
ABSTRACT
To identify novel hubs for cancer immunotherapy, we generated C57BL/6J mice with concomitant deletion of the drugable transcription factor PPARγ and transgenic overexpression of the mutant KRASG12V oncogene in enterocytes. Animals developed epithelial hyperplasia, transmural inflammation and serrated adenomas in the small intestine with infiltration of CD3+ FOXP3+ T-cells and macrophages into the lamina propria of the non-malignant mucosa. Within serrated polyps, CD3+ CD8+ T-cells and phosphorylated ERK1/2 were reduced and the senescence marker P21 and macrophage counts up-regulated, indicative of an immunosuppressive tissue microenvironment. Treatment of mutant KRASG12V mice with the PPARγ-agonist rosiglitazone augmented M1 macrophage numbers, reduced IL4 expression and diminished polyp load in mice. Rosiglitazone also promoted M1 polarisation of human THP1-derived macrophages and decreased Il4 mRNA in isolated murine lymphocytes. Thus, inhibition of the oncogenic driver mutant RAS by PPARγ in epithelial and immune cell compartments may be a future target for the prevention or treatment of human malignancies associated with intestinal inflammation.
KEYWORDS:
Introduction
Inflammatory bowel diseases (IBD), such as Morbus Crohn and Ulcerative Colitis, have been associated in human genome-wide association studies with multiple gene aberrations including those in major oncogenic drivers like KRAS.Citation1 KRAS mutations confer an increased risk for development of precancerous lesions which may progress to cancer after many years. Activating point mutations in KRAS (G12/13D, G12 V e.a.) and NRAS genes are frequently found in patients with sporadic and colitis-associated colorectal cancer (CRC), and introduction of mutated KRAS alleles in mice induces tumors in different organs of the gastrointestinal (GI) tract.Citation2
Overexpression of human mutant KRAS in enterocytes initiates the formation of two morphological subtypes of adenomatous polyps in mice defined by their histological appearance as tubularCitation3 and serratedCitation4 adenomas. The recent molecular reclassification of CRC subtypes in humans assigned the serrated morphology to RAS-pathway mutations.Citation5 A plethora of chemokines, cytokines and nuclear factor kappa B-dependent genes are involved in the initial recruitment of inflammatory cells into the lamina propria by clusters (“field effect”) of RAS-mutated epithelial cells. This fact led to the successful clinical administration of neutralizing antibodies against tumor necrosis factor-alpha (TNFα) (like infliximab).Citation1 Nonetheless, the molecular players which underlie and drive mutant RAS-driven progression of inflammation in the lamina propria to malignant transformation of epithelial cells have only been uncovered in part, and drugable targets which prevent or reverse this transition are urgently needed.
Peroxisome proliferator activated receptor-gamma (PPARγ) is a nuclear transcription factor promoting differentiation of epithelial cells and immune defence in the intestinal tract.Citation6 PPARγ is expressed throughout the GI tract with high levels in the colorectum, though defective in IBD.Citation7 PPARγ inhibits inflammation and, thereby, may prevent cancers associated with chronic inflammation. It is activated by dietary and inflammatory lipids such as nitrated linoleic acid and products derived from nitric oxide (NO) synthases. PPARγ enhances expression of mucosal defensinsCitation8 and regulates innate and adaptive immune responses in the intestine, lung and white fat, e.g. by directing macrophage (Mph/MPH) polarization (M1/M2),Citation9 dampening Th17-driven inflammationCitation10 and up-regulating suppressor cells (e.g. regulatory T-cellsCitation11).
Numerous preclinical studies evinced beneficial metabolic and anti-inflammatory effects of PPARγ, whereas the results on cell proliferation and tumor growth were less consistent.Citation6 Severe side effects led to the discontinuation of PPARγ-agonists of the glitazone class (pio/rosiglitazone) for chronic use in patients with type 2 diabetes mellitus (T2DM), while still holding the promise for applications against lethal diseases including cancer (e.g. melanoma,Citation12 prostateCitation13). Recent pilot clinical studies confirmed therapeutic efficacy of glitazones on liver disease,Citation14 cerebral strokeCitation15 and chronic myeloid leukemia,Citation16 indicative of a potential clinical benefit of combination therapies. The RAS-RAF-MEK1/2-ERK1/2 cascade leads to phosphorylation and posttranslational inactivation of PPARγ, thereby potentially counteracting its protective effect.Citation17 We therefore raised the hypothesis that constitutively active RAS-ERK1/2 signaling contributes to repression of PPARγ activity in vivo and facilitates progression of inflammation to malignancy.
Results
Generation of PPARγ-deficient mutant KRASG12V transgenic mice
To explore the functional interaction between PPARγ and RAS in the mouse intestinal epithelium in vivo, double transgenic hemizygous C57BL/6J mice for Cre-recombinase and the constitutively active human KRASG12V mutation, both under control of the murine villin promoter, were interbred with homozygous floxed Pparg mice to selectively ablate PPARγ in intestinal epithelial cells (RAS PPARΔIEC) (). Nomenclature and abbreviations are presented in Table S1. Homozygous progeny with excision of both Pparg alleles in presence of mutant KRAS was not obtained according to expected Mendelian ratios and excluded from the study (Table S2). Cre-recombinase driven by the murine lysozyme promoter yielded viable offspring with deletion of both Pparg alleles in myeloid cells (RAS PPARΔMPH). Kaplan-Meier curves () evinced that PPARγ-deficiency in myeloid cells (including macrophages) shortens the life span of mutant KRAS transgenic mice compared with WT mice (*p < 0.0001 RAS PPARΔMPH (n = 53) vs. WT (n = 149), log-rank test). Animals succumbed to massive splenomegaly and myeloproliferative disease and were not further studied. All other genotypes did not reveal any significant differences in survival.
Figure 1. Characteristics and validation of mouse genotypes. A, Breeding scheme. Nomenclature and abbreviations are presented in Table S1. B, Kaplan-Meier survival curves. PPARγ-deficiency in myeloid cells such as macrophages (MPh/MPH) but not in the intestinal epithelium (IEC) shortens the life span compared with WT mice (*p < 0.0001 RAS PPARΔMPH (n = 53) vs. WT (n = 149), log-rank test). Legend: “1” = event (death); “0” = alive or censored. C, Western blots of pull-down assays detecting active GTP-bound pan-RAS proteins in mouse whole tissue lysates compared to total pan-RAS (input) using RALGDS-GST as a bait. Representative gels are presented. KRASG12V-mutant human CRC cell line SW480 was a positive control. D, Pparg exon1/2 excision. Genomic PCRs were conducted on DNAs isolated from organs of the respective genotypes. Representative agarose gels are presented. Expected amplicon sizes are indicated.Citation49 E, PPARγ protein expression. FFPE tissue sections from WT and PPARΔIEC mice colon (Co) were stained with PPARγ Ab for immunohistochemistry (IHC). Magnification 200x. F, PPARγ target gene expression. Total RNA was extracted from snap-frozen small intestine (SI). CT-values from RT-qPCRs on acyl-CoA oxidase (Aco) were normalized to β2-microglobulin (B2 m) and calculated as -fold ± S.E. (n = 3 per genotype, *p < 0.05, Mann Whitney test). G, PPARγ and RAS target protein expression. Whole tissue lysates (from Co and SI) were subjected to Western blotting. Representative gels and quantitative analysis are shown. O.D. values of bands in gels were normalized to HSP90 and calculated as -fold ± S.E. compared with WT mice (n = 2 per genotype, *p < 0.05, Wilcoxon signed rank test).
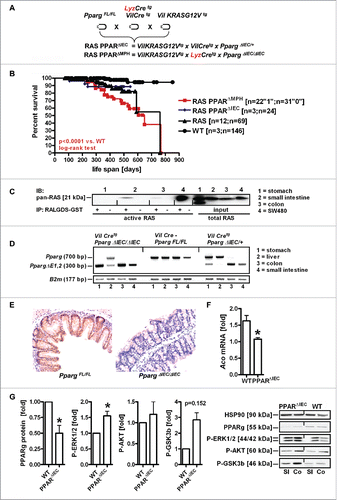
Western blot analysis of GTPase pull-down assays () detecting active GTP-bound pan-RAS proteins in whole tissue lysates using RALGDS-GST as a bait confirmed the presence of active RAS in the small intestine (SI). Genomic PCRs on DNA isolated from organs of the respective genotypes corroborated () Cre-mediated Pparg exon1/2 excision. FFPE tissue sections of the small and large intestine (colon) from WT and PPARΔIEC mice were then stained by IHC with PPARγ Ab. The strong nuclear staining positivity in enterocytes was reduced in mice with excision of Pparg exon 1/2 compared with WT animals (). RT-qPCRs of total RNA extracted from snap-frozen SI confirmed reduced mRNA expression of the PPARγ target gene acyl-CoA oxidase (Aco) ().
Western blot analysis of whole tissue lysates also demonstrated reduced PPARγ protein (). Conversely, phosphorylation of RAS target proteins, such as ERK1/2, AKT and GSK3β, was increased. Thus, this mouse strain allowed us to study the impact of active RAS on the intestinal epithelium on a background of enterocyte-specific deletion of PPARγ.
PPARγ-deficiency exacerbates pre-malignant phenotypes driven by mutant KRAS
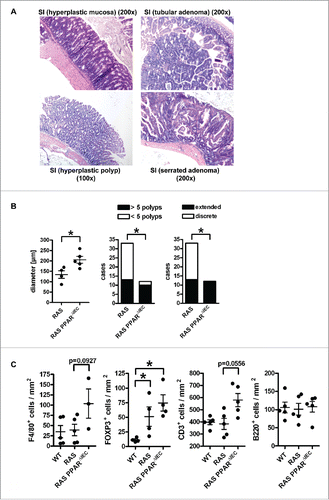
Consistent with previous reports,Citation3 carcinogenesis occurred in RAS mice with high penetrance and late onset over up to 2 years. Microscopic images of H&E staining in FFPE tissue sections () visualized the complete SI (“Swiss roll”) with hyperplastic epithelium and goblet cells. Quantitative analysis of H&E histopathology (n≥4 per genotype) was then performed (). As a surrogate indicator of transmural inflammation,Citation18,Citation19 the distance (i.e. diameter) between the crypt base until the tunica adventitia (serosa) were measured in μm (*p < 0.05, Mann Whitney test). In addition, the number of cases (mice) with more or less than 5 polyps and having discrete or extended polyps per total SI were counted (*p < 0.05, Fisher Exact test). RAS PPARΔIEC mice developed a more pronounced transmural inflammation (“bowel wall thickening”) and extended hyperplasia in the SI (mainly in the terminal ileum) and the proximal colon as compared with PPARγ-proficient littermates. Upon a latency of >12 months, hyperplastic polyps (HPP) and serrated adenomas (SA) with goblet cell hyperplasia were frequently observed together with the formation of aberrant crypt foci (ACF) and infrequent sessile serrated adenomas (SSA) with dysplasia and very few high-grade dysplastic tubular adenomas (Ad) (Table S2). Importantly, serrated-type lesions predominated in the lower SI but coexisted with single cases of tubular-type lesions in the duodenum. Mice rarely progressed to macroscopic tumors with flat white morphology at an age of 2 years (not shown). This observation was consistent with the multi-step patterns and molecular subtypes of human GI cancers,Citation5 where single mutations are insufficient to drive progression from adenoma to carcinoma.
Figure 2. PPARγ-deficiency exacerbates pre-malignant phenotypes driven by mutant KRAS in the mouse small intestine. A, Exemplary microscopic images of H&E staining in FFPE tissue sections from RAS and PPARΔIEC mice. Multiplicity and incidence of histopathological parameters in mouse genotypes are presented in Table S2. Top left: Hyperplastic epithelium with goblet cells in the lower small intestine (SI); Top right: Tubular adenoma in the duodenum; Bottom left: hyperplastic polyp (HPP); Bottom right: serrated adenoma (SA) with dysplasia. Magnification 100x and 200x. B, Quantitative analysis of H&E histopathology in A. Left: As a surrogate indicator for transmural inflammation, the distance/diameter (length) between the crypt base and the tunica adventitia (serosa) were measured (in μm). Data are means ± S.E. (n≥4 mice, *p < 0.05, Mann Whitney test). Middle: Number of cases (mice) having more or less than 5 polyps per total SI (*p < 0.05, Fisher Exact test). Right: Mice having discrete or extended polyps per total SI (*p < 0.05, Fisher Exact test). C, Mutant KRAS and PPARγ-deficiency increase immune cell numbers in the lamina propria of the non-malignant mucosa compared with WT mice. FFPE tissue sections of WT, RAS and RAS PPARΔIEC mice were stained using IHC with Abs detecting B-cells (B220/CD45R), T-cells (CD3), Tregs (FOXP3) and macrophages (F4/80). Staining intensity and frequency were scored. Data are means ± S.E. (n≥3 per genotype, *p < 0.05, Kruskal Wallis test).
PPARγ-deficiency augments mutant KRAS-mediated immune cell infiltration
To exclude the possibility that gut microbes penetrate the intestinal epithelium in mutant KRAS mice to expose the lamina propria to antigens, FFPE tissues from WT, RAS and RAS PPARΔIEC mice were stained for immunofluorescence microscopy. E-cadherin staining remained at the intercellular membrane (S1), and no nuclear translocation of β-catenin was observed (not shown). RT-qPCR analysis from snap-frozen tissues of WT and RAS mice (n≥3 per genotype, n.s., Mann Whitney test) revealed no significant changes in mRNA expression of the tight junction protein zonula occludens-1 (Zo1). Thus, mutant RAS mice seem to maintain epithelial barrier integrity.
To test whether the epithelial RAS mutation is sufficient to drive immune cell recruitment into the lamina propria, IHC on FFPE tissue sections from WT, RAS and RAS PPARΔIEC mice was done with Abs detecting major leukocyte/lymphocyte populations (n≥3 per genotype). Elevated immune cell counts in the lamina propria with single intraepithelial lymphocytes (IEL) and macrophages were recorded (S2). Quantitative analysis of staining intensity and frequency () scores revealed an increase (WT < RAS < RAS PPARΔIEC) in Tregs (FOXP3+) (*p < 0.05 vs. RAS PPARΔIEC, Kruskal Wallis test). Macrophages (F4/80+) and pan-T-cells (CD3+) displayed a similar trend, whereas pan-B-cell (CD45R/B220+) counts were unchanged. Thus, epithelial PPARγ-deficiency augmented immune cell infiltration driven by epithelial mutant KRAS.
Mutant KRAS promotes senescence and immune escape in serrated polyps
Prolonged RAS-activation can result in oncogene-induced senescence (OIS).Citation4 Since most polyps did not progress to carcinoma, we performed IHC on tissue sections of RAS and RAS PPARΔIEC mice (n≥3 per genotype) with Abs detecting P-ERK1/2 as a marker of active RAS-signaling and P21 (CIP1/WAF1) as a marker for OIS (). IHC staining underwent quantitative analysis for absolute numbers of cases having high (scores +2,+3) or low (scores 0,+1) positivity in polyps vs. the non-malignant mucosa (*p < 0.05, Fisher Exact test). Polyps were summarized from the following morphologies; hyperplastic polyp (HPP), serrated adenoma (SA) and sessile serrated adenoma (SSA). Aberrant crypt foci (ACF) and tubular adenomas (Ad) were infrequent and excluded from the analysis. Notably, the staining positivity () of P-ERK1/2 in polyps was reduced, while P21 was increased compared with the non-malignant mucosa, indicative of an arrest of RAS-ERK1/2-driven proliferation in favour of RAS-induced senescence. No difference was stated between polyps of the two genotypes (not shown). These data suggested that PPARγ is dispensable once adenoma formation has been initiated by mutant RAS, in contrast to its immunoregulatory role in the hyperplastic pre-malignant mucosa where it counteracts mutant RAS effects.
Figure 3. Mutant KRAS promotes senescence and immune escape in serrated polyps independently of PPARγ. A, IHC on tissue sections of RAS and RAS PPARΔIEC mice (n≥3 per genotype) with Abs detecting P-ERK1/2 and P21 as a marker for oncogene-induced senescence (OIS). Left: P-ERK1/2 positivity was reduced in polyps (arrow “p”) compared to the strong signal in crypts and basal regions of villi in the adjacent mucosa (arrow “m”); Right: P21 was increased in apical tips of polyps and interspersed lamina propria cells. No difference was observed between polyps of the two genotypes (not shown). B, Quantitative analysis of IHC in A. Number of mice having high (scores +2,+3) or low (scores 0,+1) positivity in polyps vs. non-malignant mucosa (*p< 0.05, Fisher Exact test). C, IHC using Abs against B-cells (B220/CD45R), T-cells (CD3), Tregs (FOXP3) and macrophages (F4/80) was calculated as in B (*p< 0.05, Fisher Exact test). Macrophages remained high in polyps, T-cells were reduced.
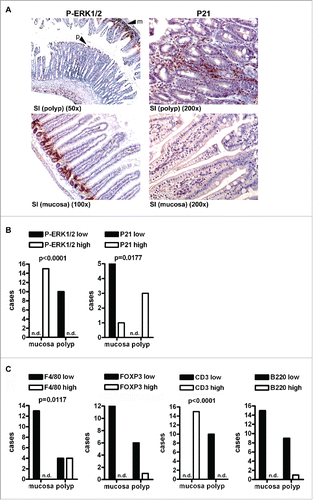
Immune evasion develops during progression of pre-malignant lesions to cancer. Therefore, we separately analyzed the immune cell infiltration of the normal non-malignant mucosa vs. polyps. Quantitative analysis of IHC () on tissue sections of RAS and RAS PPARΔIEC mice (n≥3 per genotype, *p < 0.05, Fisher Exact test) revealed that the staining positivity of macrophages (F4/80+) remained high in polyps, while T-cell (CD3+) counts were reduced therein, indicative of a potential immune evasion mechanism in the lesions (S3). Again, there was no difference between polyps of the two genotypes (not shown), indicating that PPARγ is sufficient for counteracting RAS-directed immune cell infiltration in the normal mucosa but is unable to prevent immune escape upon progression to adenoma.
RAS-mediated phenotypes are attenuated by treatment of mice with PPARγ-agonist
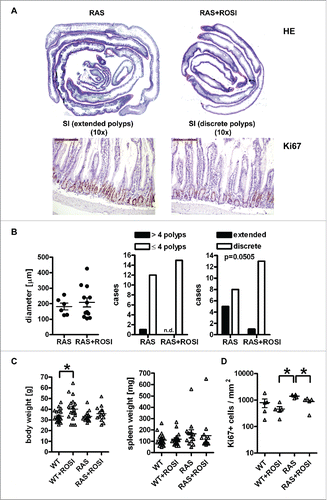
Based on the previous findings that genetic targeting of PPARγ reshapes mutant RAS-driven immune cell infiltration into the non-malignant intestinal mucosa, we next tested the reciprocal approach using pharmacological activation of PPARγ by rosiglitazone (rosi). WT and RAS mice were fed a chow diet enriched with rosi (n≥10 mice per treatment group and genotype) corresponding to an approx. dose of 25 mg/kg*day for 4 months.Citation20 Microscopic images of H&E staining in tissue sections visualized drug efficacy in “Swiss rolls” showing the complete SI with extended vs. discrete serrated polyps in chow and rosi-treated animals (). Quantitative analysis of H&E histopathology () failed to record any differences in transmural inflammation (n≥6 mice per group, n.s., Mann Whitney test). The number of cases (mice) having more or less than 4 polyps per total SI (n.s., Fisher Exact test) were not changed either. Instead, the number of mice having extended polyps per total SI (p = 0.0505, Fisher Exact test) showed a trend for reduction by rosi. Similar results were obtained if all genotypes were subjected to statistical comparison (S4 and Table S2). Notably, rosi did not unfold its weight gain adverse effect in mice with mutant RAS, as evident from the quantitative analysis of body and spleen weights (n≥10 per group, *p < 0.05 vs. ROSI, One-way ANOVA) (). IHC on tissue sections of WT, WT+ROSI, RAS and RAS+ROSI mice with Ab detecting Ki67 in the non-malignant mucosa and subsequent quantitative analysis () (n≥4 per group, *p < 0.05 vs. ROSI, Kruskal Wallis test) corroborated that PPARγ-agonist reduced proliferation in the small intestinal crypts. Conclusively, these data showed that pharmacological activation of PPARγ attenuated polyp formation in the murine SI.
Figure 4. RAS-mediated polyp load and proliferation are reduced by PPARγ-agonist. A, Microscopic images of H&E staining. Left: Swiss roll showing the complete SI from a chow-fed RAS mouse with extended serrated polyps; Right: rosi-fed RAS mouse. Magnification 10x. B, Quantitative analysis of H&E histopathology. Left: Transmural inflammation. Data are means ± S.E. (n≥6 mice per group, n.s., Mann Whitney test). Middle: Mice with more or less than 4 polyps per SI (n.s., Fisher Exact test). Right: Mice having discrete or extended polyps per SI (p = 0.0505, Fisher Exact test). C, Quantitative analysis of body and spleen weights. Data are means ± S.E. (n≥10 mice per treatment group and genotype, *p < 0.05 vs. ROSI, One-way ANOVA). D, IHC on tissue sections of RAS and RAS+ROSI mice with an Ab detecting the proliferation marker Ki67 in the non-malignant mucosa. Data are means ± S.E. (n≥4 per group, *p < 0.05 vs. ROSI, Kruskal Wallis test). Representative images are shown in A.
PPARγ-activation increases M1 macrophages in the murine small intestine
To assess the type of inflammation observed in the lamina propria of RAS mice, total RNA was extracted from snap-frozen mouse SI, cRNA was hybridized to cDNA microarrays, and differential gene expression was analyzed as described in.Citation21 Overall, genes related to inflammation and immunity were up-regulated in the SI tissue of RAS mice compared with WT animals (S5). To validate the array data, RT-qPCRs were conducted (n = 6 per genotype, *p < 0.05, Mann Whitney test) (). Notably, the M1 marker iNOS (Nos2) was decreased, while all other inflammation and immunity-related mRNAs tested were increased in RAS vs. WT mice, indicative of a chronic inflammatory microenvironment with immunosuppressive characteristics.
Figure 5. RAS-mediated phenotypes are attenuated by PPARγ-agonist. A, Immune marker gene expression in mouse tissues. Total RNA was extracted from snap-frozen SI of WT and RAS mice. CT-values from RT-qPCRs normalized to B2 m were calculated as -fold ± S.E. (n = 6 per genotype, *p < 0.05 WT vs. RAS for all genes, Mann Whitney test). B, IHC on tissue sections of RAS and RAS+ROSI mice using Abs detecting T/B cells and total macrophages in the non-malignant mucosa. Data are means ± S.E. (n = 3 per group, p = 0.100, Mann Whitney test). C, PPARγ-activation increases intestinal M1 macrophage numbers. IHC with Abs detecting subpopulations of macrophages. Data are presented as in B (n≥4 per group, *p < 0.05 vs. ROSI, Mann Whitney test).
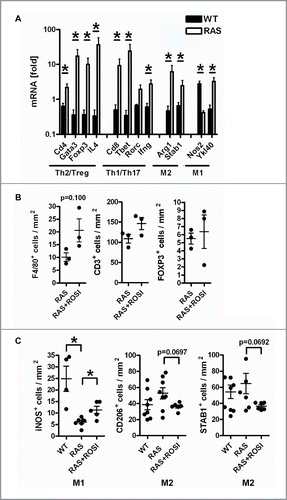
To investigate the impact of PPARγ-activation on the immune response profile, tissue sections of RAS and RAS+ROSI mice were then stained for IHC (). Quantitative analysis revealed a trend that lamina propria infiltration of macrophages (F4/80+) into the non-malignant mucosa was slightly increased by rosi (n = 3 per group, p = 0.100, Mann Whitney test), whereas T-cells remained unaffected. To further distinguish between immune cell subsets, tissue sections of RAS and RAS+ROSI mice were stained with Abs detecting CD4+ for Th and CD8+ for Tc cells and iNOS+ for M1 and STAB1+/CD206+ for M2 polarized macrophages (). Consistent with the above described RT-qPCR results, iNOS (M1) positivity was reduced in the non-malignant mucosa of RAS vs. WT mice. Conversely, lamina propria infiltration of M1 (iNOS+) macrophages into the non-malignant mucosa was increased by rosi (n≥4 per group, *p < 0.05 vs. ROSI, Mann Whitney test). Likewise, there was a trend for decreased M2 (CD206+/STAB1+) counts by rosi, whereas T-cell amounts were unchanged under all conditions. Representative images are shown in S6 and S7. These findings proposed that the RAS mutation supports an immune evasive M2-type, whereas PPARγ activation favors a “killer” M1-type microenvironment.
To determine, whether immune cell counts were also changed in mutant RAS-driven serrated polyps, quantitative analysis of IHC staining was conducted in RAS and RAS+ROSI mice (n≥3 mice per group and genotype, *p < 0.05 vs. ROSI, Mann Whitney test) (). Overall, the presence of CD8+ Tc (but not CD4+ Th) cells was reduced in polyps vs. the non-malignant mucosa, underscoring the existence of an immune evasion mechanism driven by mutant KRAS. Nonetheless, rosi could not restore T-cell numbers within polyps. Instead, a trend was stated for a rise in M1 (iNOS+) and a decrease in M2 (STAB1+) positivity by rosi. Taken together, PPARγ-agonist seems to rewire the M1/M2-type immune response by counteracting the effects of mutant KRAS.
Figure 6. Immune-related marker genes/proteins are differentially regulated by PPARγ-agonist. A, IHC on tissue sections of RAS and RAS+ROSI mice using Abs detecting subpopulations of T-cells (top) and macrophages (bottom) in polyps vs. the non-malignant mucosa. Data are means ± S.E. (n≥3 per group and genotype, *p < 0.05 vs. ROSI, Mann Whitney test). B, Total RNA was extracted from snap-frozen SI of RAS and RAS+ROSI mice. CT-values from RT-qPCRs normalized to B2 m were calculated as -fold ± S.E. (n = 9-10 per group, Mann Whitney test). Il4 (*p < 0.05) was reduced by rosi; there was a trend for Cxcl10 (p = 0.08) up-regulation.
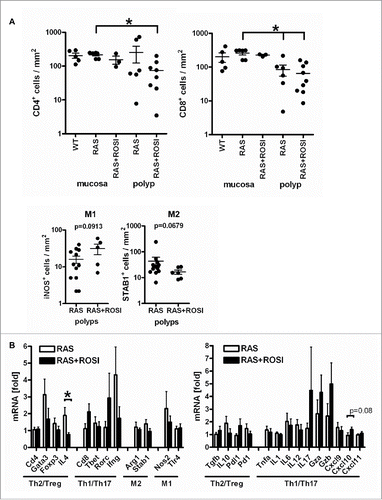
To characterize the inflammatory microenvironment in more detail, custom-made PCR arrays were designed (S8). Expression profiling of 84 genes involved in the communication between tumor cells and cells related to inflammation and immunity was conducted to reveal differential regulation of genes by the PPARγ-agonist. Total RNA was extracted from snap-frozen mouse SI from RAS and RAS+ROSI mice, and cDNA (n = 1 per group) was used as an input for the Mouse Cancer Inflammation & Immunity Crosstalk RT2 Profiler PCR Array as detailed in the Supplementary Methods. For hit validation of immune-related genes differentially regulated by PPARγ-agonist, total RNA was extracted from snap-frozen SI from RAS and RAS+ROSI mice () and subjected to RT-qPCR analysis (n = 9-10 per group, *p < 0.05, Mann Whitney test). Rosi significantly down-regulated the Th2 marker cytokine Il4 and exerted a trend to up-regulate the Th1 marker chemokine Cxcl10 (p = 0.08). As a positive control, induction of the PPARγ-target gene Aco was detectable as well (not shown). These data suggested that PPARγ participates in reprogramming of the host immune response against mutant RAS-driven oncogenic effects by enforcing a “killer”-type tumor-attacking mode of macrophages which may extend to T-cell subpopulations (please see model in ).
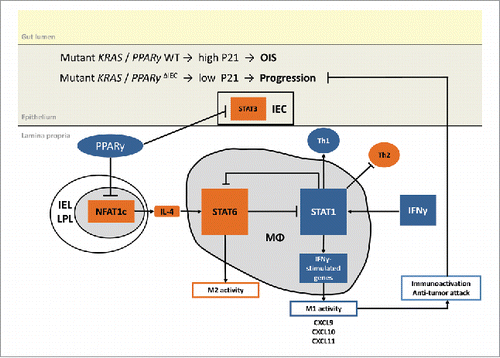
Figure 7. Proposed model of RAS and PPARγ crosstalk. Suggested mechanism based on published literature and own experimental data: PPARγ-activation reshapes the immune cell composition in the lamina propria from an immuno-suppressive towards a tumor-attacking profile. For example, PPARγ may reduce NFAT-driven Il4 (Th2/M2) gene expression to indirectly activate “killer” type responses (Th1/M1).
PPARγ-activation increases M1 markers in human THP1-derived macrophages
To test this hypothesis in an appropriate in vitro model, we employed human THP1-derived macrophages. Phorbolester (PMA) enhanced mRNA expression of markers for differentiation (CD14) and polarization to M1 (TNFA) and M2 (CD206) after treatment with cytokine cocktails as detailed in the Supplementary Methods (S9). Differentiated polarized macrophages (“M”) were then co-cultured with SW480 human CRC cells (“TU”) with and without rosi (“R”) (at 10μM) for 48h. RT-qPCRs were conducted on RNA extracted from both cell types. M1 but not M2 macrophages reduced mRNA expression for markers of proliferation (cyclin D1/CCND1), cell survival (BCL2), senescence (P21/CIP1) and metabolism (ACO) in co-cultured tumor cells (n = 3, *p < 0.05 M1 vs. M2, Two-way ANOVA). Consistent with our observation in mice, PPARγ-agonist also down-regulated M2 (CD163) but up-regulated M1(TLR4) marker expression in differentiated polarized macrophages co-cultured with tumor cells (n = 3, *p < 0.05 vs. rosi, Two-way ANOVA). In turn, tumor cells reduced M1 but not M2 marker expression in co-cultured macrophages. This reciprocal cross-talk provided additional experimental evidence that PPARγ-activation enforces an M1-dependent macrophages response leading to reduced tumor cell viability.
PPARγ-activation decreases Th2 response markers in murine primary intestinal lymphocytes
For further validation using an ex vivo primary cell model, lamina propria lymphocytes (LPLs) were isolated from SI of WT and RAS mice as detailed in the Supplementary Methods (S10). Flow cytometry (FC) confirmed the identity and purity of the LPL population. LPLs from WT mice were isolated and stained with Abs and viability dye (7AAD) as indicated in Table S3. First, lymphocytes were identified by gating based on forward and side scatter, followed by selection of single and live cells. CD3+ cells were selected, and CD4+ and CD8+ T-cell subpopulations identified and displayed as percentages from dot plots. LPLs were CD4+ (n = 7, *p < 0.05 CD4+ vs. negative, Kruskal Wallis test), whereas intraepithelial lymphocytes (IELs) were CD8+. RT-qPCRs demonstrated that LPLs from KRAS mutant animals displayed elevated marker mRNAs for the Th2 immune response compared with WT (n = 5 per genotype, *p < 0.05 WT vs. RAS, Two-way ANOVA), confirming our PCR results from mixed tissue samples. LPLs from WT mice (n = 6) were then incubated with or without rosi (10 μM) for 16h before RNA extraction. RT-qPCRs again showed that PPARγ-agonist decreased the mRNA levels of the Th2 cytokine Il4 ex vivo in isolated intestinal lymphocytes as before in situ in intestinal tissues of rosi-treated WT mice. A similar trend was obtained from lymphocytes isolated from spleens (n = 3). Taken together, PPARγ seems to be able to shape immune cell response profiles in tissue environments and isolated primary cells.
Discussion
In this study, we demonstrate that PPARγ-deficiency exacerbates mutant KRAS-driven formation of serrated polyps, a phenotype which is mitigated by treatment with PPARγ-agonist. At least one allele of intestinal epithelial PPARγ was necessary for viability of KRAS-mutant mice, underscoring the indispensable role of this nuclear receptor in embryonic development.Citation6 KO of both PPARγ alleles in myeloid cells also reduced the life span of KRAS-mutant mice, indicating that PPARγ in monocytes/macrophages and related cell types (e.g. dendritic cells and granulocytes) is important for health in adult animals. These survival data pointed at a functional crosstalk of epithelial driver mutations with drugable nuclear receptors across tissue compartments. We therefore asked whether PPARγ is able to protect against mutant KRAS-imposed oncogenic stress in the intestine.
Mutant KRAS triggers recruitment of innate immune cellsCitation22 and TregsCitation23 to mouse organs including the pancreas and lung facilitating carcinogenesis within an immunosuppressive microenvironment. Likewise, epithelialCitation24 and T cell-specific KOCitation25 of PPARγ in mice reshapes the immune response and exacerbates experimental colitis. Macrophage-specific KO of PPARγ yielded corresponding results in other tissues (e.g. liverCitation26 and lungCitation27). Consistent with these findings, we observed that KRAS-mutant mice with epithelial PPARγ-deficiency suffered from infiltration of T-cells, both total CD3+ lymphocytes and FOXP3+ Tregs, and F4/80+ macrophages into the lamina propria of the non-malignant, yet hyperplastic, mucosa of the SI, resembling an IBD-like chronic inflammation.
To test if oncogene-induced chronic inflammation can be prevented, we activated PPARγ systemically in mutant KRAS mice by a pharmacological approach as opposed to its cell-restricted genetic deletion in epithelial and myeloid compartments. Unexpectedly, long-term dietary administration of the PPARγ-ligand rosiglitazone (25mg/kg*d for 4 months) to KRAS-mutant mice did not impact on pan-T (CD3+), Th/Tc (CD4+/CD8+) or Treg (FOXP3+) cell infiltration rates, but rather augmented total (F4/80+) macrophages and M1 (iNOS+) counts at the expense of M2 (CD206+/STAB1+) cell numbers. Hence, PPARγ-activation seems to enforce an immune “switch” in the lamina propria, favoring a “tumor-attacking” rather than an immunosuppressive, tumor-promoting microenvironment.
In line with previous reportsCitation4 and the recent molecular classification of human CRC,Citation5 mutant KRAS-animals not only developed chronic inflammation but also multiple serrated polyps associated with oncogene-induced senescence (OIS). ERK1/2 phosphorylation was strongly reduced within basal (crypt) and apical (villus) serrated regions of polyps accompanied by an increase of the cyclin-dependent kinase inhibitor (CDKI) P21(WAF1/CIP1), indicative of a senescence barrier that protects against progression to carcinoma. Notably, macrophages remained within polyps and expressed both M1/M2 markers. Conversely, T-cells were excluded from serrated lesions as compared with the adjacent normal, yet hyperplastic, small intestinal mucosa. In human CRC, KRAS mutations correlate with MYC-dependent up-regulation of sirtuin-1 (SIRT1),Citation28 a deacetylase of P53 and PPARγ, leading to their inactivation. Both, mutant RASCitation4 and PPARγCitation6 up-regulate P21 and promote senescence in mice, and P53-mediated cell cycle checkpoints are often by-passed due to mutation during progression of adenoma to carcinoma. We may thus conclude that mutant KRAS-driven serrated polyps harbor a senescence barrier, which consists of OIS leading to up-regulation of CDKIs and immunological escape evident by removal of CD8+ T-cells and retention of macrophages within the lesions. However, the underlying molecular players which connect genetic and immune aspects of senescence (e.g. SASP) remain to be defined. Notably, the PPARγ-agonist did not recover T-cell counts within serrated polyps, but instead tipped the balance from a M2/Th2 towards a M1/Th1 marker profile both in polyps and the pre-malignant mucosa and reduced overall polyp load in the SI. Addressing PPARγ may thus be a strategy for prevention or intervention of inflammation-associated malignancies.
PPARγ has been firmly established as an anti-inflammatory factor, e.g. by epigenetic trans-repression of NFκB/AP1-dependent pro-inflammatory genes in immune cells,Citation29 and its agonists exert (pre)clinical benefit in IBDCitation6 and other inflammation-associated conditions (i.e. atherosclerosis, asthma, metabolic syndrome). PPARγ is also a master regulator of macrophage differentiation, polarization and function towards the M2 phenotype,Citation9 e.g. in resident macrophages in the white adipose tissue of diabetic miceCitation30 and in murine and human tumor-associated macrophages (TAMs).Citation31 The presence of TAMs usually predicts poor prognosis in cancer patients,Citation32 whereas epithelial PPARγ expression correlates with improved survival in human CRC,Citation33 indicative of compartment-specific function of PPARγ in enterocytes vs. macrophages. This concept was further supported by studies in lungCitation34 and breastCitation35 cancer mouse models.
In contrast, recent research underscored a more versatile role for PPARγ in context- and cell type-dependent reprogramming of the immune response. For example, pathogen elimination is facilitated by PPARγ-polarized macrophages via enhanced phagocytosis and release of NO, reactive oxygen species, cytotoxic cytokines and altered glucose metabolism (e.g. against Pseudomonas,Citation36 Mycobacterium,Citation37 Trypanosoma,Citation38 Leishmania,Citation39 PlasmodiumCitation40 and CandidaCitation41) in mice and in patients with chronic granulomatous disease (CGD).Citation42 Non-self recognition of tumors as an “internal pathogen” may be exploited by the same mechanism. PPARγ-activation by agonists reverses M2-mediated cytotoxic T-lymphocyte (CTL) suppressionCitation43 in favour of M1-dependent phagocytosis and killing activities. Specifically, this treatment augmented removal of apoptotic and dead cells by efferocytosis.Citation44 Conversely, inhibition of PPARγ (by overexpression of a dominant-negative mutant) in myeloid-lineage cells induced systemic inflammation, immune-suppression and tumorigenesis,Citation45 confirming our data using genetic deletion of PPARγ. This fine-tuning of challenge clearance by a delicate balance of M1/M2-type macrophages activities contributes to the resolution of inflammation and prevents chronic tissue damage, and may thus counteract transition to malignancy, which can otherwise be regarded as “a wound that never heals”.
Based on the existing literature and our data (), we draw the following model: PPARγ activation reduces expression of the Th2 cytokine IL4 (e.g. via transrepression of NFATCitation29) in LPLs leading to defective STAT3/6 signaling in IL4-target cells, impairing Th2/M2 differentiation and polarisation, thereby indirectly favoring the development of Th1/M1 “killer” cell phenotypes. PPARγ in IECs inhibits proliferation and promotes senescence together with RAS in a cell-intrinsic fashion,Citation4 thus counteracting the progression of benign adenomas to cancer. The cross-talk between macrophages and enterocytes may comprise additional soluble factors such as CXCL10, a marker chemokine of the host's Th1-orientated immune response induced by PPARγ-agonist and a prognostic factor in cancer patients.Citation46 Pro- and anti-inflammatory cytokines in turn regulate PPARγ expression and activities, corroborating mutual cellular communication networks.Citation29
Additional experiments have to specify the nature of PPARγ-target cell subpopulations for anti-diabetic insulin-sensitizers of the glitazone class within the complex mucosa-associated intestinal immune system that differs considerably between rodents and humans. In sum, our preclinical data propose that PPARγ-activation reprograms the host's immune response against mutant KRAS-driven oncogenic effects by enforcing “killer”-type macrophages. The results in mice were statistically significant, but the magnitude of the biologic effect renders the biologic significance for translation to humans uncertain and may limit applications in patients. Nonetheless, our work may relate to a possible clinical relevance. Cancer immunotherapies are designed to restructure the immunosuppressive tumor microenvironment towards a tumor-attacking mode. A high “immunoscore”, i.e. infiltration of CTLs and immunogenicity markers (e.g. MHC and B7 molecules), predicts favorable prognosis in CRC.Citation47 However, clear endpoints and response prediction markers for checkpoint inhibitor Abs are lacking. Current strategies against T-cell/tumor interfaces may be complemented with macrophage-based therapies, and repurposing and combination with established drugs already approved for other diseases is encouraged.Citation48 Therein, PPARγ-agonists may have a perspective as potential candidates for future immunotherapies.
Methods
Animals
Wildtype (WT) C57BL6/J and transgenic (tg) mice with an activating human KRASG12V mutation were obtained from Charles River (Lyon, France) and KPJ,Citation3 respectively. Floxed and Cre transgenic mice were from Jackson Labs (Bar Harbor, ME). Intestine-specific disruption of Pparg gene function was achieved by excision of loxp sites (abbrev. FL) flanking exons 1 and 249 followed by interbreeding with mice transgenic for Cre recombinaseCitation50 and KRASG12VCitation3 both under the control of the murine villin promoter in intestinal epithelial cells (enterocytes) [C57BL6/J-Tg(Vil-KRAS*G12V)Robine/J x B6.129-Ppargtm2Rev/JxC57 BL/6-Tg(Vil-cre)Robine/J]. Macrophage-specific deletion of Pparg was achieved by using mice expressing Cre recombinase under the murine lysozyme promoterCitation51 [C57BL6/J-Tg(Vil-KRAS*G12V)Robine/J x B6.129-Ppargtm2Rev/J x B6.129 P2-Lyz2tm1(cre)Ifo/J]. Nomenclature and abbreviations for genotype combinations are listed in Table S1. Mice were housed in a specific pathogen-free animal facility on a 12 h day/night light cycle and fed water and chow diet ad libitum. For experimental studies, chow diet was supplemented with 0.02% (w/w) rosiglitazone maleate (Altromin, Lage, Germany) for 4 months (∼25 mg/kg*day) as published previously.Citation20 All experiments were conducted in agreement with ethical approvals from the governmental authorities of Baden-Württemberg, Karlsruhe, Germany (35-9185.82/G-176/12, 35–9185.81/G-146/15).
Reagents, primers and antibodies
Additional information is provided in Supplementary Methods and Tables S3/S4.
Immunohistochemistry (IHC)
Ab and hematoxylin & eosin (H&E) stainings on formalin-fixed paraffin-embedded (FFPE) tissue sections were done as detailed in.Citation52 Antigen retrieval was performed by heating in antigen unmasking solution H-3000 (Vector Laboratories, Burlingame, USA) and blocking with H2O2, if not stated otherwise. Abs were diluted as detailed in Table S3, and staining was processed as recommended by the Vectastain ABC kit (Vectorlabs). The substrate 3,3′-diamino benzidine (DAB) (Vectorlabs) was employed for detection followed by hematoxylin counterstaining. Frequency and intensity of staining positivity was determined in epithelial and lamina propria (stroma) cells. The staining scores were defined as: 0+ = negative (0–25%), 1+ = weak (25–50%), 2+ = moderate (50–75%), 3+ = strong (75–100%). Percentage was calculated as IHC positive cells divided by total number of cells within the field. Signals were quantified observer-blinded at a standard bright-field microscope using Image J (imagej.nih.gov/ij) (n >20 signals per field; n = 5 fields per image). The field area was defined by the morphology of a given crypt-villus unit and measured in square mm (mm2).
Software and statistics
Data from in vivo and in vitro studies are displayed as means ± S.E. from at least 3 independent experiments from different cell passages or individuals (frozen samples from mice). The cut-off values for dichotome analysis of IHC staining scores (0 to 3+) were calculated as ≥2.0 for tumor and stroma positivity.Citation52 Optical densities (OD) of bands in gels from Western blots and PCRs were measured using automated imaging devices and quantified with Image J (imagej.nih.gov/ij). Data were normalized to house keeping genes or proteins as indicated in the legends to figures and calculated as -fold or % compared to control. Statistical analysis was done with Graphpad Prism (version 4.0, La Jolla, CA). All tests were unpaired and two-sided if not stated otherwise. P-values < 0.05 were considered significant (*).
Abbreviations
| Ab | = | antibody |
| ACF | = | aberrant crypt foci |
| Ad | = | adenoma |
| APC | = | allophycocyanin |
| BSA | = | bovine serum albumin |
| CD | = | cluster of differentiation |
| Cre | = | recombinase |
| CTL | = | cytotoxic T-lymphocyte |
| CRC | = | colorectal cancer |
| ERK | = | extracellular signal-regulated protein kinase |
| FC | = | flow cytometry |
| FCS | = | fetal calf serum |
| FL | = | floxed gene (loxp site inserted) |
| FFPE | = | formalin-fixed and paraffin-embedded |
| FITC | = | fluorescein isothiocyanate |
| G | = | tumor grade |
| GI | = | gastrointestinal |
| GSEA | = | gene set enrichment analysis |
| H&E | = | hematoxylin and eosin |
| HPP | = | hyperplastic polyp |
| IEC | = | intestinal epithelial cells |
| IEL | = | (intestinal) intraepithelial lymphocytes |
| IL | = | interleukin |
| IFN | = | interferon |
| IHC | = | immunohistochemistry |
| LPL | = | lamina propria lymphocytes |
| Lyz | = | lysozyme |
| LPS | = | lipopolysaccharide |
| M1/2 | = | macrophage polarization type 1/2 |
| NT | = | normal (intestinal) tissue |
| MAPK | = | mitogen-activated protein kinase |
| MEK | = | MAPK kinase |
| MPH/Mph | = | macrophage |
| OD | = | optical density |
| OIS | = | oncogene-induced senescence |
| P21 | = | cyclin-dependent kinase inhibitor (WAF1/CIP1) |
| PE | = | phycoerythrin |
| PMA | = | phorbol 12-myristate 13-acetate |
| PPARγ | = | peroxisome proliferator-activated receptor gamma |
| PPARΔIEC | = | Pparg FL/+//FL/FL x VilCretg |
| PPARΔMPH | = | Pparg FL/+//FL/FL x LyzCretg |
| RAS (gene name) | = | Rous sarcoma oncogene |
| RAS | = | (mouse genotype) VilKRASG12Vtg |
| RAS | = | PPARΔIEC VilKRASG12Vtg x Pparg FL/+ x VilCretg |
| RAS | = | PPARΔMPH VilKRASG12Vtg x Pparg FL/+//FL/FL x LyzCretg |
| RAS+ROSI | = | RAS mice treated with rosi ROSI rosiglitazone |
| RTK | = | receptor tyrosine kinase |
| SASP | = | senescence-associated secretory phenotype |
| SCF | = | subcellular fractionation |
| SI | = | small intestine |
| (S)SA | = | (sessile) serrated adenoma |
| T2DM | = | type 2 diabetes mellitus |
| TCL | = | total cell lysate |
| tg | = | transgene |
| Tc | = | cytotoxic T-cells |
| Th | = | helper T-cells |
| Treg | = | regulatory T-cells |
| TNF | = | tumor necrosis factor |
| TU | = | tumor tissue |
| Vil | = | Villin |
| WT | = | wildtype |
| WT | = | (mouse genotype) Pparg +/+//FL/+//FL/FL |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Grant support
EB received funding from the Deutsche Krebshilfe (#108287, #111086), the Deutsche Forschungsgemeinschaft (Bu2285) and the German Cancer Research Center (DKFZ-MOST, Ca158). TG was funded by the intramural “Translational Physician Scientist” (TraPS) program (Medical Faculty Mannheim, University Heidelberg). ME was supported by the State of Baden-Württemberg for “Center of Geriatric Biology and Oncology (ZOBEL) – Perspektivförderung” and “Biology of Frailty – Sonderlinie Medizin”.
Transcript profiling
Data from cDNA microarrays were deposited at Gene Expression
Omnibus (GEO) with the accession number GSE47772 under the following link:https://www.ncbi.nlm.nih.gov/gds/?term = GSE47772[Accession]
Author contributions
All authors cooperated and contributed to, critically reviewed and approved the manuscript. EB, WR and ME defined the research theme. TG, CW, PW, FH, SH, TF, SY and KPJ designed methods and carried out the experiments. EB, TG, CW, PW, SH and KPJ analyzed the data and interpreted the results. EB wrote the paper. KPJ provided mouse strains and performed bioinformatic analysis. TiG and JK provided, analyzed and interpreted immunostainings.
supp_data_1423168.docx
Download MS Word (45 KB)Acknowledgments
We want to thank Sandra Schneider and Karin Kaiser for excellent technical assistance.
Additional information
Funding
References
- de Souza HS, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. 2016;13:13–27. doi:10.1038/nrgastro.2015.186. PMID:26627550
- Normanno N, Tejpar S, Morgillo F, De Luca A, Van Cutsem E, Ciardiello F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat Rev Clin Oncol. 2009;6:519–27. doi:10.1038/nrclinonc.2009.111. PMID:19636327
- Janssen KP, el-Marjou F, Pinto D, Sastre X, Rouillard D, Fouquet C, Soussi T, Louvard D, Robine S. Targeted expression of oncogenic K-ras in intestinal epithelium causes spontaneous tumorigenesis in mice. Gastroenterology. 2002;123:492–504. doi:10.1053/gast.2002.34786. PMID:12145803
- Bennecke M, Kriegl L, Bajbouj M, Retzlaff K, Robine S, Jung A, Arkan MC, Kirchner T, Greten FR. Ink4 a/Arf and oncogene-induced senescence prevent tumor progression during alternative colorectal tumorigenesis. Cancer Cell. 2010;18:135–46. doi:10.1016/j.ccr.2010.06.013. PMID:20708155
- Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–6. doi:10.1038/nm.3967. PMID:26457759
- Peters JM, Shah YM, Gonzalez FJ. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat Rev Cancer. 2012;12:181–95. PMID:22318237
- Bouguen G, Langlois A, Djouina M, Branche J, Koriche D, Dewaeles E, Mongy A, Auwerx J, Colombel JF, Desreumaux P. Intestinal steroidogenesis controls PPARgamma expression in the colon and is impaired during ulcerative colitis. Gut. 2015;64:901–10. doi:10.1136/gutjnl-2014-307618. PMID:25053717
- Peyrin-Biroulet L, Beisner J, Wang G, Nuding S, Oommen ST, Kelly D, Parmentier-Decrucq E, Dessein R, Merour E, Chavatte P. Peroxisome proliferator-activated receptor gamma activation is required for maintenance of innate antimicrobial immunity in the colon. Proc Natl Acad Sci U S A. 2010;107:8772–7. doi:10.1073/pnas.0905745107. PMID:20421464
- Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–61. doi:10.1038/nri3088. PMID:22025054
- Klotz L, Burgdorf S, Dani I, Saijo K, Flossdorf J, Hucke S, Alferink J, Nowak N, Beyer M, Mayer G. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J Exp Med. 2009;206:2079–89. doi:10.1084/jem.20082771. PMID:19737866
- Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, Mathis D. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486:549–53. doi:10.1038/nature11132. PMID:22722857
- Luo C, Lim JH, Lee Y, Granter SR, Thomas A, Vazquez F, Widlund HR, Puigserver P. A PGC1alpha-mediated transcriptional axis suppresses melanoma metastasis. Nature. 2016;537:422–426. doi:10.1038/nature19347. PMID:27580028
- Torrano V, Valcarcel-Jimenez L, Cortazar AR, Liu X, Urosevic J, Castillo-Martin M, Fernández-Ruiz S, Morciano G, Caro-Maldonado A, Guiu M. The metabolic co-regulator PGC1alpha suppresses prostate cancer metastasis. Nat Cell Biol. 2016;18:645–56. doi:10.1038/ncb3357. PMID:27214280
- Geach T. NASH: Pioglitazone safe and effective for treating T2DM in patients with NASH. Nat Rev Endocrinol. 2016;12:498.
- Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M, Guarino PD, Lovejoy AM, Peduzzi PN, Conwit R. Pioglitazone after ischemic stroke or Transient Ischemic attack. N Engl J Med. 2016;374:1321–31. doi:10.1056/NEJMoa1506930. PMID:26886418
- Prost S, Relouzat F, Spentchian M, Ouzegdouh Y, Saliba J, Massonnet G, Beressi JP, Verhoeyen E, Raggueneau V, Maneglier B. Erosion of the chronic myeloid leukaemia stem cell pool by PPARgamma agonists. Nature. 2015;525:380–3. doi:10.1038/nature15248. PMID:26331539
- Banks AS, McAllister FE, Camporez JP, Zushin PJ, Jurczak MJ, Laznik-Bogoslavski D, Shulman GI, Gygi SP, Spiegelman BM. An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature. 2015;517:391–5. doi:10.1038/nature13887. PMID:25409143
- Kim JJ, Shajib MS, Manocha MM, Khan WI. Investigating intestinal inflammation in DSS-induced model of IBD. J Vis Exp. 2012;60:3678–83. doi:10.3791/3678. PMID:22331082
- Erben U, Loddenkemper C, Doerfel K, Spieckermann S, Haller D, Heimesaat MM, Zeitz M, Siegmund B, Kühl AA. A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int J Clin Exp Pathol. 2014;7:4557–76. PMID:25197329
- Friedrich T, Richter B, Gaiser T, Weiss C, Janssen KP, Einwächter H, Schmid RM, Ebert MP, Burgermeister E. Deficiency of caveolin-1 in Apc(min/+) mice promotes colorectal tumorigenesis. Carcinogenesis. 2013;34:2109–18. doi:10.1093/carcin/bgt142. PMID:23640045
- Abal M, Obrador-Hevia A, Janssen KP, Casadome L, Menendez M, Carpentier S, Barillot E, Wagner M, Ansorge W, Moeslein G. APC inactivation associates with abnormal mitosis completion and concomitant BUB1B/MAD2L1 up-regulation. Gastroenterology. 2007;132:2448–58. doi:10.1053/j.gastro.2007.03.027. PMID:17570218
- Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–47. doi:10.1016/j.ccr.2012.04.024. PMID:22698407
- Granville CA, Memmott RM, Balogh A, Mariotti J, Kawabata S, Han W, Lopiccolo J, Foley J, Liewehr DJ, Steinberg SM. A central role for Foxp3+ regulatory T cells in K-Ras-driven lung tumorigenesis. PLoS One. 2009;4:e5061. doi:10.1371/journal.pone.0005061. PMID:19330036
- Adachi M, Kurotani R, Morimura K, Shah Y, Sanford M, Madison BB, Gumucio DL, Marin HE, Peters JM, Young HA. Peroxisome proliferator activated receptor gamma in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut. 2006;55:1104–13. doi:10.1136/gut.2005.081745. PMID:16547072
- Guri AJ, Mohapatra SK, Horne WT, 2nd, Hontecillas R, Bassaganya-Riera J. The role of T cell PPAR gamma in mice with experimental inflammatory bowel disease. BMC Gastroenterol. 2010;10:60. doi:10.1186/1471-230X-10-60. PMID:20537136
- Moran-Salvador E, Titos E, Rius B, González-Périz A, García-Alonso V, López-Vicario C, Miquel R, Barak Y, Arroyo V, Clària J. Cell-specific PPARgamma deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells. J Hepatol. 2013;59:1045–53. doi:10.1016/j.jhep.2013.06.023. PMID:23831119
- Malur A, McCoy AJ, Arce S, Barna BP, Kavuru MS, Malur AG, Thomassen MJ. Deletion of PPAR gamma in alveolar macrophages is associated with a Th-1 pulmonary inflammatory response. J Immunol. 2009;182:5816–22. doi:10.4049/jimmunol.0803504. PMID:19380830
- Kriegl L, Vieth M, Kirchner T, Menssen A. Up-regulation of c-MYC and SIRT1 expression correlates with malignant transformation in the serrated route to colorectal cancer. Oncotarget. 2012;3:1182–93. doi:10.18632/oncotarget.628. PMID:23045412
- Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10:365–76. doi:10.1038/nri2748. PMID:20414208
- Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–20. doi:10.1038/nature05894. PMID:17515919
- Pello OM, De Pizzol M, Mirolo M, Soucek L, Zammataro L, Amabile A, Doni A, Nebuloni M, Swigart LB, Evan GI. Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood. 2012;119:411–21. doi:10.1182/blood-2011-02-339911. PMID:22067385
- Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, Zhao YW, Wei YQ. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One. 2012;7:e50946. doi:10.1371/journal.pone.0050946. PMID:23284651
- Ogino S, Shima K, Baba Y, Nosho K, Irahara N, Kure S, Chen L, Toyoda S, Kirkner GJ, Wang YL. Colorectal cancer expression of peroxisome proliferator-activated receptor gamma (PPARG, PPARgamma) is associated with good prognosis. Gastroenterology. 2009;136:1242–50. doi:10.1053/j.gastro.2008.12.048. PMID:19186181
- Li H, Sorenson AL, Poczobutt J, Amin J, Joyal T, Sullivan T, Crossno JT Jr, Weiser-Evans MC, Nemenoff RA. Activation of PPARgamma in myeloid cells promotes lung cancer progression and metastasis. PLoS One. 2011;6:e28133. doi:10.1371/journal.pone.0028133. PMID:22145026
- Avena P, Anselmo W, Whitaker-Menezes D, Wang C, Pestell RG, Lamb RS, Hulit J, Casaburi I, Andò S, Martinez-Outschoorn UE. Compartment-specific activation of PPARgamma governs breast cancer tumor growth, via metabolic reprogramming and symbiosis. Cell Cycle. 2013;12:1360–70. doi:10.4161/cc.24289. PMID:23574724
- Bedi B, Yuan Z, Joo M, Zughaier SM, Goldberg JB, Arbiser JL, Hart CM, Sadikot RT. Enhanced clearance of Pseudomonas aeruginosa by Peroxisome Proliferator-Activated receptor Gamma. Infect Immun. 2016;84:1975–85. doi:10.1128/IAI.00164-16. PMID:27091928
- Kogiso M, Shinohara T, Dorey CK, Shibata Y. Role of PPARgamma in COX-2 activation in mycobacterial pulmonary inflammation. Inflammation. 2012;35:1685–95. doi:10.1007/s10753-012-9486-x. PMID:22696146
- Penas F, Mirkin GA, Vera M, Cevey Á, González CD, Gómez MI, Sales ME, Goren NB. Treatment in vitro with PPARalpha and PPARgamma ligands drives M1-to-M2 polarization of macrophages from T. cruzi-infected mice. Biochim Biophys Acta. 2015;1852:893–904. doi:10.1016/j.bbadis.2014.12.019. PMID:25557389
- Lefevre L, Gales A, Olagnier D, Bernad J, Perez L, Burcelin R, Valentin A, Auwerx J, Pipy B, Coste A. PPARgamma ligands switched high fat diet-induced macrophage M2b polarization toward M2 a thereby improving intestinal Candida elimination. PLoS One. 2010;5:e12828. doi:10.1371/journal.pone.0012828. PMID:20877467
- Serghides L, Kain KC. Peroxisome proliferator-activated receptor gamma-retinoid X receptor agonists increase CD36-dependent phagocytosis of Plasmodium falciparum-parasitized erythrocytes and decrease malaria-induced TNF-alpha secretion by monocytes/macrophages. J Immunol. 2001;166:6742–8. doi:10.4049/jimmunol.166.11.6742. PMID:11359831
- Diaz-Gandarilla JA, Osorio-Trujillo C, Hernandez-Ramirez VI, Talamás-Rohana P. PPAR activation induces M1 macrophage polarization via cPLA(2)-COX-2 inhibition, activating ROS production against Leishmania mexicana. Biomed Res Int. 2013;2013:215283. doi:10.1155/2013/215283. PMID:23555077
- Fernandez-Boyanapalli RF, Frasch SC, Thomas SM, Malcolm KC, Nicks M, Harbeck RJ, Jakubzick CV, Nemenoff R, Henson PM, Holland SM. Pioglitazone restores phagocyte mitochondrial oxidants and bactericidal capacity in chronic granulomatous disease. J Allergy Clin Immunol. 2015;135:517–527 e12. doi:10.1016/j.jaci.2014.10.034. PMID:25498313
- Van Ginderachter JA, Meerschaut S, Liu Y, Brys L, De Groeve K, Hassanzadeh Ghassabeh G, Raes G, De Baetselier P. Peroxisome proliferator-activated receptor gamma (PPARgamma) ligands reverse CTL suppression by alternatively activated (M2) macrophages in cancer. Blood. 2006;108:525–35. doi:10.1182/blood-2005-09-3777. PMID:16527895
- Fernandez-Boyanapalli RF, Falcone EL, Zerbe CS, Marciano BE, Frasch SC, Henson PM, Holland SM, Bratton DL. Impaired efferocytosis in human chronic granulomatous disease is reversed by pioglitazone treatment. J Allergy Clin Immunol. 2015;136:1399–401 e1-3. doi:10.1016/j.jaci.2015.07.034.
- Wu L, Yan C, Czader M, Foreman O, Blum JS, Kapur R, Du H. Inhibition of PPARgamma in myeloid-lineage cells induces systemic inflammation, immunosuppression, and tumorigenesis. Blood. 2012;119:115–26. doi:10.1182/blood-2011-06-363093. PMID:22053106
- Karin N, Wildbaum G. The role of Chemokines in shaping the balance between CD4(+) T cell subsets and its Therapeutic implications in Autoimmune and cancer diseases. Front Immunol. 2015;6:609. doi:10.3389/fimmu.2015.00609. PMID:26648938
- Dolgin E. Cancer immunology community seeks better end points. Nat Rev Drug Discov. 2016;15:807–809. doi:10.1038/nrd.2016.254. PMID:27895329
- Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, Mueller B, Quaratino S, Sabatos-Peyton C, Petruzzelli L. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer. 2017;17:286–301. doi:10.1038/nrc.2017.17. PMID:28338065
- He W, Barak Y, Hevener A, Olson P, Liao D, Le J, Nelson M, Ong E, Olefsky JM, Evans RM. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A. 2003;100:15712–7. doi:10.1073/pnas.2536828100. PMID:14660788
- el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–93. doi:10.1002/gene.20042. PMID:15282745
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–77. doi:10.1023/A:1008942828960. PMID:10621974
- Friedrich T, Sohn M, Gutting T, Janssen KP, Behrens HM, Röcken C, Ebert MPA, Burgermeister E. Subcellular compartmentalization of docking protein-1 contributes to progression in colorectal cancer. EBioMedicine. 2016;8:159–72. doi:10.1016/j.ebiom.2016.05.003. PMID:27428427