreading. Surprisingly, significant heterogeneity was observed in the TCR repertoires of individual mice, with the expanded clones being almost entirely unique for each tumor even when a population of transgenic T-cells specific for a dominant OVA epitope was adoptively transferred. Collectively these results suggest that dual checkpoint blockade may be necessary to enhance clinical activity in some tumor types as strategies that release local immune suppression, like anti-PD1, may not be effective without a broadening of the T-cell repertoire by an agent like anti-CTLA4.
Results
Immunologic profile of pre-clinical TNBC model
To investigate the impact of checkpoint antibodies in TNBC, we first established an orthotopic TNBC model using the ER-/PR-/HER2-, p53 mutated, basal-like murine breast cancer cell line E0771.Citation32,Citation33 These cells were derived from a spontaneous medullary breast cancer in C57/BL6 mice and readily form aggressive tumors when implanted into the mammary fat pad of mice (). We further evaluated these cells by RNAseq and confirmed that in addition to p53 being mutated (one stop codon mutation at G32* and another R334P tetramerization mutation, similar to those seen in Li-Fraumeni syndrome and breast cancer patientsCitation34-36), there are approximately 5 mutations per megabase of the genome. This mutation rate puts this cell line within the upper range of reported mutational load seen in human breast tumors and would be predicted to regularly form neoantigens.Citation14 More than 7% of the mutations found in this cell line are predicted to have a moderate to high impact on the affected proteins and over 44% are predicted to cause missense or nonsense proteins (Sup table 1 and 2). Taken together, this RNAseq data indicates that this cell line is predicted to have a clinically relevant number of neoantigens present. In addition, our previous research shows that human TNBCs secrete multiple inflammatory factors, leading to significant infiltration by immune cells.Citation37
Figure 1. Characterization of immune infiltration in E0771 TNBC tumor model. (A) E0771 cells were orthotopically implanted into the mammary fat pad of C57 BL/6 mice and tumor volume was measured biweekly. (B) Tumors were enzymatically digested and analyzed for immune cell infiltration when tumors reached terminal endpoint volume. Gating as follows: CD45+CD11-CD4+ or CD8+ T cells, CD45+CD11b+Ly6ChiLy6G- monocytes, CD45+CD11b+Ly6Ghi neutrophils, CD45+CD11b+Ly6 C-Ly6G-F480+ macrophages (C) CD45+CD11b-CD8β-CD4+FoxP3+ Tregs from spleens and tumors were quantified by flow cytometry when tumors reached terminal endpoint volume. (D) Representative histograms from tumors and spleens pregated on live, CD45+, CD11b-, CD8β-, CD4+, FoxP3+ or FoxP3- (top) and quantification (bottom). (E) CD45+CD11b-CD8β+ or CD45+CD11b-CD4+ cells from the spleen or tumors were analyzed for expression of PD1 when tumors reached terminal volume. (F) E0771 cells grown in culture, E0771 cells grown in culture with 10ng/ml recombinant IFNγ for 48 hours, or taken ex vivo from tumors grown orthotopically in the mammary fat pad of C57 BL/6 mice when tumors reached terminal endpoint volume were stained for PDL1 expression. Error bars indicate SEM. Representative of 3 experiments. n = 5 mice per group; *P < 0.05; **P < 0.01; ***P < 0.001 by paired t-test.
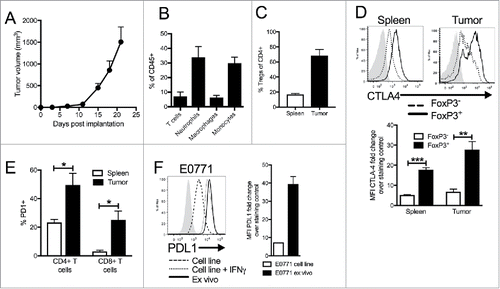
To determine if this model would exhibit immune infiltration in vivo, E0771 cell were injected and tumors analyzed at the terminal endpoint. These analyses demonstrated that tumors arising from E0771 cells contained large populations of lymphoid and myeloid cells, similar to human TNBC (). Substantial numbers of inflammatory monocytes and neutrophils were present in these tumors, but macrophages could also be found (). There were both CD4+ and CD8+ T-cells in the tumors, with the majority of intratumoral CD4+ T-cells being FoxP3+ Treg cells (). Evaluation of expression of immune checkpoint molecules revealed that TILs had high expression of both CTLA4 and PD1 compared to the spleen, with Tregs having the highest expression of CTLA4 ( and ). Analysis of the E0771 tumor cells revealed expression of PDL1 (, dashed line) that was significantly increased on tumor cells in vivo (, solid line), similar to levels induced in vitro after IFN-γ stimulation (, dotted line). Collectively, these results suggest that the E0771 pre-clinical model possesses many of the immunologic prerequisites identified in human TNBCs that would predict responsiveness to ICB antibody therapies.
The combined impact of anti-PD1 and anti-CTLA4 on anti-tumor immunity in TNBC
Based on our immunologic characterization of E0771 TNBCs in vivo, we hypothesized that this model would be particularly susceptible to ICB targeting PD1 and/or CTLA4. E0771 tumor bearing (>100 mmCitation3) mice were treated with anti-PD1, anti-CTLA4, or a combination of both. Control mice were treated with isotype matched irrelevant antibodies. Surprisingly, treatment with anti-PD1 and anti-CTLA4 alone elicited only moderate anti-tumor effects in this model while their combination resulted in a profound anti-tumor response, with 80% of mice demonstrating complete tumor regression (). Critically, we found that these responses were not limited to our E0771 model, as this treatment was highly effective against other TNBC cell lines (4T1 and JC) (Sup Fig. 1A and ) and led to long term survival of highly aggressive spontaneously occurring triple-negative breast tumors in MMTV-PYMT transgenic mice (Sup Fig. 1C).Citation38 This suggested that anti-CTLA4/anti-PD1 dual ICB was having a synergistic impact on anti-tumor immune responses.
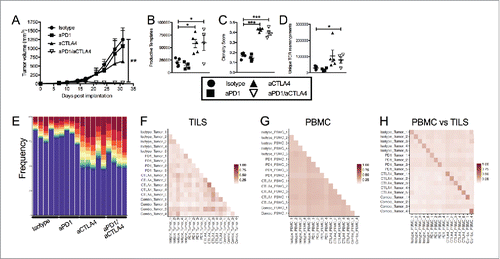
Figure 2. Dual blockade of PD1 and CTLA4 has profound anti-tumor effect on E0771 TNBC tumors and TCR repertoire. (A) E0771 cells were orthotopically implanted into the mammary fat pad of C57 BL/6 mice and measured biweekly. Mice were randomized into groups and treated with anti-PD1 and/or anti-CTLA4 (IgG2 a) antibodies biweekly beginning when tumors measured >100 mmCitation3 or were treated with isotope controls antibodies. (B-D) High-throughput quantitative sequencing of the rearranged TCR β genes of tumor or PBMC samples. Analyses were performed using immunoSEQ analyzer software (Adaptive Biotechnologies) and represent a single experiment. (B) Number of total productive TCR templates present in tumors. (C) Clonality score of TCRs present in tumors. (D) Number of unique TCR rearrangements present in tumors. (E) Frequency of the top ten TCR clones found in individual tumor samples. Each color (red through green) at the top of the bars represents the top 10 individual clones. The blue bar represents all remaining clones present in the sample. (F-H) Similarity heat map between individual tumor and PBMC samples. Dark red score of 1 is exactly the same and white score of 0 is completely dissimilar. (F) Similarity between tumor samples. (G) Similarity between PBMC samples. (H) Similarity between tumor and PBMC samples. Error bars indicate SEM. n = 5 per group for A; n = 3-5 per group for B-E; n = 2-5 per group for F; *P < 0.05; **P < 0.01, ***P < 0.001 by one-way ANOVA with Bonferroni's multiple comparisons test to isotype control group.
Changes in the TCR repertoire of TNBC TILs and PBMCs following checkpoint blockade
To further understand the mechanism of action of these ICBs on the immune response to TNBC, we evaluated their impact on the intratumoral and systemic TCR repertoire. In these experiments, we treated mice as before (Sup Fig. 2A and ) and after 1 week, we sequenced the CDR3 region of the β-chain of T-cell receptors present in peripheral blood and tumors and performed paired analysis of available samples.Citation39 TCR sequence analysis of TILs demonstrated a significant increase in the number of productive TCR templates present in the tumors of mice treated with anti-CTLA4 alone and the anti-PD1/anti-CTLA4 combination, but not in mice treated with anti-PD1 alone (). A clonality score was used in which 0 represented a perfectly polyclonal population and 1 represented a perfectly monoclonal population. Treatment with anti-CTLA4 led to an increase in this clonality score, indicating that the T-cell populations were becoming more mono- or oligoclonal (). While this indicates an expansion of several dominant clones, the number of unique TCR rearrangements was also increased following treatment with anti-CTLA4, demonstrating a broadening of the repertoire (). This is further demonstrated by graphing the proportion of total TCR transcripts that are accounted for by the top 10 clones, with each of the top 10 being shown in a different color (). The blue bar represents the fraction of the repertoire that is accounted for by all remaining clones not in the top 10. This analysis highlights the shift that occurs in the repertoire of anti-CTLA4 treated tumors, with approximately half of the total population being represented by the top 10 clones. Thus, anti-CTLA4 treatment resulted in a simultaneous expansion of dominant clones while also increasing the diversity of the overall repertoire in the tumor. Similar trends were seen in the PBMCs, however the overall clonality score did not increase significantly and the outgrowth of hyperexpanded clones was not as evident (Sup Fig. 2C and 2D).
We further compared the TCR repertoires based on overall similarity of the CDR3 sequences within the repertoires, with a score of 0 being completely dissimilar and a score of 1 being identical. When tumor samples from all mice were compared, the TIL TCRs between mice were largely dissimilar (F). TCRs in PBMCs showed an intermediate level of similarity between mice, consistent with the randomly generated, broad population of T-cells generally expected in circulation (). Finally, the repertoires that showed the highest degree of similarity were the matched blood and tumor samples from the same mouse, showing that the outgrowth of T cell clones in any given tumor were also found in systemic circulation (). These studies demonstrate the individual nature of the TCR repertoire present in each mouse, independent of treatment and in spite of the homogenous nature of the experimental system.
Using an OVA expressing TNBC model to evaluate checkpoint mechanisms of action
Overall, the TCR sequencing analysis revealed that anti-CTLA4 treatment leads to broad repertoire changes, regardless of the presence of anti-PD1. However, the profound anti-tumor response is only seen when both therapies are combined, suggesting unique, complimentary mechanisms of action for each therapy. To probe these mechanisms, we modified our E0771 cell line to express a non-self gene, ovalbumin (OVA) (Sup Fig. 3A). OVA has a 76% protein homology overlap with its nearest murine homolog, serpinb3, and therefore contains many neoantigenic determinants to facilitate immunological tracking and potentially homogenize immune responses between mice. Critically, we found that inclusion of OVA did not alter growth kinetics of the parental E0771 line in vitro or in vivo when implanted in SCID/beige mice (Sup Fig. 3B and C). In immunocompetent mice, this cell line allowed for tracking of tumor specific T-cell responses by restimulating splenocytes with an immunodominant OVA peptide (SIINFEKL) and assessing IFN-γ production by ELISPOT (Sup Fig. 3D) or B cell responses by quantitating OVA specific antibodies in the serum by ELISA (Sup Fig. 3E). We confirmed that despite a detectable anti-OVA response in these mice, expression of the immune target (OVA) was retained in tumors ex vivo from C57BL/6 mice, although levels were decreased relative to the initial cell line or tumors grown in SCID/beige mice (Sup Fig. 3F). This suggests that immune responses to OVA neoepitopes occurred, but that they only elicited a modest anti-tumor response.
To evaluate a potential role for non-self neoepitope-specific T-cells in controlling tumor growth, we adoptively transferred OVA-specific, TCR transgenic T-cells (OTI cells) intravenously (IV) 3 days after implantation of E0771-OVA cells. Cells were transferred at this early timepoint to allow time for in vivo activation, expansion, and infiltration into tumors before they became too large. Surprisingly, we found that transfer of 1 × 106 OTI cells had no effect on tumor growth and 5 × 106 OTI cells had only a minor, non-statistically significant, impact (Sup Figure 3G), despite substantial infiltration of tumors by OTI T-cells at the terminal endpoint shown (Sup Figure 3H). Taken together, we hypothesized that the tumor microenvironment of E0771-OVA tumors was immunosuppressive and targeting these pathways could enhance responses.
Subsequently, we tested our checkpoint inhibitors in this model and again saw that while either checkpoint inhibitor alone was effective, only the combination of anti-PD1 and anti-CTLA4 led to complete tumor regression (). To determine if these changes were the result of altered tumor-specific immunity, we assessed systemic anti-OVA responses. Notably, enhanced tumor-specific adaptive responses were only observed when both antibodies were used ( and ). Additionally, we observed a significant increase in the frequency of TILs in dual treated mice at the terminal endpoint shown that was not seen with either therapy alone (). Moreover, the proportion of live cells that were CD45+ cells increased in dual treated tumors (data not shown) and the frequency of Tregs present in treated tumors decreased, significantly altering the effector:Treg ratio in favor of effector T-cells ( and ). These results suggest that dual ICB promotes an effective T-cell response, possibly dominated by a T-cell clonal expansion to an OVA neoepitope.
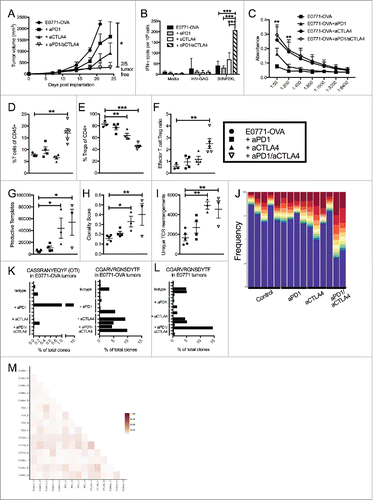
Figure 3. Dual blockade of PD1 and CTLA4 has profound anti-tumor effect on neoantigen expressing E0771-OVA tumors and has same effect on TCR repertoire. (A) E0771-OVA cells were grown subcutaneously in C57 BL/6 mice and tumor volume was measured biweekly. Mice were randomized into groups and treated with anti-PD1 and/or anti-CTLA4-IgG2 a antibodies biweekly beginning when tumors measured >100 mmCitation3 or left untreated. (B) Splenocytes from mice in (A) taken at the terminal endpoint were stimulated as indicated and IFN-γ producing cells were analyzed by ELISPOT. (C) Serum from mice in (A) taken at the terminal endpoint was analyzed by ELISA for anti-OVA antibodies. (D) CD4+ and CD8+ T cells from tumors of mice in (A) taken at the terminal endpoint were quantified by flow cytometry. (E) CD4+ FoxP3+ Tregs from tumors of mice in (A) taken at the terminal endpoint were quantified by flow cytometry. (F) Effector T cell: Treg ratio from (D) and (E) was calculated. A-E representative of 3 experiments. (G-M) High-throughput quantitative sequencing of the rearranged TCR β genes. Analyses were performed using immunoSEQ analyzer software (Adaptive Biotechnologies) and represent a single experiment. (G) Number of total productive TCR templates present in tumors. (H) Clonality score of TCRs present in tumors. (I) Number of unique TCR rearrangements present in tumors. (J) Frequency of the top ten TCR clones found in individual tumor samples. Each color (red through green) at the top of the bars represents the top 10 individual clones. The blue bar represents all remaining clones present in the sample. (K) Frequency of the top clone (left) and the OTI TCR clone (right) in each E0771-OVA tumor (L) Frequency of the top clone in each E0771 tumor (M) Similarity heat map between individual tumor samples. Dark red score of 1 is exactly the same and white score of 0 is completely dissimilar. Error bars indicate SEM. n = 5 per group for A-F; n = 3-5 per group for G-M; *P < 0.05; **P < 0.01; ***P < 0.001 by one-way ANOVA with Bonferroni's multiple comparisons test to E0771-OVA control group; P value in (C) are for both aCTLA4 and aCTLA4/aPD1 groups.
TCR repertoire changes in the E0771-OVA model are identical to those found in E0771 model
To determine the nature of T-cell expansion in the context of a commonly expressed non-self antigen, we repeated our TCR sequencing studies in this OVA-expressing model to confirm the effect of treatment and observe the impact of OVA on the TIL TCR repertoire. Given OVA expression, we hypothesized that the repertoire would be more consistent between individual mice, as it would be heavily biased towards an anti-OVA response. To further test this hypothesis, we transferred a modest number of OVA-specific OTI cells (105) into all mice the day after tumor implantation to serve as an immunodominant, trackable tumor-specific T-cell clone that we predicted would be highly expanded in ICB treated mice. We then treated established E0771-OVA tumors with our previous antibody combinations and sequenced the CDR3 region of the β-chain of T-cell receptors present in tumors. In this experiment, mice were either sacrificed when tumors began to regress or when they reached a terminal endpoint volume (Sup Figure 3I).
As with the parental E0771 tumors, anti-CTLA4 treatment led to a significant increase in the productive TCR templates, the clonality score, and the number of unique TCR rearrangements present in TILs (-). Similarly, we again saw outgrowth of hyperexpanded clones within the TILs such that the top 10 clones accounted for more than 50% of the total repertoire (). Unexpectedly, the OTI TCR sequence (CASSRANYEQYF) represented an expanded clone in only one of the samples (1 of 15) and, while present in some, it was absent from most of the tumor samples (8/15, ). Critically, this clone was not found in any of the mice when the tumors were not expressing OVA (data not shown). These data indicate that OT-I expansion in the tumor microenvironment was heterogeneous among mice and that endogenous T-cells targeting other tumor epitopes were dominant. Interestingly, a single clone (CDR sequence CGARVRGNSDYTF) was present in a majority of the samples and represented one of the expanded clones in nearly half of the samples regardless of whether the tumor expressed OVA or not ( and ). This provides further evidence that the expression of OVA did not overshadow the response to other tumor antigens. Contrary to our prediction, evaluation of individual clone sequences again revealed very little similarity between mice despite a non-self gene expressing multiple neoantigens (). Likewise, when the similarity of repertories were compared between OVA and non-OVA expressing tumors, a similar lack of overlap was seen between tumors regardless of OVA expression (Sup Fig. 3J).
It is also important to note that the inclusion of OVA did not alter the overall response of mice to various ICB therapies (A). These results demonstrate that while OVA-specific immune responses occurred in these mice, the transfer of OVA specific T-cells did not enhance responsiveness or constitute a significant portion of expanded TILs in most mice. Furthermore, OVA expression did not elicit more homogenous responses in ICB treated mice or predict responsiveness to ICB therapies.
The role of CTLA4 and Tregs in TNBC immunity
To better understand the underlying mechanisms of the anti-tumor responses elicited by dual ICB, we focused our attention on understanding the T-cell expansion mediated by ICB antibodies in TNBC. The TCR repertoire analysis revealed that anti-CTLA4 treatment leads to broad repertoire changes, regardless of the presence of anti-PD1. Having established a system that allows us to track anti-tumor immune responses and recapitulates the repertoire changes seen with the parental TNBC cell line, we wanted to examine mechanistically how each antibody was impacting tumor growth. We have previously shown that the majority of CD4+ T-cells in E0771 TNBC tumors are FoxP3+ Treg cells that express high levels of CTLA4 ( and ), and this pattern is also true in our E0771-OVA tumors (). We and others have previously noted a reduction in tumor-infiltrating Tregs following treatment with anti-CTLA4 (). To understand the impact of CTLA4 antibodies on this population in TNBC, we utilized different isotypes of anti-CTLA4 that would either primarily block receptor function (IgG1) or could additionally elicit antibody-dependent cell-mediated cytotoxicity (ADCC) (IgG2a).Citation40-43 This activity was confirmed by in vitro ADCC assays of CTLA4+ cells using both antibody isotypes (Sup. Fig 4A).
Figure 4. Targeting CTLA4 depletes intratumoral Tregs and enhances anti-tumor responses. (A) Flow cytometry intracellular staining of CTLA on CD4+ FoxP3- or CD4+ FoxP3+ T cells from the spleens of E0771-OVA tumor bearing mice and E0771-OVA tumors when tumors had reached a terminal endpoint. Representative histograms and summary data shown. Gray histogram represents staining control. (B) E0771-OVA cells were grown subcutaneously in C57 BL/6 mice and tumor volume was measured biweekly. Mice were randomized into groups and treated with anti-CTLA4 antibodies biweekly beginning when tumors measured >100 mmCitation3 or left untreated. (C) CD45+CD11b-CD4+FoxP3+ Tregs in tumors from mice in (B) at the terminal endpoint were quantified by flow cytometry. (D) Splenocytes from mice in (B) at the terminal endpoint were stimulated as indicated and IFN-γ producing cells were analyzed by ELISPOT. A-D representative of 3 experiments. Error bars indicate SEM. n = 5 per group for A-E; *P < 0.05; **P < 0.01; ***P < 0.001 by one-way ANOVA with Bonferroni's multiple comparisons test to E0771-OVA control group.
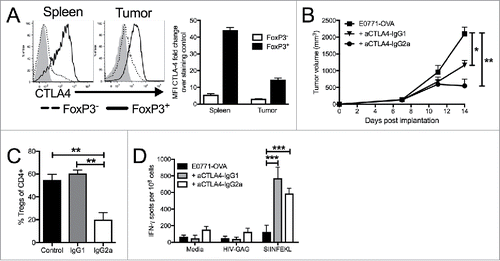
We administered these antibodies to E0771-OVA tumor-bearing mice and found that while both isotypes had an anti-tumor effect, the effect seen with anti-CTLA4-IgG2 a was more profound than with anti-CTLA4-IgG1 (). To confirm whether this anti-tumor effect resulted from a reduction of FoxP3+ Treg cells, we evaluated their splenic and tumor Treg populations. We noted as before a significant decrease in the frequency of Tregs present in anti-CTLA4-IgG2 a treated tumors (). Interestingly, these effects were specific to the tumor microenvironment as Treg ratios in the spleens of anti-CTLA4-IgG2a mice remained unchanged compared to anti-CTLA4-IgG1 and control mice (Sup Fig. 4B). Additionally, we found that anti-CTLA4-IgG2a elicited significantly weaker antibody-dependent cell phagocytosis (ADCP) compared to anti-CTLA4-IgG1, further suggesting that elimination of Tregs was mediated by ADCC (Sup Figure 4C). These findings are consistent with other reports of tumor specific Treg depletion by anti-CTLA4 in other tumor models.Citation40,Citation44,Citation45 However, we noted comparable increases in anti-OVA T-cell responses in both CTLA4 treatment groups (). This suggests that CTLA4 blockade is responsible for enhanced systemic anti-OVA immunity, but that Treg depletion within the tumor microenvironment is critical in allowing these augmented immune responses to directly suppress tumor growth. Therefore, local depletion of CTLA4+ Tregs and blockade of inhibitory CTLA4 signaling may have a tandem impact in enhancing T-cell infiltration into the TNBC tumor microenvironment.
TNBC express PDL1 and suppress PD1+ T-cells and anti-tumor immunity in vivo
While anti-CTLA4 antibodies were critical for T-cell expansion, the combination with anti-PD1 antibodies had a synergistic effect and greatly enhanced anti-tumor immunity. In our E0771 TNBC tumors, PDL1 is highly expressed on tumor cells, while PD1 is highly expressed on various T-cell populations and ), although it is unclear what role tumor expressed PDL1 plays in TNBC. A recent study suggests that tumor-specific PDL1 expression is critical for immunosuppression of certain immunogenic tumors, but may be less critical in other types of cancer.Citation46 To understand how PDL1 expression by TNBC cells impacts tumor-specific T-cell function, we first compared the IFN-γ response of OVA-specific OTI cells to three different TNBC tumor stimulator cells in an ELISPOT assay: E0771 cells, E0771-OVA cells, and E0771-OVA expressing cells that were engineered to overexpress PDL1 (PDL1-OE) (Sup Figure 5A). E0771-OVA cells elicited IFN-γ secretion from a significant number of OTI cells; however, 4-fold fewer T-cells secreted IFN-γ in response to E0771-OVA-PDL1-OE (). We next assayed the cytolytic activity of OTI cells against the same three populations of tumor cells. Although 75% of control OVA+ cells were killed by OTI cells, E0771-OVA-PDL-OE cells were largely protected from this OTI T-cell mediated killing (). Taken together, these in vitro assays establish the negative impact of PDL1 expression by E0771 cells on effector T-cell function.
Figure 5. Manipulation of PDL1/PD1 enhances tumor targeting by immune cells. (A) ELISPOT assay for IFN-γ producing cells with OTI T cells as responders and E0771 parental, E0771-OVA or E0771-OVA PDL1 OE tumor cells as stimulators. (B) E0771 cell lines expressing luciferase were incubated with indicated ratios of OTI T cells and % of live cells as measured by luciferase expression compared to no OTI control is shown. (C) E0771-OVA or E0771-OVA PDL1-OE tumors were grown subcutaneously in C57 BL/6 mice and measured biweekly. (D) E0771-OVA GFP-CRISPR control or E0771-OVA PDL1 KO tumors were grown subcutaneously in C57 BL/6 mice and measured biweekly. (E) E0771-OVA GFP-CRISPR control or E0771-OVA PDL1 KO tumors were grown as before and on day 3 post tumor implantation, 1 × 106 OTI T cells were transferred IV into a cohort of each group. (F) Serum from terminal bleed of mice in (E) was analyzed by ELISA for anti-OVA antibodies. (G) Splenocytes from mice in (E) at the terminal endpoint were stimulated as indicated and IFN-γ production was analyzed by ELISPOT. A-B representative of 3 experiments. C-G representative of 2 experiments. Errors bars indicate SEM. n = 5 per group for A-F; *P < 0.05; **P < 0.01; ***P < 0.001 by 2-tailed Student's t test or one-way ANOVA with Bonferroni's multiple comparisons test to E0771-OVA control group.
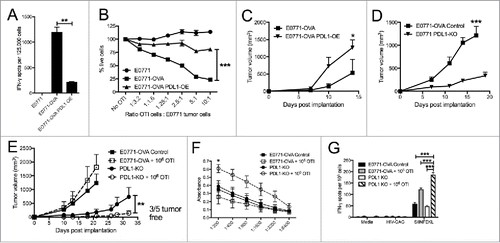
As PDL1 expression suppressed T-cell cytotoxicity, we also wanted to determine if it had a direct impact on TNBC growth, as has been reported in other cancers.Citation47,Citation48 We found that although overexpression of PDL1 did not alter the growth of E0771-OVA cells in vitro, there was a small enhancement of tumor growth when these cells were implanted in immunodeficient SCID-beige mice (Sup Figure 5B and C). However, overexpression of PDL1 significantly enhanced growth kinetics compared to the unmanipulated cell line in immunocompetent mice (), supporting a dominant role for PDL1 in TNBC immune evasion.
We also examined the impact of the loss of PDL1 on tumor growth using a CRISPR/Cas9 system to knock-out PDL1 in TNBC cells. Single clones were selected and evaluated for expression of PDL1 by flow cytometry (Sup Figure 5D). We found clones with both partial and full KO of PDL1 utilizing different CRISPR sgRNA targeting sequences. We observed a profound decrease in tumor growth of full PDL1-KO E0771-OVA cells compared to control lines in immunocompetent animals (). Implantation of the partial PDL1-KO demonstrated an intermediate reduction in growth, corresponding to the intermediate level of PDL1 expression (Sup Figure 5E). Interestingly, PDL1 expression was selected for in vivo, as ex vivo analysis of tumor PDL1 showed selection for a PDL1hi population (Sup Fig. 5F). Growth of PDL1-KO cells in immunodeficient SCID mice was also slightly reduced, which was consistent with the enhanced growth seen by PDL1-OE (Sup Fig. 5G). Many of the CD45+ cells within the tumor also express high levels of PDL1 (data not shown), however the dramatic impact of knocking out PDL1 in the tumor cells specifically demonstrates the vital role for PDL1 on tumor cells rather than CD45+ cells in altering tumor growth.
To determine if this growth difference of PDL1-KO cells was due to an increased functionality of T-cells within the tumor, we again utilized the OTI T-cell transfer system. We implanted control or full PDL1 knockout cells and transferred 1 × 106 OTI T-cells IV 3 days post tumor implantation. As before, the OTI transfer had minimal impact on tumor growth of control tumors but transfer of 1 × 106 OTI T-cells was highly effective at stopping the growth of PDL1-KO tumors, with several mice remaining tumor free after OTI transfer (). Notably, analysis of serum showed high titers of anti-OVA antibodies in this group (), suggesting that the destruction of OVA+ tumors by OTI cells triggers systemic antibody responses to OVA. Notably, OVA is not expressed on the cell surface of our tumor cells and anti-OVA antibodies generated in tumor bearing mice do not bind tumor cells directly (data not shown). Additionally, splenic analysis demonstrated an increased response to the OVA peptide SIINFEKL, with no increase in background responses (). These findings of immune evasion and decreased CD8 T-cell function as the result of tumor specific PDL1 expression are consistent with other recent work using CRISPR/Cas9 to knock out PDL1 in colorectal tumor models.Citation46
We had previously tested how effective treatment with anti-PD1 was in this model and saw a partial anti-tumor response (). It is interesting to note the incomplete effect seen with anti-PD1 treatment compared to PDL1 knockout, which is most likely due to a deficiency in the local dosage and tissue penetration achieved with antibody treatment. Altogether,the moderate impact seen with treatment of tumor bearing mice with PD1 blocking antibodies highlights the role TNBC specific PDL1 expression plays in locally suppressing tumor-specific T-cells within the tumor and the need for additional T-cell activation systemically by another agent like adoptive cell transfer or anti-CTLA4 treatment in this TNBC model.
Heterogeneity of anti-tumor immunity in OVA+ TNBC model
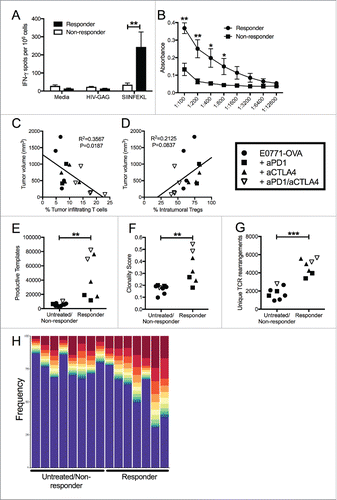
Given the use of genetically identical animals implanted with a TNBC cell line expressing the same non-self gene, the lack of repertoire similarity between mice was surprising, but reflective of the tumor-specific systemic adaptive immune responses observed in individual mice. While the group averages reflected significant differences between treatment strategies, treated mice largely grouped into responders and non-responders. This divergence persisted across all (OVA and non-OVA) experiments despite identical treatment strategies. We reanalyzed immune responses from mice bearing E0771-OVA tumors whose response to treatment resulted in tumor regression (responders) versus those whose tumors continued to grow while being treated (non-responders) (Sup Fig. 6). This response grouping analysis demonstrated that responsive mice had significantly higher systemic tumor-specific adaptive immune responses compared to non-responders ( and ). Despite the correlation of anti-OVA antibodies with responsiveness, we could not detect any surface expression of OVA in these tumors (data not shown). This suggests that their presence is a better indicator of immune responses primed against tumor cells rather than direct anti-tumor effects of the anti-OVA antibodies themselves. In addition, when the percentage of TILs was graphed against tumor size, increased infiltration correlated with smaller tumors () while increased frequency of Tregs within tumors correlated with increased tumor size (D). Analysis of individual TCR repertoires demonstrated that the repertoires of responsive mice had a higher clonality score and an increased number of TCR templates present in their tumors (). The presence of expanded clones is also a hallmark of tumors that responded to treatment (). These data demonstrate the critical impact of checkpoint inhibition in expanding primed adaptive responses to achieve anti-tumor immunity and suggest that these immune correlates may be an effective means to predict patient responses.
Figure 6. Unique patterns seen in mice that respond to treatment, regardless of what treatment. (A) E0771-OVA cells were grown subcutaneously in C57 BL/6 mice. Mice were randomized into groups and treated with anti-PD1 and anti-CTLA4-IgG2 a antibodies biweekly beginning when tumors measured >100 mmCitation3 or left untreated. Splenocytes from these mice were stimulated as indicated and IFN-γ production was analyzed by ELISPOT. (B) Serum from mice treated as in (A) was analyzed by ELISA for anti-OVA antibodies. Data shown in (A) and (B) is pooled from 2 independent experiments. (C) Correlation of tumor volume with the % of TILs. (D) Correlation of tumor volume with the % of Tregs. (E-H) Regrouping of data presented in . (E) Number of total productive TCR templates present in tumors. (F) Clonality score of TCRs present in tumors. (G) Number of unique TCR rearrangements present in tumors. (H) Frequency of the top ten TCR clones found in individual tumor samples. Each color (red through green) at the top of the bars represents the top 10 individual clones. The blue bar represents all remaining clones present in the sample. Error bars indicate SEM. Data are grouped as responders (tumors decreasing in size post treatment) and untreated/non-responders (tumors continuing to grow post treatment or left untreated). n = 6-9 per group for A and B; n = 3-5 per group for C-H; * P < 0.05; ** P < 0.01; *** P < 0.001 by two-tailed Student's t-test.
Discussion
Despite the promising clinical outcomes of ICB targeting PD1/PDL1 in TNBC in clinical trials, the phase I clinical data suggests that the use of these antibodies in isolation will be ineffective for the majority (∼80%) of TNBC patients.Citation16-20 These modest clinical outcomes are in spite of the fact that most TNBCs are characterized by high levels of TILs, neoantigen expression, and elevated immunosuppressive molecules; which are currently thought to be the immunologic prerequisites for responsiveness to ICB therapies.Citation13-15 This discrepancy highlights the need for additional mechanistic study into the use of these ICB antibodies in TNBC, as well as how their combination might allow for enhanced efficacy. To address these gaps, we investigated the combined use of anti-PD1 and anti-CTLA4 antibodies in TNBC using an immunologically relevant model to human TNBC that containing elevated TILs and expression of neoepitopes and immune checkpoint genes. Using this model, we demonstrate that treatment with a combination of antibodies targeting CTLA4 and PD1 stimulates an effective anti-tumor immune response better than either antibody alone, an effect that we confirmed in several other TNBC models. We further used this model to dissect the immunologic contribution and mechanisms underlying each antibody, as well as their combination (). Finally, an analysis of difference between responsive and unresponsive mice revealed that different immune correlates could predict responsiveness in individual mice.
Figure 7. Mechanism of action for dual checkpoint blockade. (A) T cell expansion in the lymphoid tissue is hampered by negative signals from CTLA4. (B) Intratumoral Tregs and PDL1 expression by tumor cells dampen T cell responses within the tumor. (C) Treatment with anti-CTLA4 blocks engagement with CD80/86 and allows for expansion of T cells. (D) Binding of anti-CTLA4 to Tregs allows for blockade of contact mediated suppression of T cell responses and antibody-dependent cell-mediated cytotoxicity (ADCC) of Tregs by NK cells and/or monocytes. Anti-PD1 blocks engagement of PD1 with tumoral PDL1 and leads to enhanced cytokine production by CD4 and CD8 T cells and increased tumor cell killing by CD8 T cells.
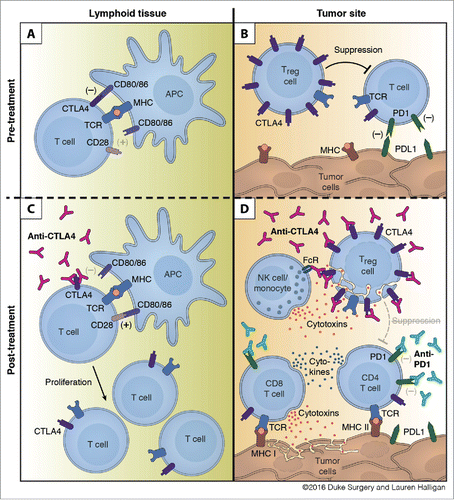
We show that CTLA4 antibodies dramatically altered the systemic T-cell response, causing an increased clonality due to expansion of immunodominant clones but also a broadening of the number of unique clones present in the tumors. In addition, anti-CTLA4 led to a depletion of Tregs specifically in the tumor microenvironment (D). While it is widely accepted that anti-CTLA4 functions at the level of T-cell priming, its impact on CTLA4+ Tregs is only now beginning to be appreciated. Consistent with work from several other groups, we have shown that treatment with anti-CTLA4 results in the depletion of Tregs specifically from the TNBC tumor microenvironment.Citation40,Citation44,Citation45 Treg elimination with the tumor and systemic blockade of CTLA4+ Tregs may also explain the large number of immune-related adverse events (irAEs) seen in clinical studies of CTLA4 antibodies (ipilimumab and tremelimumab), where Grade III toxicities were observed in up to 25% of treated patients.Citation49,Citation50 While we did not observe these overt toxicities in our studies, this may relate to the short duration of our treatment, incomplete blockade of CTLA4, or relative naïve nature of the specific pathogen-free mouse immune system. A recent study did show that treatment of human monocytes with ipilimumab results in ADCC of Tregs ex vivo, but additional clinical studies will be needed to define the role that tumor Treg depletion or systemic blockade of CTLA4+ Treg functionality is playing in these irAEs.Citation51
Treatment with aPD1 had a modest anti-tumor effect but no enhancement of systemic anti-tumor responses or expansion of TCR repertoire were observed at the terminal endpoint. We further investigated if anti-PD1/PDL1 inhibition had a direct impact on TNBC tumor growth, independent of T-cell responses. Although several studies have identified a tumor-specific signaling role for PDL1 in certain cancer lines,Citation47,Citation48 we did not observe any growth differences in PDL1-KO or PDL1 overexpressing TNBC cells in vitro. However, we observed a modest growth reduction from PDL1-KO TNBC cells and a modest growth advantage of PDL1 overexpressing TNBC cells in mice. Unlike some reports in melanoma,Citation52 we were unable to detect PD1 expression in our tumor population in vitro or ex vivo (data not shown). These results suggest that PD1-PDL1 interactions may mediate a growth pathway between tumor cells and other non-T-cells in the tumor microenvironment, possibly through PD1 expressing dendritic cells, tumor-associated macrophages, or another myeloid derived population.Citation53-57
However, we observed the greatest anti-tumor activity following combination of anti-PD1 and anti-CTLA4 in all models of TNBC that we tested, characterized by an expanded TCR repertoire. Consistent with this, analysis of the TCR repertoire following radiation and dual checkpoint inhibition in B16F10 melanoma tumors also revealed an increased maximal clonal frequency.Citation58 As we saw in our mouse model, examination of the TCR repertoire of breast cancer patients demonstrated a significant increase in the number of unique TCR clones following anti-CTLA4 treatment but not after treatment with anti-PD1.Citation59-62 These findings are consistent with a model in which CTLA4 blockade enhances T-cell priming, allowing for an increased number of unique clones while also allowing for the expansion of immune dominant clones (). In contrast, anti-PD1 functions locally within the tumor, enhancing T-cell function and potentially increasing T-cell retention as has been seen in other cancersCitation63 ().
Critically, the TCR repertoire analysis revealed a surprising lack of similarity between mice, even when tumors expressed the non-self neoantigen OVA. Despite transfer of immunodominant OVA-specific OTI T-cells after OVA+ tumor implantation, this clone was expanded in only one of the tumors and was undetectable in over half of the tumors examined (). While OTI is a widely recognized immunodominant OVA-specific T-cell clone in C57/BL6 mice, it is possible that some of the other expanded clones could be OVA specific. However, this seems unlikely given that a single clone was identified in a majority of the mice regardless of whether the tumor cells expressed OVA, suggesting that this dominant clone was not OVA specific. Moreover, the expression of OVA in TNBC cells did not significantly alter response rates to ICB therapies. Based on these findings, we suspect that the unique nature of TCR expansion, rather than the expression of specific neoantigens, may explain the majority of variability in response to treatment (). This is supported by a recent study demonstrating no significant association between the level of neoantigen burden and T-cell ‘inflamed’ microenvironment across most cancers (including breast cancer).Citation64 The striking individual heterogeneity in our studies are reminiscent of clinical trial data from patient responses to checkpoint inhibition, with patients either seeing a durable clinical benefit or having no response at all.Citation65 This shared feature may make our model ideal to further probe for pre-clinical biomarkers for response.
In our analysis of responsive mice, we observed that systemic OVA-specific T-cell and B-cell responses strongly correlated with anti-tumor responses. This occurred in spite of the lack of expansion of OTI T-cells in tumors after adoptive transfer. This may suggest that effective T-cell responses in the TNBC TME precipitate adaptive epitope spreading to other antigens to potentially augment anti-tumor immunity. This seems particularly relevant for anti-OVA antibodies, given their inability to bind the surface of E0771-OVA cells (data not shown). Consistent with our data, a recent longitudinal study found that signatures of adaptive immune activation present in tumor biopsy samples obtained early during the course of treatment are highly predictive of eventual response to immune checkpoint blockade.Citation13 Based on these findings, we predict that monitoring overall immune activation, systemic T-cell and B-cell responses to common cancer antigens, and general characteristics of the TCR repertoire breadth may be a better indicator of individual responsiveness. Supportive of this prediction, the ability of PBMCs to produce IFN-γ following ex vivo restimulation correlated with favorable survival outcomes in response to dual checkpoint therapy.Citation66 Furthermore, a recent clinical study determined that systemic T-cell repertoires of complete responders to PD-1 antibody therapies of virally induced cancers had the largest expansion of T-cells clones specific to self-antigens and not to non-self viral neoantigens.Citation67 As such, the further identification of reliable biomarkers that target treatment to those who are most likely to see clinical benefit is critically important given the significant toxicities associated with these therapies.Citation68,Citation69
Collectively, our study demonstrates that PD1 and CTLA4 checkpoint blockade inhibits immunosuppression of TILs through distinct, complementary mechanisms to expand unique T-cell repertoires and enhance their effectiveness in TNBC. These findings suggest that these combinations may be effective in treating TNBC and that their inclusion may also enhance the efficacy of adoptive T-cell and CAR T-cell therapies.Citation70,Citation71 Moreover, our results highlight that the expansion of effector TILs is unique to each tumor, despite the expression of a common non-self gene with multiple neoantigenic epitopes and the transfer of T-cells specific for the immunodominant neoantigenic epitope. These surprising results suggest that the expression of neoantigens may not be adequate to select immune checkpoint blockade responsive patient populations in all tumor types. However, our studies also revealed that systemic tumor-specific adaptive immunity and TCR expansion in peripheral blood strongly correlates with the anti-tumor response. These findings indicate the potential utility of monitoring systemic immune response and TCR expansion of TILs as potentially the most useful correlates in clinical studies utilizing CTLA4 and PD1 antibodies in TNBC.
Materials and methods
Cell lines
E0771 medullary breast adenocarcinoma cells were a kind gift from Dr. Erik Nelson originally isolated from a spontaneous cancer in C57 BL/6 mice at the Jackson Laboratory in 1939.Citation32 E0771 cells were maintained in DMEM with 10% FBS and cell lines generated through lentiviral transduction using OVA and Firefly Luciferase containing viruses (cloning details available upon request). 4T1 and JC cells were obtained from the American Tissue Culture Collection.
RNA sequencing (RNAseq) and analysis
E0771 cells were purchased from CH3 Biosystems (Amherst, New York), and cultures were maintained with 10% FBS (Atlanta Biologicals), 1% penicillin-streptomycin (ThermoFisher), and 10 mM HEPES (ThermoFisher) in DMEM. Total RNA was extracted from E0771 cultures using the Qiagen RNeasy Mini Kit according to the manufacturer's protocol. RNA-sequencing libraries were prepared with 3 μg total RNA input using the Illumina TruSeq Stranded mRNA Library Prep Kit according to the manufacturer's protocol. Sequencing was performed on the Illumina NextSeq500 with the help of the Cedars-Sinai Medical Center Genomics Core generating approximately 60 M and 23 M reads. Reads were aligned to the mm9 version of the mouse reference genome using the STAR aligner. The GATK pipeline was used to call variants from the aligned data to generate a VCF file, which was subsequently annotated using SnpEff and filtered for Trp53 using SnpSift.
Mice
Female C57 BL/6 (Jackson Labs, Bar Harbor, MA), SCID-beige (Jackson Labs, Bar Harbor, MA), OTI (B6.129S6-Rag2tm1Fwa-Tg(TcraTcrb)1100Mjb; Jackson Labs, Bar Harbor, MA), OTI-GFP/CD45.1 (C57 BL/6-Ly5.2/Cr-RAG2KO-Tg(TcraTcrb) 1100Mjb/J; from Dr. Phillip Scott, University of Pennsylvania), MMTV-tTA (from Dr. Kay Wagner, University of Nebraska Medical Center, Omaha, NebraskaCitation72) and PYMT (from Dr. Bill Muller McGill University, Montreal, Quebec, CanadaCitation38) mice between the ages of 6 and 12 weeks old were used for all experiments. All mice were maintained, bred, and used in accordance with Duke IACUC-approved protocols.
Tumor cell implantation
E0771 and 4T1 cells were injected subcutaneously into the mammary fat pad or flank of mice (1 × 105 cells per animal) and measured biweekly. Tumor measurements were made using calipers and volumes calculated using the formula (v = width*width*(length/2)).
Antibody/DT treatment
Antibodies against PD1 were given IP 200 μg/mouse bi-weekly (Clone RMP1-14; BioxCell, West Lebanon, NH) and CTLA4 were given IP 200 μg/mouse bi-weekly (Clone 9D9 IgG1 and IgG2 a; kind gift from Bristol Myers Squibb). Diphtheria toxin (Sigma, St. Louis MO) was given IP 1 μg/mouse bi-weekly.
Flow cytometry
For flow cytometry, cells were isolated from spleens, lymph nodes or tumors. Unless indicated, all flow cytometry was done on spleens and tumors from mice when tumors reached a terminal endpoint volume (∼2000 mm3). Prior to staining, tumors were digested using a mix of collagenase (1 mg/mL), DNAse (20 U/mL), and hyaluronidase (100 μg/mL) for 90 minutes at 37°C. Digested tumors, spleens, and LNs were mechanically dissociated by smashing through a 40-µm cell strainer (Greiner Bio-One). Red blood cells were lysed with RBC lysing buffer (Sigma). Fixable Aqua dye (Invitrogen) was added to assess cell viability. Cells were incubated with fluorochrome-conjugated antibodies and fixed with 1% formalin (Sigma). For intracellular staining, a FoxP3 Fix/Perm kit was used according to the manufacturer's instructions (eBioscience). Antibodies used include: PD1 (29F.1A12or RMP1-30), PDL1 (10F.9G2), CD45 (30F11), CD8β (YTS156.7.7), CD4 (GK1.5), NK1.1 (PK136), CD44 (IM7), CD11b (M1/70), Ly6C (HK1.4), F480 (BM8), Ly6G (1A8), FoxP3 (FJK-16S) and/or CTLA4 (UC10-4B9) (all Biolegend). Data were collected using an LSR II flow cytometer (BD Bioscience) and analyzed with FlowJo software (Tree Star).
ELISA
Plates were coated with 50 μg/ml OVA protein (BioLegend, San Diego, CA) overnight, washed with PBS + 0.05% Tween 20, and blocked with PBS + 1% BSA (Sigma). A serial dilution of serum was added, followed by an anti-mouse IgG streptavidin-HRP conjugated secondary (1:2000; 7076; Cell Signaling Technology, Danvers, MA). Plates were developed with TMB substrate (Biolegend) and absorbance determined using a Bio-Rad Model 680 microplate reader (Bio-Rad, Hercules, CA, USA). Data points were plated with technical duplicates.
ELISPOT
Mouse IFN-γ ELISPOT assay (Mabtech Inc., Cincinnati, OH) was performed according to according to manufacturer's instructions. Briefly, splenocytes (500,000 cells/well) were incubated in RPMI-1640 medium (Invitrogen) with 10% fetal bovine serum for 24 hours. Cells were stimulated with OVA peptide (SIINFEKL; 1 μg/ml; Sigma) or irrelevant HIV-gag peptide mix (2.6 μg/ml: JPT, Germany). PMA (50 ng/ml) and Ionomycin (1 μg/ml) (Sigma) were used as positive controls.
CRISPR gene targeting
Gene targeting of PDL1 and control GFP by CRISPR/Cas9 was accomplished through the use of pLentiCRISPRv2 (a gift from Feng Zhang, Addgene plasmid # 52961).Citation73 Genes were targeted using the guide sequences (GTACACCACTAACGCAAGC and TGGTTGATTTTGCGGTATG) and (AGTACACCACTAACGCAAGC and GGACTTGTACGTGGTGGAGTA) for PDL1 or (GGGCGAGGAGCTGTTCACCG) for the GFP control and selection of cells using puromycin selection. Successful targeting of PDL1 was determined by flow cytometry screening after single cell clonal selection. Control cells were made using guide sequence targeting GFP rather than PDL1.
Luciferase killing assay
E0771-OVA cells were stably infected with a luciferase expressing lentivirus (GreenFire-1, SBI Palo Alto, CA) and selected for Luciferase expression. These cells were plated 5,000-7,000 cells/well in 96 well plates with various concentrations of OTI cells. Each condition was plated with 12 replicates. Plates were incubated for 24–72 hours before tumor cell death was measured after lysing cells in a Tritonx100 lysis buffer. Luciferase content was measured using a Veritas microplate luminometer (Turner Biosystems). Fraction of live cells at each OTI concentration was calculated as a percent of the signal measured in control wells that received no OTI cells.
TCR sequencing
A >10 mg portion of each tumor was flash frozen. Peripheral blood was collected into 4% sodium citrate buffer before PBMCs were isolated using Histopaque-1083 (Sigma). All TCR-β characterization was performed by Adaptive Biotechnologies using the ImmunoSeq TCR-β ‘survey level’ mouse assay for tumors and ‘deep level’ mouse assay for PBMC.Citation39 OTI TCR tracking was based on the OTI sequence CASSRANYEQYF.
Quantitative rt-PCR
Real-time PCR was performed using an ABI 7300 system using standard methods and intron spanning primers for OVA (forward 5’-CCCCATTGCCATCATGTCAG-3’ and reverse 5’-TGCCACACTGAGCTTCAATACTG-3’). Expression differences were assessed using the comparative cycle threshold (CT) method against GAPDH control gene.
MTT assay
A total of 5000 cells were plated in a final volume of 0.2 ml in 96-well flat bottom plates with indicated concentrations of Trastuzumab-DM1. After 3 days, 20 μl of a 5 mg/ml MTT solution in phosphate-buffered saline were added to each well for 4 h. After removal of the medium, 100 μl of dimethylsulfoxide were added to each well. The absorbance at 540 nm was determined using a Bio-Rad Model 680 microplate reader (Bio-Rad, Hercules, CA, USA). At minimum, triplicate wells were assayed for each condition.
ADCC/ADCP luciferase assay
We used CytoTox-Glo™ Cytotoxicity Assay (ADCC) or ADCP FcγRIIa-H Bioassay (Promega, Madison WI) according to the manufacturer's instructions. Briefly for ADCP, we plated 25 k target cells (MDA-MB-231-CTLA4+) the day before and incubated the cells with the indicated concentration of antibody for 30 minutes. After that time, effector cells were added at a 2:1 ratio and the assay was read at 6 hours post-incubation.
Statistical analysis
Data are presented as mean ± SEM. Tumor volumes, flow cytometry, ELISA, and ELISPOT data from experiments with 3 or more treatment groups were analyzed by 1-way ANOVA with Bonferroni's multiple comparisons test. A 2-tailed, unpaired Student's t test was used for experiments with only 2 groups. Tumor volumes were analyzed at the terminal endpoint only, unless otherwise indicated. Statistical analysis was performed using Prism (GraphPad). P values of 0.05 or less were considered statistically significant. Not all significant differences are shown in every graph. *P < 0.05; **P < 0.01; ***P < 0.001
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
2017ONCOIMM0888R-s01.docx
Download MS Word (1.6 MB)Acknowledgments
We thank members of the Duke Surgery Center for Applied Therapeutics and all of our colleagues for stimulating discussions. We also acknowledge the helpful input about the analysis of the TCR receptor sequencing data from Dr. Kevin Eng, Roswell Park Cancer Institute. We want to acknowledge the talent and skill of our medical illustrator, Lauren Halligan, MSMI, who created the final mechanistic figure.
Additional information
Funding
References
- Hoos A. Development of immuno-oncology drugs – from CTLA4 to PD1 to the next generations. Nat Rev Drug Discovery. 2016;15:235–247. doi:10.1038/nrd.2015.35.
- Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T-cell activation. Immunity. 1994;1:405–413. doi:10.1016/1074-7613(94)90071-X. PMID:7882171.
- Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–9553. doi:10.1128/MCB.25.21.9543-9553.2005. PMID:16227604.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi:10.1038/nrc3239. PMID:22437870.
- Egen JG, Kuhns MS, Allison JP. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat Immunol. 2002;3:611–618 doi:10.1038/ni0702-611. PMID:12087419.
- Walker LSK. Treg and CTLA-4: Two intertwining pathways to immune tolerance. J Autoimmunity. 2013;45:49–57. doi:10.1016/j.jaut.2013.06.006.
- Tai XG, et al. Basis of CTLA-4 function in regulatory and conventional CD4(+) T cells. Blood. 2012;119:5155–5163. doi:10.1182/blood-2011-11-388918. PMID:22403258.
- Pusztai L, Karn T, Safonov A, Abu-Khalaf MM, Bianchini G. New strategies in breast cancer: Immunotherapy. Clin Cancer Res. 2016;22:2105–2110. doi:10.1158/1078-0432.CCR-15-1315.
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Eng J Med. 2010;363:711–723. doi:10.1056/NEJMoa1003466.
- Poole RM. Pembrolizumab: First global approval. Drugs. 2014;74:1973–1981. doi:10.1007/s40265-014-0314-5. PMID:25331768.
- Gunturi A, McDermott DF. Nivolumab for the treatment of cancer. Exp Opin Invest Drugs. 2015;24:253–260. doi:10.1517/13543784.2015.991819.
- Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33:1974–1982. doi:10.1200/JCO.2014.59.4358.
- Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discovery. 2016;6:827–837. doi:10.1158/2159-8290.CD-15-1545. PMID:27301722.
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi:10.1126/science.aaa4971. PMID:25838375.
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi:10.1126/science.aaa1348. PMID:25765070.
- Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Dolled-Filhart M, Emancipator K, Gonzalez EJ, et al. Abstract S1-09: A phase Ib study of pembrolizumab (MK-3475) in patients with advanced triple-negative breast cancer. Cancer Res. 2015;75:S1–09. doi:10.1158/1538-7445.SABCS14-S1-09.
- Emens LA, Braiteh FS, Cassier P, Delord J, Eder JP, Fasso M, Xiao Y, Wang Y, Molinero L, Chen DS, et al. Inhibition of PD-L1 by MPDL3280 A leads to clinical activity in patients with metastatic triple-negative breast cancer (TNBC). Cancer Res. 2015;75:2859. doi:10.1158/1538-7445.AM2015-2859.
- Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Pathiraja K, Aktan G, Cheng JD, et al. Pembrolizumab in patients with advanced triple-negative breast cancer: Phase Ib KEYNOTE-012 study. J Clin Oncol. 2016;34(21):2460–7 doi:10.1200/JCO.2015.64.8931. PMID:27138582.
- Adams S, Schmid P, Rugo HS, Winer EP, Loirat D, Awada A, Cescon DW, Iwata H, Campone M, Nanda R, et al. Phase 2 study of pembrolizumab (pembro) monotherapy for previously treated metastatic triple-negative breast cancer (mTNBC): KEYNOTE-086 cohort A. J Clin Oncol. 2017;35:1008–1008
- Adams S, Loi S, Toppmeyer D, Cescon DW, De Laurentiis M, Nanda R, Winer EP, Mukai H, Tamura K, Armstrong A, et al. Phase 2 study of pembrolizumab as first-line therapy for PD-L1–positive metastatic triple-negative breast cancer (mTNBC): Preliminary data from KEYNOTE-086 cohort B. J Clin Oncol. 2017;35:1088–1088.
- West NR, Milne K, Truong PT, Macpherson N, Nelson BH, Watson PH. Tumor-infiltrating lymphocytes predict response to anthracycline-based chemotherapy in estrogen receptor-negative breast cancer. Breast Cancer Res. 2011;13:R126. doi:10.1186/bcr3072. PMID:22151962
- Hornychova H, Melichar B, Tomsova M, Mergancova J, Urminska H, Ryska A. Tumor-infiltrating lymphocytes predict response to neoadjuvant chemotherapy in patients with breast Carcinoma. Cancer Invest. 2008;26:1024–1031. doi:10.1080/07357900802098165. PMID:19093260.
- Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, Shulman LN, Martino S, Wang M, Jones VE, Saphner TJ, et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol. 2014;32:2959–66. doi:10.1200/jco.2013.55.0491.
- Adams S, Goldstein LJ, Sparano JA, Demaria S, Badve SS. Tumor infiltrating lymphocytes (TILs) improve prognosis in patients with triple negative breast cancer (TNBC). Oncoimmunology. 2015;4:e985930. doi:10.4161/2162402X.2014.985930. PMID:26405612.
- Miyashita M, Sasano H, Tamaki K, Hirakawa H, Takahashi Y, Nakagawa S, Watanabe G, Tada H, Suzuki A, Ohuchi N, et al. Prognostic significance of tumor-infiltrating CD8(+) and FOXP3(+) lymphocytes in residual tumors and alterations in these parameters after neoadjuvant chemotherapy in triple-negative breast cancer: a retrospective multicenter study. Breast Cancer Res. 2015;17:13. doi:10.1186/s13058-015-0632-x. PMID:25633049.
- Denkert, C, von Minckwitz G, Brase JC, Sinn BV, Gade S, Kronenwett R, Pfitzner BM, Salat C, Loi S, Schmitt WD, et al. Tumor-infiltrating Lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol. 2015;33:983–91. doi:10.1200/JCO.2014.58.1967. PMID:25534375.
- Stagg J, Allard B. Immunotherapeutic approaches in triple-negative breast cancer: latest research and clinical prospects. Therapeutic Adv Medical Oncol. 2013;5:169–181. doi:10.1177/1758834012475152.
- Hendrickx W, Simeone I, Anjum S, Mokrab Y, Bertucci F, Finetti P, Curigliano G, Seliger B, Cerulo L, Tomei S, et al. Identification of genetic determinants of breast cancer immune phenotypes by integrative genome-scale analysis. Oncoimmunology. 2017;6:e1253654. doi:10.1080/2162402X.2016.1253654. PMID:28344865.
- Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, Diaz LA, Jr, Papadopoulos N, Kinzler KW, Vogelstein B, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. 2014;111:11774–11779. doi:10.1073/pnas.1410626111. PMID:25071169.
- Gao L, et al. Enhanced anti-tumor efficacy through a combination of integrin alpha v beta 6-targeted photodynamic therapy and immune checkpoint inhibition. Theranostics. 2016;6:627–637. doi:10.7150/thno.14792. PMID:27022411.
- Ott PA, Hodi FS, Kaufman HL, Wigginton JM, Wolchok JD. Combination immunotherapy: a road map. J Immunotherapy Cancer. 2017;5:16. doi:10.1186/s40425-017-0218-5.
- Dunham LJ, Stewart HL. A survey of transplantable and transmissible animal tumors. J Natl Cancer Inst. 1953;13:1299–1377. PMID:13035452.
- Johnstone CN, Smith YE, Cao Y, Burrows AD, Cross RSN, Ling X, Redvers RP, Doherty JP, Eckhardt BL, Natoli AL, et al. Functional and molecular characterisation of EO771.LMB tumours, a new C57 BL/6-mouse-derived model of spontaneously metastatic mammary cancer. Dis Models Mechan. 2015;8:237–251. doi:10.1242/dmm.017830.
- Lomax ME, Barnes DM, Hupp TR, Picksley SM, Camplejohn RS. Characterization of p53 oligomerization domain mutations isolated from Li-Fraumeni and Li-Fraumeni like family members. Oncogene. 1998;17:643–649. doi:10.1038/sj.onc.1201974. PMID:9704930.
- Lomax ME, Barnes DM, Gilchrist R, Picksley SM, Varley JM, Camplejohn RS. Two functional assays employed to detect an unusual mutation in the oligomerisation domain of p53 in a Li-Fraumeni like family. Oncogene. 1997;14:1869–1874. doi:10.1038/sj.onc.1201133. PMID:9150393.
- Smardova J, Nemajerova A, Trbusek M, Vagunda V, Kovarik J. Rare somatic p53 mutation identified in breast cancer: a case report. Tumour Biol. 2001;22:59–66 doi:10.1159/000050597. PMID:11125276.
- Hartman ZC, Poage GM, den Hollander P, Tsimelzon A, Hill J, Panupinthu N, Zhang Y, Mazumdar A, Hilsenbeck SG, Mills GB, et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013;73:3470–3480. doi:10.1158/0008-5472.CAN-12-4524-T. PMID:23633491.
- Rao T, Ranger JJ, Smith HW, Lam SH, Chodosh L, Muller WJ. Inducible and coupled expression of the polyomavirus middle T antigen and Cre recombinase in transgenic mice: an in vivo model for synthetic viability in mammary tumour progression. Breast Cancer Res. 2014;16:R11. doi:10.1186/bcr3603. PMID:24457046
- Carlson CS, Emerson RO, Sherwood AM, Desmarais C, Chung MW, Parsons JM, Steen MS, LaMadrid-Herrmannsfeldt MA, Williamson DW, Livingston RJ, et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun. 2013;4:2680. doi:10.1038/ncomms3680. PMID:24157944.
- Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ. Anti-CTLA-4 antibodies of IgG2 a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1:32–42. doi:10.1158/2326-6066.CIR-13-0013. PMID:24777248.
- Fossati-Jimack L, Ioan-Facsinay A, Reininger L, Chicheportiche Y, Watanabe N, Saito T, Hofhuis FM, Gessner JE, Schiller C, Schmidt RE, et al. Markedly different pathogenicity of four immunoglobulin G isotype-switch variants of an antierythrocyte autoantibody is based on their capacity to interact in vivo with the low-affinity Fc gamma receptor III. J Exp Med. 2000;191:1293–1302. doi:10.1084/jem.191.8.1293.
- Hamaguchi Y, Xiu Y, Komura K, Nimmerjahn F, Tedder TF. Antibody isotype-specific engagement of Fc gamma receptors regulates B lymphocyte depletion during CD20 immunotherapy. J Exp Med. 2006;203:743–753. doi:10.1084/jem.20052283. PMID:16520392.
- Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. Fc gamma RIV: A novel FcR with distinct IgG subclass specificity. Immunity. 2005;23:41–51. doi:10.1016/j.immuni.2005.05.010.
- Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, Wilson NS, Dranoff G, Brogdon JL. Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med. 2013;210:1685–1693. doi:10.1084/jem.20130573. PMID:23897982.
- Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma. J Exp Med. 2013;210:1695–1710. doi:10.1084/jem.20130579. PMID:23897981.
- Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, Freeman GJ, Sharpe AH. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. 2017;214:895–904. doi:10.1084/jem.20160801. PMID:28302645.
- Clark CA, Gupta HB, Sareddy G, Pandeswara S, Lao S, Yuan B, Drerup JM, Padron A, Conejo-Garcia J, Murthy K, et al. Tumor-Intrinsic PD-L1 signals regulate cell growth, pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer Res. 2016;76:6964–6974. doi:10.1158/0008-5472.CAN-16-0258. PMID:27671674.
- Gupta HB, Clark CA, Yuan B, Sareddy G, Pandeswara S, Padron AS, Hurez V, Conejo-Garcia J, Vadlamudi R, Li R, et al. Tumor cell-intrinsic PD-L1 promotes tumor-initiating cell generation and functions in melanoma and ovarian cancer. Signal Transduction Targeted Therapy. 2016;1:16030. doi:10.1038/sigtrans.2016.30. PMID:28798885
- Kaehler KC, Hauschild A. Treatment and side effect management of CTLA-4 antibody therapy in metastatic melanoma. J Der Deutschen Dermatologischen Gesellschaft. 2011;9:277–285. doi:10.1111/j.1610-0387.2010.07568.x.
- Bertrand A, Kostine M, Barnetche T, Truchetet ME, Schaeverbeke T. Immune related adverse events associated with anti-CTLA-4 antibodies: systematic review and meta-analysis. Bmc Med. 2015;13:211. doi:10.1186/s12916-015-0455-8. PMID:26337719
- Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, Michielin O, Weide B, Romero P, Speiser DE. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A. 2015;112:6140–6145. doi:10.1073/pnas.1417320112. PMID:25918390.
- Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, Elco CP, Lee N, Juneja VR, Zhan Q, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. 2015;162:1242–1256. doi:10.1016/j.cell.2015.08.052. PMID:26359984.
- Cho HY, Choi EK, Lee SW, Jung KO, Seo SK, Choi IW, Park SG, Choi I, Lee SW Programmed death-1 receptor negatively regulates LPS-mediated IL-12 production and differentiation of murine macrophage RAW264.7 cells. Immunol Letters. 2009;127:39–47. doi:10.1016/j.imlet.2009.08.011.
- Said EA, Dupuy FP, Trautmann L, Zhang Y, Shi Y, El-Far M, Hill BJ, Noto A, Ancuta P, Peretz Y, et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4(+) T cell activation during HIV infection. Nat Med. 2010;16:452–U136. doi:10.1038/nm.2106. PMID:20208540.
- Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495–499. doi:10.1038/nature22396. PMID:28514441.
- Lim TS, Chew V, Sieow JL, Goh S, Yeong JP, Soon AL, Ricciardi-Castagnoli P. PD-1 expression on dendritic cells suppresses CD8(+) T cell function and antitumor immunity. Oncoimmunology. 2016;5:e1085146. doi:10.1080/2162402X.2015.1085146. PMID:27141339.
- Yao S, Wang S, Zhu Y, Luo L, Zhu G, Flies S, Xu H, Ruff W, Broadwater M, Choi IH, et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood. 2009;113:5811–5818. doi:10.1182/blood-2009-02-203141. PMID:19339692.
- Twyman-Saint\sVictor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–377. doi:10.1038/nature14292. PMID:25754329.
- Robert L, Harview C, Emerson R, Wang X, Mok S, Homet B, Comin-Anduix B, Koya RC, Robins H, Tumeh PC, et al. Distinct immunological mechanisms of CTLA-4 and PD-1 blockade revealed by analyzing TCR usage in blood lymphocytes. Oncoimmunology. 2014;3:2. doi:10.4161/onci.29244.
- Robert L, Tsoi J, Wang X, Emerson R, Homet B, Chodon T, Mok S, Huang RR, Cochran AJ, Comin-Anduix B, et al. CTLA4 blockade Broadens the peripheral T-cell receptor repertoire. Clinical Cancer Res. 2014;20:2424–2432. doi:10.1158/1078-0432.CCR-13-2648.
- Page DB, Diab A, Yuan J, Dong Z, Soloman SB, Patil S, Hudis CA, Wolchok JD, Norton L, McArthur HL Pre-operative immunotherapy with tumor cryoablation (cryo) plus ipilimumab (ipi) induces potentially favorable systemic and intratumoral immune effects in early stage breast cancer (ESBC) patients. 2015;3:O6. doi:10.1186/2051-1426-3-s1-06 PMCID: PMC4547170.
- Page DB, Yuan J, Redmond D, Wen YH, Durack JC, Emerson R, Solomon S, Dong Z, Wong P, Comstock C, et al. Deep Sequencing of T-Cell Receptor DNA as a biomarker of clonally expanded TILs in breast cancer after immunotherapy. Cancer Immunol Res. 2016;4(10):835–844. doi:10.1158/2326-6066.CIR-16-0013. PMID:27587469.
- Noguchi T, Ward JP, Gubin MM, Arthur CD, Lee SH, Hundal J, Selby MJ, Graziano RF, Mardis ER, Korman AJ, et al. Temporally distinct PD-L1 expression by tumor and host cells contributes to immune escape. Cancer Immunol Res. 2017;5:106 doi:10.1158/2326-6066.CIR-16-0391. PMID:28073774.
- Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, Gajewski AP, Andrade J, Gajewski TF. Density of immunogenic antigens does not explain the presence or absence of the T-cell–inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci. 2016;113:E7759–E7768. doi:10.1073/pnas.1609376113. PMID:27837020.
- Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD. Pooled analysis of long-term survival data from phase II and phase III trials of Ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33:1889–U1848. doi:10.1200/JCO.2014.56.2736. PMID:25667295.
- McNamara MJ, Hilgart-Martiszus I, Barragan Echenique DM, Linch SN, Kasiewicz MJ, Redmond WL. Interferon-gamma production by peripheral lymphocytes predicts survival of tumor-bearing mice receiving dual PD-1/CTLA-4 blockade. Cancer Immunol Res. 2016;4:650–657. doi:10.1158/2326-6066.CIR-16-0022. PMID:27262113.
- Stevanović S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, Robins HS, Robbins PF, Klebanoff CA, Rosenberg SA, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science. 2017;356:200. doi:10.1126/science.aak9510. PMID:28408606.
- Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, et al. Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma. N Eng J Med. 2015;372:2006–2017. doi:10.1056/NEJMoa1414428.
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N Eng J Med. 2013;369:122–133. doi:10.1056/NEJMoa1302369.
- Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123;2625–2635. doi:10.1182/blood-2013-11-492231. PMID:24578504.
- Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–281. doi:10.1038/nri3191. PMID:22437939.
- Sakamoto K, Schmidt JW, Wagner KU. Generation of a novel MMTV-tTA transgenic mouse strain for the targeted expression of genes in the embryonic and postnatal mammary gland. Plos One. 2012;7:e43778. doi:10.1371/journal.pone.0043778.
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nature Methods 2014;11:783–784. doi:10.1038/nmeth.3047. PMID:25075903.